

Abstract
Circulating levels of soluble forms of urokinase-type plasminogen activator receptor (suPAR) are generally elevated in sera from children and adults with FSGS compared with levels in healthy persons or those with other types of kidney disease. In mice lacking the gene encoding uPAR, forced increases in suPAR concentration result in FSGS-like glomerular lesions and proteinuria. However, whether overexpression of suPAR, per se, contributes to the pathogenesis of FSGS in humans remains controversial. We conducted an independent set of animal experiments in which two different and well characterized forms of recombinant suPAR produced by eukaryotic cells were administered over the short or long term to wild-type (WT) mice. In accordance with the previous study, the delivered suPARs are deposited in the glomeruli. However, such deposition of either form of suPAR in the kidney did not result in increased glomerular proteinuria or altered podocyte architecture. Our findings suggest that glomerular deposits of suPAR caused by elevated plasma levels are not sufficient to engender albuminuria.
Keywords: podocyte, glomerulosclerosis, pathophysiology of renal disease and progression, nephrotic syndrome, proteinuria
During the 1970s, the first cases of kidney transplantation revealed some peculiar forms of idiopathic FSGS, the prognosis of which was worsened by the immediate occurrence or recurrence of nephrotic syndrome.1 Forty years later, the precise molecular mechanisms responsible for the recurrence of FSGS in kidney transplant recipients remain essentially unknown. In the Buffalo/Mna rat strain, recurrence is germline-encoded.2,3 In humans, circulating permeability factors have been proposed as causative agents for the recurrence of FSGS in the kidney allografts.4,5 Recently, a soluble form of the urokinase-type plasminogen activator receptor (suPAR) has been implicated in the pathogenesis of FSGS.4,5 This original proposition is founded on the following key observations: (1) serum concentration of suPAR is higher in patients with FSGS, including those with recurrent FSGS, than in healthy controls or patients with other types of kidney diseases5 and (2) an induced increase in the concentration of suPAR in mice results in suPAR deposition in the glomeruli, activation of β3 integrin on podocytes, albuminuria, and foot process effacement.5 These processes were also reported to be independent of the cognate protease ligand of urokinase-type plasminogen activator receptor (uPAR), urokinase.5
A recent retrospective analysis of two independent patient cohorts in North America and in Europe has substantiated the association between high circulating levels of suPAR and FSGS.6 High circulating levels of suPAR, however, have been observed in other human diseases, but without the FSGS phenotype (e.g., elevated suPAR is a general biomarker for poor prognosis in many pathologic conditions, including some cancers).7,8 In addition, Maas et al. compared serum uPAR levels in patients with FSGS with levels in minimal change diseases and failed to identify a significant association between levels and diagnosis.9 Although this investigation may have lacked statistical power,10 an independent study by Bock et al.11 recently reached the same conclusion. In view of the conflicting evidence in the literature, the involvement of suPAR in FSGS is controversial.10
The putative role of suPAR in the pathogenesis of FSGS is clinically relevant. In an attempt to substantiate this causal relationship, we established robust mouse models using well defined purified murine suPAR preparations and investigated the effect of administration of suPAR on glomerular pathology and induction of albuminuria.
Results
We infused two different soluble forms of uPAR in wild-type (WT) C57BL/6J mice: a structurally and functionally well characterized monomeric mouse uPAR produced in eukaryotic S2-cells12,13 and a commercially available uPAR-Fc chimera (mouse uPAR linked to a human IgG1, purchased from R&D Systems). The uPAR-Fc used in this study is identical to the one originally used to demonstrate the effect of administered suPAR on glomerular pathology and induction of proteinuria.5 We also used doses of suPAR that included the 20-µg dose used in that study,5 but we also escalated the dosing by several orders. We delivered suPAR via intravenous boluses of suPAR (20 µg per mouse; Figure 1B), far exceeding the normal physiologic range in humans (median range, 2 ng/ml). Although we did observe suPAR deposition in the glomeruli using confocal microscopy (Figure 1E), our administration of suPAR failed to induce an increase in proteinuria in mice at 24 hours as evaluated by ELISA (Figure 1C) or by polyacrylamide gel electrophoresis (Figure 1, A, B, and D). Similar lack of effect of suPAR administration was also observed when 129S2svPas mice were subjected to this treatment or when the intravenously administered dose was escalated to 100 µg of suPAR and the effect there of was monitored noninvasively (Figure 1A, left). Results were similar when more invasive monitoring was used (Figure 1D) spanning a 3-hour period. Of note, podocyte structure (in C57BL6J mice) remained pristine 24 hours after suPAR administration of either of the two purified recombinant uPAR forms, as evaluated by electron microscopy (Figure 1, F and G).
