

Abstract
Significance: Voltage-gated K+ channels are a large family of K+-selective ion channel protein complexes that open on membrane depolarization. These K+ channels are expressed in diverse tissues and their function is vital for numerous physiological processes, in particular of neurons and muscle cells. Potentially reversible oxidative regulation of voltage-gated K+ channels by reactive species such as reactive oxygen species (ROS) represents a contributing mechanism of normal cellular plasticity and may play important roles in diverse pathologies including neurodegenerative diseases. Recent Advances: Studies using various protocols of oxidative modification, site-directed mutagenesis, and structural and kinetic modeling provide a broader phenomenology and emerging mechanistic insights. Critical Issues: Physicochemical mechanisms of the functional consequences of oxidative modifications of voltage-gated K+ channels are only beginning to be revealed. In vivo documentation of oxidative modifications of specific amino-acid residues of various voltage-gated K+ channel proteins, including the target specificity issue, is largely absent. Future Directions: High-resolution chemical and proteomic analysis of ion channel proteins with respect to oxidative modification combined with ongoing studies on channel structure and function will provide a better understanding of how the function of voltage-gated K+ channels is tuned by ROS and the corresponding reducing enzymes to meet cellular needs. Antioxid. Redox Signal. 21, 933–952.
Introduction
Voltage-gated potassium (K+) channels form a large family of ion channels with the common properties of strong selectivity for K+ and a dependence of their open probability (Po) on the membrane voltage; at resting voltages of −70 to −90 mV such channels are typically closed, and they open when the membrane depolarizes. In excitable cells, these features enable voltage-gated K+ channels to counteract electrical excitation; they may contribute to setting and stabilizing the resting potential or they help to regulate action potential shape and frequency. In nonexcitable cells, such as in the epithelial cells of the inner ear or the kidney, they serve roles as K+ transport vehicles. To suit to a wide range of physiological functions, voltage-gated K+ channels are diverse in their primary structure and the channel complexes often include auxiliary subunits as detailed below. In addition, through co- and post-translational protein modifications these channels are tuned to the specific cellular needs. A subgroup of such modifications is changes in channel function induced by reactive oxygen species (ROS) or reactive oxygen intermediates. While various reviews focusing on potential roles of channel modification by ROS in diseases or degenerative phenomena have appeared [e.g., (4, 86, 117)], we will focus here on mechanistic insights underlying the oxidative modulation of voltage-gated K+ channels.
The focus on molecular mechanism has become feasible because voltage-gated K+ channels have a long history of in-depth research. Stimulated by the first cloning of cDNAs of the Shaker channel from Drosophila melanogaster (55, 99, 101, 133), many other genes in various species were to follow. Numerous channel-related inherited diseases underscore a range of physiological functions [e.g., (1)], and the progress in X-ray crystallography has provided us with detailed information on the atomic structure of some of these channels [e.g., (74, 75)]. Last but not least, the functional properties of voltage-gated K+ channels can be evaluated with electrophysiological methods to a very high precision because their voltage-dependence allows the correction of leak and capacitive currents when measuring ionic currents in the voltage-clamp mode. Therefore, high-quality electrical signals combined with detailed kinetic modeling report on functional features of the channels and provide access to the investigation of even minute but potentially important modulating influences.
Voltage-gated K+ channels are formed by α-subunits with six transmembrane segments (6TM, S1–S6; Fig. 1). Four α-subunits assemble to build a channel complex in which the narrow part is lined by the pore loops connecting S5 and S6. Some α-subunits can coassemble with different types of α-subunits thus creating diverse heterotetramers.
FIG. 1.

Voltage-gated K+ channels. (A) Structure of a Kv1.2/Kv2.1 chimeric channel according to Long et al. (74) (KCNA2/KCNB1; PDB 2R9R) with docked Kvβ2 subunits. (B) Structural elements covering S1–S6 of a single subunit of the Kv1.2/2.1 chimera. Two K+ ions at the selectivity filter are also indicated. (C) Topological model of a 6TM α-subunit of voltage-gated K+ channels illustrating the voltage-sensor domain (S1–S4), the pore/gate domain (S5–S6), and an N-terminal domain that functions in some channel types as inactivation “ball” domain. 6TM, six transmembrane segments. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
To become selective for K+, adjacent carbonyl oxygens located in the pore loops are directed into the center of the channel—a feature involving either G-Y-G or G-F-G in the sequence of the selectivity filter region (31). Projecting toward the cytosolic side, a water-filled cavity completes the channel pore. This is a site where various drugs bind to K+ channels to occlude the pore or to interfere with the gating, that is, with the opening and closing of the ion conduction gate.