Figure 1.

Intravenous injections of two different forms of eukaryotic recombinant mouse suPAR in wild-type (WT) mice (C57BL/6J or 129S2SvPasCrl, females 8–12 weeks old) do not cause foot process effacement or significant proteinuria despite the deposition of both recombinant proteins on glomerular structures. (A) Left: SDS-PAGE migration of purified recombinant mouse uPAR proteins (1 µg per lane) expressed by eukaryote cells: monomeric mouse uPAR produced in S2 cells (m-suPAR) and a commercially available uPAR-Fc chimera used in the study by Wei et al.5 The electrophoretic mobility of m-suPAR and m-suPARFc correspond to their expected molecular masses. Right: Injection of massive doses of these recombinant suPARs (100 µg) does not induce significant proteinuria in mice after 24 hours. LC, loading control. (B) Representative SDS-PAGE analyses of void urine normalized for creatinine content (1 mg per lane enables a strict comparison of the albuminuria development following injection of native [m-suPAR] or chimeric [m-suPARFc] soluble uPAR [20 µg equals the dose used in Wei et al.5]). Urine from a normal subject (NS) without albuminuria serves as negative control, and urine specimens from a patient with IgA nephropathy (Berger's disease) and non–nephrotic-range proteinuria (15 mg/g creatinine) and from an LPS-treated mouse served as positive controls. The arrow indicates the electrophoretic mobility of albumin. (C) ELISA assessment of quantity of albuminuria 24 hours after uPAR administration (n=5–8 per group). CTL, control. (D) Invasive monitoring of bladder urine for development of proteinuria on a shorter time scale (2 hours) excludes the existence of transient albuminuria caused by circulating suPAR. Comparison between urine collected before suPAR administration (time zero, U0) and 120 minutes after injection (U120) are shown. Surgical stress and catheterization lead to the induction of spurious albuminuria, which is not related to the anesthesia procedure (urine before [UbA] and after [UaA] anesthesia). Image representative of four catheterized mice. On the left, 1 μg of either BSA or m-suPAR have been deposited as positive controls. (E) Confocal microscopy showing suPAR deposits on normal glomerular structures at 24 hours from mice depicted in B (suPAR in red and podocin in green). (F and G) Transmission electronic microscopy of mouse glomeruli after native suPAR (right) or chimeric suPAR (left) injection as depicted in A and B. No features of podocyte dedifferentiation are observed at 24 hours despite suPAR or chimeric suPAR deposits in E. All results are expressed as mean±SD (n=5–8 per group).
A similar deposition of suPAR in the kidney glomeruli was observed in two different experimental settings: first testing the administration of suPAR in LPS-treated mice (Figure 2D) and second testing the effect of prolonged delivery of suPAR (200 µg subcutaneously delivered via mini-osmotic pump over 7 days on mice not challenged with LPS), as shown in Figure 3D. These deposits are in accordance with the original observation obtained with use of Plaur−/− mice or a Plaur−/− hybrid kidney mouse model challenged by LPS, in which suPAR deposits and proteinuria were evaluated 24 hours after the last LPS injection.5
Figure 2.

Intraperitoneal administration of two distinct forms of recombinant mouse suPAR 24 hours after the last LPS insult fails to exacerbate LPS-associated albuminuria. (A) Injection of 25 µg of either form of suPAR does not lead to further increases in albuminuria, as evaluated by density scanned SDS-PAGE gel (B) or ELISA (C) (n=8 per group). CTL, control. (D) Confocal microscopy demonstrates the increase in glomeruli expressed uPAR or sequestered soluble uPAR following LPS injection (suPAR/uPAR in red and podocin in green). On the right, CTL indicates staining without primary uPAR antibody, showing the absence of a nonspecific staining. All results are expressed as mean±SD (n=8 per group); *P<0.05 (one way ANOVA).
Figure 3.
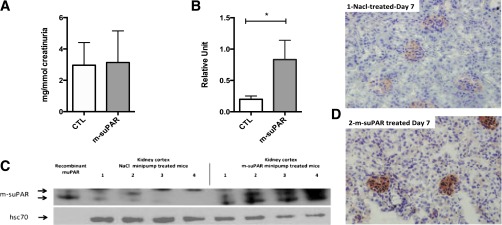
Long-term delivery of suPAR in WT mice. Seven-day sustained delivery of recombinant mouse suPAR (200 µg) by mini-osmotic pumps in normal WT mice (C57BL6/J). (A) Albuminuria (ELISA) evaluated after 7 days of mouse suPAR delivery; absence of any induction of albuminuria between NaCl-treated control (CTL) group versus the suPAR-delivered group (n=8–10 mice per group). (B and C) Western blot detection of uPAR in lysates of kidney cortex 7 days after mini-pump installation showing a massive accumulation of suPAR in the kidney (n=4 per group). The arrows indicate suPAR, and the double band aspect represents the endogenous glycosylation heterogeneity of mouse uPAR. (D) Immunohistochemical detection of suPAR deposited in the glomeruli of mouse kidneys 7 days after mini-pump installation. Deposits are found essentially on glomerular structures (magnification ×400). Overall, the subcutaneous delivery of 200 µg of recombinant suPAR over a week failed to induce any albuminuria in normal mice despite suPAR deposits/accumulation on glomerular structures. All results are expressed as mean±SD (n=8 per group); *P<0.05 (one-way ANOVA).
At this time point, we nevertheless find that the magnitude of albuminuria was not exacerbated by administration of either type of suPAR (25 µg per mouse) (Figure 2, A–C), arguing against any bystander effect elicited by the exogenously added soluble mouse uPAR forms.
Similarly, 1-week sustained exposure to the recombinant nonchimeric purified suPAR was not accompanied by the onset of any albuminuria in C57BL6/J mice (Figure 3A), despite the massive accumulation of suPAR in normal kidney cortex at day 7, demonstrated by both Western blot (Figure 3, B and C) and immunohistochemistry (Figure 3D).
Discussion
The uPAR is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein expressed in numerous tissues, including podocytes.4 Being devoid of any intracellular signaling domain per se, uPAR has to operate through bona fide transmembrane signaling receptors, such as integrins, to modulate outside-in signaling.14 Another important function of uPAR is its role in focalizing and modulating pericellular proteolysis in relation to remodeling of the extracellular matrix, including fibrinolysis.12 Combined, these properties assist in promoting the adhesion and migration of uPAR-expressing cells.14
Shedding of suPAR to the circulation may represent an unavoidable bystander effect associated with the high local proteolytic activity in areas undergoing active tissue remodeling or chronic inflammation. This is exemplified by the reactive tumor-stroma microenvironment in the invading front of solid carcinomas.15–18 The protein architecture of human uPAR reveals the linker region connecting domain I and II to be highly solvent exposed and unstructured,19 which renders it very susceptible to proteolytic attack by several proteases, such as uPA, plasmin, matrix metallopeptidase 12, kallikrein 4, leukocyte elastase, and cathepsin G.20–22 Another modus operandi for the occurrence of soluble uPAR is the numerous alternative splicing events reported for human and mouse uPAR on the mRNA level.23–25 For example, one of the more interesting splice variants reported for human uPAR results in an mRNA transcript in which exons 4 and 5 are deleted, thus encoding a receptor variant with no domain II.24,26 This truncated receptor may, nevertheless, still be GPI-anchored on the cell surface as the signal peptide and the GPI anchorage site remain intact.27,28 At present, however, it remains to be proven whether this variant is expressed in vivo at the protein level and is secreted in detectable amounts into the circulation.