Voltage sensing is mediated by an action of the voltage-sensor domain (VSD, S1–S4), with S4 carrying the major component of the gating charge, and the pore/gate domain (PGD, S5-pore-S6) (Fig. 1B, C). Conformational changes of the VSD induced by changes in the transmembrane electric field are coupled by means of the cytosolic S4–S5 linker to the pore/gate module (58, 75). The latter then results in a movement of the S6 segments to open the pore at the intracellular side. This combination of VSD and PGD defines the fundamental features of voltage-gated K+ channels. However, as described below, extended N- and C-terminal protein domains contribute many additional features such as the ability of the channels to undergo inactivation or to be modulated by second messengers such as Ca2+ or cyclic nucleotides.
Association of auxiliary protein subunits (β-subunits) further increases the diversity of K+ channels. Some of them are cytosolic proteins that dock to the N-terminal domains of the α-subunits (Kvβ) (74, 103) (Fig. 1A). Others are membrane proteins with one or two transmembrane segments like members of the KCNE or KCNMB family, respectively. In addition, Ca2+-binding proteins such as calmodulin (115), K channel-interacting proteins (KChiPs) (3), or dipeptidyl aminopeptidase-like protein (DPPX) (87) may bind to the channels. In all cases, β-subunits equip the K+ channel complexes with additional features.
Various nomenclatures exist to describe voltage-gated K+ channels or the corresponding genes. Just concentrating on α-subunits, there is the HUGO nomenclature of human genes, the Kv notation according to the protein function, and a long list of traditional names. In Figure 2A, a phylogenetic tree illustrates the relatedness of genes coding for α-subunits of voltage-gated K+ channels using their HUGO names. The linear sequence is thus the basis for the formation of subfamilies within the family of voltage-gated K+ channels (Fig. 2B). The largest one is the so-called “Kv family” (KCNA-KCND), which subdivides into Kv1 (Shaker, KNCA), Kv2 (Shab, KCNB), Kv3 (Shaw, KCNC), and Kv4 (Shal, KCND) channels. While coassembly within the subfamily is observed, subunits coded by these genes typically do not form heterooligomers with subunits from other subfamilies. There are more genes that belong to this subfamily (e.g., Kv5-Kv9, KCNF, KCNG, KCNV, and KCNS) whose gene products can modulate the previously described subunits. Further subfamilies are ether à go-go (EAG) channels (Kv10-Kv12, KCNH) and KCNQ channels (Kv7, some of them also referred to as M-channels). Here, we focus on the Kv, EAG, and KCNQ subfamilies. In addition, we will discuss voltage- and Ca2+-activated big conductance K+ channels (BK channels, SLO1, KCa1.1, and KCNMA1); they are activated by cytosolic Ca2+ and possess an additional S0 transmembrane segment placing the N terminus to the extracellular side. Other members of this group of KCa or Slo channels also have a large conductance and are voltage dependent, but they are co-activated by Na+ (KCNT1), Cl−(KCNT2) or protons (KCNMA3).
FIG. 2.

Nomenclature of voltage-gated K+ channels. (A) Phylogenetic tree of human genes coding for voltage-gated K+ channel α-subunits of the 6TM family in HUGO nomenclature. The major subfamilies are indicated. (B) Names of proteins according the Kv nomenclature, genes in HUGO nomenclature, and some traditional names of voltage-gated K+ channels. Names of subfamilies (black) and groups (gray) are also indicated. Note that KCNQ1/Kv7.1 is also known as KCNA8, KCNA9, and Kv1.9.
Given the large number of K+ channel complexes and considering that exposure to ROS often has an impact on channel function—sometimes in a prominent manner, sometimes more subtle—the first step in analyzing the underlying molecular mechanisms is to identify the biochemical effector sites of ROS. On the level of gene transcription, ROS-dependent regulation of the expression of genes coding for K+ channels can take place. Acute and direct ROS effects may be grouped into those affecting the channel protein itself, affecting some auxiliary subunits, or membrane components. If a known protein is affected, specific residues that are modified by ROS (typically cysteine, methionine, tyrosine, or histidine) may be identified to provide further insight. Some typical ROS molecules, their conversion, and select protein modifications often observed are presented in Figure 3. Rather than modifying the channel protein, alterations of the cellular ROS level may have an impact on the balance of redox couples such as Fe2+/Fe3+, GSH-red/GSH-ox, nicotinamide adenine dinucleotide phosphate (NADPH)/NADP+, etc. and those may be the molecules modulating channel function.