With a view to the proposition that long-term exposure to elevated levels of soluble uPAR will initiate FSGS lesions in vivo, it is interesting to consider the clinical features of an acquired hematologic stem cell disorder, paroxysmal nocturnal hemoglobinuria (PNH). This disease is caused by the clonal expansion of bone marrow stem cells carrying somatic mutations in the early components of the biosynthetic pathway responsible for the GPI anchors. As a consequence, the affected leukocytes are unable to tether proteins destined to become GPI-anchored to the cell membrane and as a default are secreted after removal of the C-terminal signal sequence authorizing this post-translational modification.29 Accordingly, uPAR is absent from the cell surface of leukocytes affected by the PNH driver mutations; these abnormal cells respond by secreting a soluble intact receptor, thus sustaining chronically elevated levels of suPAR in the plasma of patients with PNH.29–32 Despite these abnormally high levels of suPAR in the plasma of patients with PNH, to the best of our knowledge, FSGS is not one of the prevalent renal lesions observed in these patients.33 Renal dysfunction does occur in patients with PNH, but most of these clinical features can be alleviated by eculizumab treatment, highlighting chronic complement-mediated hemolysis as the driver of renal pathogenesis.34,35
In view of the above considerations and the lack of effect of administration of suPAR on podocyte structure and protein excretion that we observed in our mouse model, it is important to re-evaluate the original evidence that leads to the proposal of circulating uPAR being the causative serum factor for induction of FSGS.5 As opposed to the present study, soluble recombinant mouse uPAR was administered in the original study to C57BL/6 mice carrying a specific ablation of the Plaur gene encoding mouse uPAR, but the significance of this difference for the subsequent development of proteinuria is at present unclear. The Plaur−/− mice were shown to be protected from an LPS-induced proteinuria compared with their WT counterpart.4 Furthermore, a similarly challenged Plaur−/− kidney transplanted into a WT mice recipient developed similar podocytic lesions to the contralateral WT kidney.5 Here we performed an analogous experiment on WT mice and found that a superimposed injection of recombinant suPAR failed to increase the albuminuria induced by LPS treatment. These experiments constituted an excellent positive control of LPS-induced albuminuria in our hands, and our experimental data argue against an albuminuric effect of suPAR in WT mice. Nevertheless and overall, if the genetic ablation of Plaur (in the kidney in particular) is the cause of the effect observed on proteinuria, it is difficult to translate this finding to the pathogenesis of FSGS in humans.
The second line of evidence presented in the original report on a causative correlation between FSGS/proteinuria and circulating soluble uPAR is based on genetically engineered WT mice, in which sustained levels of plasma uPAR are achieved by electroporation of the skin with a plasmid encoding a truncated mRNA splice variant of mouse uPAR covering the first 133 residues of the full-length receptor (GenBank: BC010309, cDNA clone IMAGE: 3158012).5,23 Although the mRNA encoding this truncated muPAR splice variant is indeed expressed in vivo in the small intestine of the mouse, as verified by in situ hybridization,23 we are not aware of any solid evidence that a corresponding folded protein is secreted in vivo or in vitro. In hindsight, we speculate that this particular splice mRNA transcript may not be translated into a folded protein product in vivo because it encodes only one and one half LU-domain. The fully matured uPAR is a modular protein composed of three homologous LU domains (Ly-6/uPAR), which are encoded by separate exon sets and are characterized by a central β-sheet and four to five intradomain disulfide bonds.19 The truncated uPAR encoded by the muPAR splice variant in question terminated in the middle of the second LU-domain, thus violating all reasonable considerations concerning the maintenance of protein architecture and disulfide connectivity of this multidomain receptor, as defined by the numerous crystal structures we have solved for mouse and human uPAR.13,30,36–38
In summary, we demonstrate that both short-term and prolonged administration of suPAR and its deposition on glomerular structures are not sufficient per se to cause proteinuria in mice. Further investigation on the pathogenesis of FSGS is therefore needed to reconcile previous data showing an association with high circulating levels of suPAR and albuminuria.
Concise Methods
Animals and Induction of Experimental LPS Nephritis
All mice (C57BL/6J, 129S2SvPas) were provided by the Charles Rivers Laboratory (Charles River Laboratories, L'Arbresle, France). Eight- to 12-week-old female mice were used in all experiments, with five to eight mice per group in each experiment. The LPS model was established as previously described.4,39 Renal injury was evaluated at multiple time points from 2 to 48 hours as described.5 Control mice received PBS or BSA (50 µg total) for experiments using LPS.
Soluble uPAR Administration and Albuminuria Quantification
Administration of suPAR by the intravenous or intraperitoneal route gave similar results. For the short-term evaluation (<120 minutes), urine output was invasively monitored by bladder catheterization to detect development of proteinuria on a very short time scale. BP was also invasively monitored by femoral catheterization. SuPAR injection was performed via femoral intravenous infusion of 50 µg of m-suPAR. This recombinant form of mouse suPAR is described in detail elsewhere.13 In a second step, urine was collected 24 hours after intravenous injection of m-suPAR or the chimeric m-suPARFc (a mouse suPAR linked to the human Fc IgG1, commercially available and provided by R&D Systems, 531-PA-100); both are synthesized by eucaryotic cells.
In the LPS model, m-suPAR or m-suPARFc was coinjected intraperitoneally, with the second LPS dose corresponding 24 hours after the first LPS dose and 24 hours before urine collection, as described.5
The continuous delivery of 200 µg of recombinant suPAR by an osmotic pump over a week was done as follows. Briefly, Alzet mini-pumps (model 1007D; Charles River Laboratories) were subcutaneously inserted under light general anesthesia (xylazine/ketamine). Mini-pumps loaded only by NaCl were used for the control group. Eight to 10 mice per group were used in this experiment (C57BL6/J, female, 10 weeks old). Void urine samples were collected on day 7.
Western Blot Analysis of Renal Cortex
Proteins were extracted from renal cortex using radio-immunoprecipitation assay lysis buffer supplemented with sodium orthovanadate, phenylmethanesulfonyl fluoride, and protease inhibitor cocktail (Tebu Bio, Le Perray-en-Yvelines, France), and 10 mM sodium fluoride. After centrifugation at 10,000 rpm for 10 minutes at 4°C, protein concentrations were determined from the supernatant using the Bradford assay. Aliquots of 20 μg of protein were run on NuPAGE 4/12% electrophoresis gels (Invitrogen) and then transferred onto a polyvinylidene difluoride membrane (Immobilon-p; Millipore, Saint-Quentin-en-Yvelines, France). Immunoblotting was performed using goat-specific primary antibodies anti-mouse uPAR biotinylated (uPAR antibody from R&D [BAF534; R&D Systems Europe, Lilles, France]). Then, the membrane was incubated with extravidin–horseradish peroxidase (Sigma-Aldrich, France). The revelation was performed with the ECL plus kit (GE Healthcare). Densitometric analysis on ImageJ software (National Institutes of Health, Bethesda, MD) was then performed for quantification.
SDS-PAGE Migration of Void Urine Samples
Urine deposits normalized to creatinuria for a comparable creatinine content (1 mg per lane) enabled accurate investigation of the albuminuria after injection of native (m-suPAR) or chimeric (m-suPARFc) soluble uPAR. In comparison, normalized urine from a normal subject served as a negative control, and urine from a patient with IgA nephropathy (Berger disease) and non–nephrotic-range proteinuria (15 mg/g creatinine) and urine from an LPS-treated mouse served as positive controls. Relative quantification was done using Image J software. A mouse antialbumin ELISA kit (Albuwell; Exocell, Inc.) was also used to assess the extent of albuminuria and yielded similar results.
Transmission Electron Microscopy
Mice were perfused with 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer at a pH of 7.4. Kidneys were removed, cut into small pieces, and immersed in 2.5% glutaraldehyde containing 1% tannic acid in 0.1 M PBS for 2 hours at 4°C. Samples were postfixed with 1% OsO4, dehydrated, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and then examined under a Philips CM10 electron microscope.