FIG. 3.

ROS processing and protein modification. Modification of cysteine, methionine, tryrosine, histidine, and tryptophan often observed in proteins upon oxidative stress. Top box: Formation of glutathione dimers (GSSG), catalyzed by glutathione peroxidase (GPx) and recycling to reduced glutathione (GSH) via the NADPH system. Bottom box: Removal and formation of reactive species via superoxide dismutase (SOD), catalase (Cat), and the Fenton reaction involving transition metals (here: ferrous iron, Fe2+). H2O2, hydrogen peroxide; O2•−, superoxide anion; OH•, hydroxyl radical; OH−, hydroxyl anion; Fe3+, ferric iron; MsrA/B, methionine sulfoxide reductase A/B; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species.
Finally, it needs to be assessed whether a ROS effect on a channel is the result of irreversible protein degradation (e.g., via ubiquitination) or a reversible ROS-dependent regulation of channel function. As we will see below, in many cases a dependence of K+ channels on exogenously applied oxidative modifiers has been demonstrated, but we often lack information on the physiological stimuli and the molecular mechanisms underlying the responses elicited by various ion channel types.
The Kv Subfamily
Voltage-gated K+ channels of the Kv subfamily activate on membrane depolarization and give rise to so-called delayed-rectifier currents. Some of them, in particular Kv1.4, Kv3, and Kv4 channels, can undergo rapid inactivation, an aspect leading to A-type currents, and this will be discussed below.
Kv1.1 and Kv1.2
Kv1.1 and Kv1.2 channels are found in the brain (77) and involved in regulation of neurotransmitter release (29, 54). Studying the impact of nitric oxide (NO) on paraventricular nucleus cells from rat slices, Yang et al. (150) observed that this challenge increased spontaneous miniature inhibitory postsynaptic currents. Using specific Kv channel blockers, they further found that Kv1.1 and Kv1.2 serve as downstream effectors of NO-dependent GABA release in these neurons, possibly by a combination of NO and ROS forming very reactive but short-lived peroxynitrite (ONOO−) (150).
An influence of peroxynitrite was also shown for Kv channels in small coronary arteries, where they play a role in vasodilation (70). An elevated glucose level attenuated the dilator function of Kv channels, possibly by nitration of a tyrosine residue. Peroxynitrite stress during hyperglycemia thus impairs Kv channel availability and may influence coronary blood flow and myocardial perfusion (70).
Conforti and Millhorn (22) showed that Kv1.2 channels could play a role in O2 chemoreception in pheochromocytoma 12 (PC-12) cells derived from adrenal chromaffin cells. Following prolonged hypoxia (18 h, 10% O2), the expression of Kv1.2 increased and the cells became more responsive to short-term hypoxia suggesting that Kv1.2 might modulate an O2-sensitive signaling pathway (22, 23). While anoxia decreased the current through Kv1.2 channels, no impact on Kv2.1 was observed. A strategy based on this finding, involving Kv1.2/Kv2.1 channels and point mutations, revealed that a methionine in the S5 region of Kv1.2 (M380) is necessary for the response of Kv1.2 channels to anoxia (25).
Kv1.3
Kv1.3 channels are predominantly expressed in T- and B-lymphocytes and play a vital role in regulation of membrane potential and Ca2+ signaling (146). Inhibition of Kv1.3 channel activity leads to suppression of Ca2+ signaling, cytokine production, and T-lymphocyte activation and proliferation (16). Exposure of freshly isolated human T-lymphocytes and leukemic Jurkat T-cells to hypoxia inhibited Kv1.3 currents by up to 47% (24) and this effect could be attributed to a decreased Kv1.3 surface expression (19). This dependence of Kv1.3 on hypoxia may provide an explanation for the failure of T-cell activation in proliferating cancer tissue, which often provides hypoxic conditions.
Szigligeti et al. (126) provided a mechanistic link for the O2 sensitivity of Kv1.3 channel in T-cells: Kv1.3 channels were resistant to hypoxia in Jurkat T-cells deficient in the src protein tyrosine kinase p56Lck and they gained O2 sensitivity when constitutively active Lck (Y505FLck) was transfected to Lck-deficient cells (126).