Immunofluorescence and Confocal Analysis of uPAR Deposits in Glomeruli
Glomerular uPAR deposition was assessed using a rat anti-mouse uPAR antibody (MAB531; R&D Systems Europe) on cryosections revealed by a coupled Alexafluor 546 nm anti-rat IgG. Deposits localized in podocytes were detected using a guinea pig anti-mouse nephrin antibody (Progen, Heidelberg, Germany), followed by an Alexafluor 488 nm coupled anti–guinea pig secondary antibody. All secondary antibodies were purchased from Life Technologies (Saint-Aubin, France). Confocal microscope protocol was used as previously described.40
Immunohistochemistry Analysis of uPAR Deposits in Kidney Cortex
Kidneys were snap-frozen in liquid nitrogen, and 5-μm acetone-fixed cryostat sections were assessed for mouse uPAR deposition using a goat anti-mouse uPAR antibody detected by secondary antibodies purchased from Nichirei-Histofine Simple Stain Mouse MAX PO (goat) (Tokyo, Japan) and then visualized with 3-amino-9-ethylcarbazole AEC (Dako, Carpinteria, CA). The tissue sections were counterstained with hematoxylin and coverslipped.
Statistical Analyses
A total of five to eight mice per experimental group were analyzed in two separate experiments at least, and the results are expressed as mean±SD. Differences were compared using one-way ANOVA. When variance differed significantly, a Mann–Whitney test was performed. All statistical analyses were performed using GraphPad Prism 5.0. Differences were considered statistically significant when the P value was <0.05.
NOTE
During the editing of the present paper, three articles appeared demonstrating that increased levels of soluble shed uPAR is not a specific biomarker for human focal segmental glomerulosclerosis.41–43
Disclosures
None.
Acknowledgments
We thank Caroline Martin and Claude Kitou for providing animal housing. We thank Mark Crutchfield for English editing advice.
This work was financially supported by INSERM and by the Faculté de Médecine Pierre et Marie Curie. D.C. is the recipient of a European Renal Association–European Dialysis and Transplant Association grant.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Soluble Urokinase-Type Plasminogen Activator Receptor in FSGS: Stirred but Not Shaken,” on pages 1611–1613.
References
- 1.Hoyer JR, Vernier RL, Najarian JS, Raij L, Simmons RL, Michael AF: Recurrence of idiopathic nephrotic syndrome after renal transplantation. Lancet 2: 343–348, 1972 [DOI] [PubMed] [Google Scholar]
- 2.Le Berre L, Godfrin Y, Perretto S, Smit H, Buzelin F, Kerjaschki D, Usal C, Cuturi C, Soulillou JP, Dantal J: The Buffalo/Mna rat, an animal model of FSGS recurrence after renal transplantation. Transplant Proc 33: 3338–3340, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Le Berre L, Godfrin Y, Günther E, Buzelin F, Perretto S, Smit H, Kerjaschki D, Usal C, Cuturi C, Soulillou JP, Dantal J: Extrarenal effects on the pathogenesis and relapse of idiopathic nephrotic syndrome in Buffalo/Mna rats. J Clin Invest 109: 491–498, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J: Modification of kidney barrier function by the urokinase receptor. Nat Med 14: 55–63, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap HK, Saleem M, Zhang Q, Nikolic B, Chaudhuri A, Daftarian P, Salido E, Torres A, Salifu M, Sarwal MM, Schaefer F, Morath C, Schwenger V, Zeier M, Gupta V, Roth D, Rastaldi MP, Burke G, Ruiz P, Reiser J: Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17: 952–960, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei C, Trachtman H, Li J, Dong C, Friedman AL, Gassman JJ, McMahan JL, Radeva M, Heil KM, Trautmann A, Anarat A, Emre S, Ghiggeri GM, Ozaltin F, Haffner D, Gipson DS, Kaskel F, Fischer DC, Schaefer F, Reiser J, PodoNet and FSGS CT Study Consortia : Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol 23: 2051–2059, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobsen B, Ploug M: The urokinase receptor and its structural homologue C4.4A in human cancer: Expression, prognosis and pharmacological inhibition. Curr Med Chem 15: 2559–2573, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Lund IK, Illemann M, Thurison T, Christensen IJ, Høyer-Hansen G: uPAR as anti-cancer target: Evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr Drug Targets 12: 1744–1760, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Maas RJ, Wetzels JF, Deegens JK: Serum-soluble urokinase receptor concentration in primary FSGS. Kidney Int 81: 1043–1044, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Maas RJ, Deegens JK, Wetzels JF: Serum suPAR in patients with FSGS: Trash or treasure? Pediatr Nephrol 28: 1041–1048, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Bock ME, Price HE, Gallon L, Langman CB: Serum soluble urokinase-type plasminogen activator receptor levels and idiopathic FSGS in children: A single-center report. Clin J Am Soc Nephrol 8: 1304–1311, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly BM, Choi EY, Gårdsvoll H, Bey AL, Currie BM, Chavakis T, Liu S, Molinolo A, Ploug M, Leppla SH, Bugge TH: Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood 116: 1593–1603, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin L, Gårdsvoll H, Huai Q, Huang M, Ploug M: Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: Implication for preclinical cancer therapy. J Biol Chem 285: 10982–10992, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith HW, Marshall CJ: Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol 11: 23–36, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Høyer-Hansen G, Behrendt N, Ploug M, Danø K, Preissner KT: The intact urokinase receptor is required for efficient vitronectin binding: Receptor cleavage prevents ligand interaction. FEBS Lett 420: 79–85, 1997 [DOI] [PubMed] [Google Scholar]
- 16.Montuori N, Ragno P: Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front Biosci (Landmark Ed) 14: 2494–2503, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Henic E, Borgfeldt C, Christensen IJ, Casslén B, Høyer-Hansen G: Cleaved forms of the urokinase plasminogen activator receptor in plasma have diagnostic potential and predict postoperative survival in patients with ovarian cancer. Clin Cancer Res 14: 5785–5793, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Rasch MG, Lund IK, Almasi CE, Hoyer-Hansen G: Intact and cleaved uPAR forms: Diagnostic and prognostic value in cancer. Front Biosci 13: 6752–6762, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Kjaergaard M, Hansen LV, Jacobsen B, Gardsvoll H, Ploug M: Structure and ligand interactions of the urokinase receptor (uPAR). Front Biosci 13: 5441–5461, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Høyer-Hansen G, Rønne E, Solberg H, Behrendt N, Ploug M, Lund LR, Ellis V, Danø K: Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain. J Biol Chem 267: 18224–18229, 1992 [PubMed] [Google Scholar]
- 21.Koolwijk P, Sidenius N, Peters E, Sier CF, Hanemaaijer R, Blasi F, van Hinsbergh VW: Proteolysis of the urokinase-type plasminogen activator receptor by metalloproteinase-12: Implication for angiogenesis in fibrin matrices. Blood 97: 3123–3131, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Beaufort N, Debela M, Creutzburg S, Kellermann J, Bode W, Schmitt M, Pidard D, Magdolen V: Interplay of human tissue kallikrein 4 (hK4) with the plasminogen activation system: hK4 regulates the structure and functions of the urokinase-type plasminogen activator receptor (uPAR). Biol Chem 387: 217–222, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Kristensen P, Eriksen J, Blasi F, Danø K: Two alternatively spliced mouse urokinase receptor mRNAs with different histological localization in the gastrointestinal tract. J Cell Biol 115: 1763–1771, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pliyev BK: Activated human neutrophils rapidly release the chemotactically active D2D3 form of the urokinase-type plasminogen activator receptor (uPAR/CD87). Mol Cell Biochem 321: 111–122, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Suh TT, Nerlov C, Danø K, Degen JL: The murine urokinase-type plasminogen activator receptor gene. J Biol Chem 269: 25992–25998, 1994 [PubMed] [Google Scholar]
- 26.Luther T, Kotzsch M, Meye A, Langerholc T, Füssel S, Olbricht N, Albrecht S, Ockert D, Muehlenweg B, Friedrich K, Grosser M, Schmitt M, Baretton G, Magdolen V: Identification of a novel urokinase receptor splice variant and its prognostic relevance in breast cancer. Thromb Haemost 89: 705–717, 2003 [PubMed] [Google Scholar]