Kv1.5
Kv1.5 channels contribute to the repolarizing ultra-rapid delayed-rectifier current IKur in the heart, and aberrations in Kv1.5 channels can lead to atrial fibrillation (91) and sudden cardiac death (90). Svoboda et al. (124) demonstrated that a global increase in sulfenic acid-modified proteins is associated with atrial fibrillation. Kv1.5 channels may be important players in this process because sulfenic acid modification appears to take place at a conserved cysteine at the C terminus (C581). Under prolonged oxidative stress, sulfenic acid modification diminished Kv1.5 channel surface expression. In freshly isolated rat hearts, acute sulfenic acid modification also inhibited Kv currents and induced sustained arrhythmia. This finding may provide a therapeutic clue for the treatment of atrial fibrillation (124).
Kv2.1
In neuronal tissue, Kv2.1 appears to be involved in modulation of neuronal apoptosis; using a model of experimentally induced apoptosis, Pal et al. (94) showed that cortical neurons expressing dominant-negative Kv2.1 became insensitive while Kv2.1-overexpressing CHO cells became susceptible to apoptosis induction. Since oxidative stress-induced apoptosis is a major etiology of neurodegenerative disorders and aging, suppression of Kv2.1 could be a protective strategy. A recent report by Dallas et al. (27) showed that carbon monoxide (CO), an endogenous signaling molecule typically produced during the degradation of heme, inhibited native Kv2.1 channels in hippocampal neurons and recombinant Kv2.1 in HEK 293 cells. Application of the membrane-permeant oxidizing agent DTDP increased the surface expression of Kv2.1 and induced apoptosis while the CO donor CORM-2 diminished this vulnerability. This observation suggests a mechanism of neuroprotection, where CO provides resistance against oxidant-induced apoptosis via Kv2.1 suppression (27).
Kv2.1 expression has also been linked to Alzheimer's disease progression. A study by Cotella et al. (26) found that Kv2.1 forms oligomers in the presence of oxidizing agents, and old mice showed ∼10-fold higher oligomer formation than young mice. Further, it was revealed that a conserved cysteine in the cytoplasmic side (C73) was the target of oxidation while mutant C73A was functionally indistinguishable from wild-type channels. In Caenorhabditis elegans it was found that β-amyloid facilitates oxidation of the Kv2.1 paralog KvS-1 and induces neuronal apoptosis while the mutant homolog to Kv2.1-C73A rendered the cells resistant to β-amyloid-induced oxidation and apoptosis. This study might provide a therapeutic clue for the treatment of age-related degenerative diseases (26). This hypothesis received support from the results of Yuan et al. (152) who reported that Donepezil, a commercially available drug that improves cognitive and global functions in Alzheimer's disease patients and for those suffering from vascular dementia (33), provides protection against oxygen/glucose deprivation-induced cell apoptosis by inhibiting Kv2.1 currents. Although the exact mechanism is not known, Donepezil may prevent the oxidation of Kv2.1 channels and thereby protect against apoptosis (152).
In pancreatic β-cells, Kv2.1 regulates glucose-dependent insulin secretion (81, 83), and insulin secretion also depends on the cytosolic status of the NADPH/NADP+ ratio (2). At body temperature, Kv2.1 undergoes some inactivation: the time course of inactivation was slowed down upon oxidation and depended on the NADPH/NADP+ ratio suggesting that oxidative control of Kv2.1 function is a key mechanism in the control of insulin release (82).
Oxygen sensing in the lung
Several Kv channels are involved in the regulation of the pulmonary circulation and implicated to be oxygen sensitive (86). A physiological response to hypoxia in the lung is hypoxic pulmonary vasoconstriction (HPV), which redirects the blood flow to better-ventilated areas of the lung, thus increasing the total area involved in gaseous exchange. Patients suffering from hypoxic pulmonary diseases and residents of high altitude have sustained HPV, which can lead to vascular remodeling, pulmonary hypertension, and possibly fatal heart failure (6, 86, 125). As illustrated in Figure 4A, the Kv-related redox regulation of HPV starts with a block of Kv channels because of ROS produced during hypoxia. K+ channel block results in cell depolarization that activates voltage-gated Ca2+ (CaV) channels, which are also directly activated by hypoxia (38). Finally, intracellular Ca2+ triggers contraction and induces HPV (6, 86, 125). It should be noted, though, that the extent of ROS production during acute and sustained hypoxia and reoxygenation remains controversial (39) and certainly further studies aiming at the quantitative ROS monitoring under such conditions are required.
FIG. 4.