- 27.Wilhelm OG, Wilhelm S, Escott GM, Lutz V, Magdolen V, Schmitt M, Rifkin DB, Wilson EL, Graeff H, Brunner G: Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J Cell Physiol 180: 225–235, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Sato S, Kopitz C, Grismayer B, Beaufort N, Reuning U, Schmitt M, Luther T, Kotzsch M, Krüger A, Magdolen V: Overexpression of the urokinase receptor mRNA splice variant uPAR-del4/5 affects tumor-associated processes of breast cancer cells in vitro and in vivo. Breast Cancer Res Treat 127: 649–657, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Ploug M, Eriksen J, Plesner T, Hansen NE, Danø K: A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem 208: 397–404, 1992 [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Gårdsvoll H, Yuan C, Lin L, Ploug M, Huang M: Crystal structure of the urokinase receptor in a ligand-free form. J Mol Biol 416: 629–641, 2012 [DOI] [PubMed] [Google Scholar]
- 31.Sloand EM, Pfannes L, Scheinberg P, More K, Wu CO, Horne M, Young NS: Increased soluble urokinase plasminogen activator receptor (suPAR) is associated with thrombosis and inhibition of plasmin generation in paroxysmal nocturnal hemoglobinuria (PNH) patients. Exp Hematol 36: 1616–1624, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rønne E, Pappot H, Grøndahl-Hansen J, Høyer-Hansen G, Plesner T, Hansen NE, Danø K: The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol 89: 576–581, 1995 [DOI] [PubMed] [Google Scholar]
- 33.Luzzatto L, Gianfaldoni G, Notaro R: Management of paroxysmal nocturnal haemoglobinuria: A personal view. Br J Haematol 153: 709–720, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Hill A, Rother RP, Khursigara G, Fu CL, Omine M, Browne P, Rosse W: Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol 85: 553–559, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Takahashi K, Yoshimura A, Inoue Y, Takahashi N, Sugenoya Y, Morita H, Kinugasa E, Ideura T: A case of paroxysmal nocturnal hemoglobinuria combined with focal segmental glomerular sclerosis. Nippon Jinzo Gakkai Shi 43: 39–43, 2001 [PubMed] [Google Scholar]
- 36.Mertens HD, Kjaergaard M, Mysling S, Gårdsvoll H, Jørgensen TJ, Svergun DI, Ploug M: A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J Biol Chem 287: 34304–34315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gårdsvoll H, Kjaergaard M, Jacobsen B, Kriegbaum MC, Huang M, Ploug M: Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor. J Biol Chem 286: 43515–43526, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gårdsvoll H, Jacobsen B, Kriegbaum MC, Behrendt N, Engelholm L, Østergaard S, Ploug M: Conformational regulation of urokinase receptor function: impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J Biol Chem 286: 33544–33556, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Comper WD, Mundel P: Is the LPS-mediated proteinuria mouse model relevant to human kidney disease? Nat Med 15: 133–134, author reply 133–134, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Letavernier E, Bruneval P, Vandermeersch S, Perez J, Mandet C, Belair MF, Haymann JP, Legendre C, Baud L: Sirolimus interacts with pathways essential for podocyte integrity. Nephrol Dial Transplant 24: 630–638, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Meijers B, Maas RJ, Sprangers B, Claes K, Poesen R, Bammens B, Naesens M, Deegens JK, Dietrich R, Storr M, Wetzels JF, Evenepoel P, Kuypers D: The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis. Kidney Int 10.1038/ki.2013.505, 2014 [DOI] [PubMed] [Google Scholar]
- 42.Wada T, Nangaku M, Maruyama S, Imai E, Shoji K, Kato S, Endo T, Muso E, Kamata K, Yokoyama H, Fujimoto K, Obata Y, Nishino T, Kato H, Uchida S, Sasatomi Y, Saito T, Matsuo S: A multicenter cross-sectional study of circulating soluble urokinase receptor in Japanese patients with glomerular disease. Kidney Int 10.1038/ki.2013.544, 2014 [DOI] [PubMed] [Google Scholar]
- 43.Sinha A, Bajpai J, Saini S, Bhatia D, Gupta A, Puraswani M, Dinda AK, Agarwal SK, Sopory S, Pandey RM, Hari P, Bagga A: Serum-soluble urokinase receptor levels do not distinguish focal segmental glomerulosclerosis from other causes of nephrotic syndrome in children. Kidney Int 10.1038/ki.2013.546, 2014 [DOI] [PubMed] [Google Scholar]