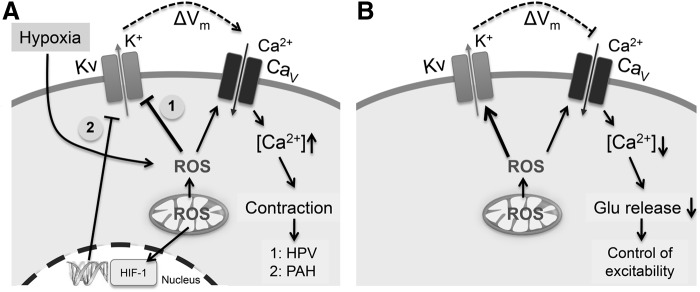
Impact of ROS-dependent K+ channel modulation on cell functions. (A) Examples of how redox-dependent inhibition of Kv channels in pulmonary smooth muscle cells may change cell function: inhibition of Kv channels directly by ROS [1] or downregulation via an impact on gene transcription [2] may lead to membrane depolarization, activation of CaV channels, influx of Ca2+, and subsequent hypoxic pulmonary vasoconstriction (HPV, 1) or pulmonary arterial hypertension (PAH, 2), respectively. (B) In neurons, ROS-mediated activation of Kv channels, such as by removal of inactivation in A-type channels, may limit the influx of Ca2+ through depolarization-dependent CaV channels leading to decreased electrical excitability because of diminished transmitter release. CaV, voltage-gated Ca2+; HIF, hypoxia-inducible factor; Glu, glutamate.
Kv channels are also involved in vascular remodeling by modulating cell proliferation and apoptosis in pulmonary artery smooth muscle cells. Under chronic hypoxia, Kv channels in these cells, such as Kv1.5 and Kv2.1, are downregulated by the activation of transcription factors hypoxia-inducible factor-1 (HIF-1) and nuclear factor-activating T (NFT), which results in the pathogenesis of pulmonary arterial hypertension, a disease with 15% mortality rate (8). The loss of Kv channels causes membrane depolarization and a rise in intracellular [Ca2+] (Fig. 4A) that ultimately stimulates proliferation; the loss of Kv channels also leads to accumulation of intracellular K+, which inhibits caspases and reduces apoptosis, an event often related to the pathogenesis of cancer (7, 86).
Inactivating Kv Channels
The term “A-type currents” refers to depolarization-activated K+ currents that rapidly inactivate. In neurons, these currents play a vital role in the control of excitability, regulation of presynaptic spike duration, Ca2+ entry, and neurotransmitter release (72). In cardiac muscle, the transient outward K+ current (Ito) contributes to the initial repolarization phase of cardiac action potentials (138). Genetic analysis has revealed that A-type currents are mediated by voltage-gated K+ channels of the Kv subfamily (Fig. 2), but the molecular mechanisms leading to inactivation are diverse.
Two inactivation processes, fast and slow, are readily recognized (50, 51). Fast inactivation follows the “ball-and-chain” mechanism in which one of the four N-terminal ends plug the channel cavity (50, 51, 155). This N-type inactivation is abolished by genetic deletion or enzymatic removal of the N terminus, and it can be restored by intracellular application of peptides derived from the N terminus (5, 155). Mammalian Kv channel α-subunits capable of generating A-type currents are divided into three major classes: (i) Kv1—Shaker-related: Kv1.4, (ii) Kv3—Shaw-related: Kv3.3, Kv3.4, and (iii) Kv4—Shal-related: Kv4.1-3. A special case is the Drosophila Shaker channel, for which various splice variants are equipped with N-terminal inactivation domains. Although only a limited number of Kv α-subunits possess N-terminal structures capable of inducing N-type inactivation, some auxiliary cytoplasmic β-subunits of the Kvβ family (Fig. 5) interact with Kv1 α-subunits to create N(β)-type inactivation by contributing their N-terminal inactivation domains (44, 45, 103).
FIG. 5.

N-type inactivation. (A, B) Current traces at 40 mV from Xenopus oocytes expressing Kv1.4 (A) and Kv1.5+Kvβ1.1 (B) in the on-cell mode (black) and upon excision to the inside-out configuration (gray) indicating the redox dependence of fast inactivation. (C) Alignment of N-terminal sequences of α-subunits (top) and Kvβ subunits (bottom) that induce N-type inactivation. Cysteine (C) and methionine (M) residues are highlighted.
For historical reasons, slow inactivation is termed “C-type inactivation” (50, 51). The molecular/atomic mechanism is not well understood but the pore structure and the permeating ions play a vital role [for review see Hoshi and Armstrong (47)]; therefore, the term C/P-type inactivation is also used (76). Nearly all voltage-gated K+ channels can undergo C-type inactivation; typically this is slow but in some cases it can be on the order of 100 ms (98).
In either case, regulation of A-type currents is a very powerful tool because even a minor slowdown of inactivation or an increased steady-state noninactivating component constitutes a gain of function phenomenon, which typically has a stronger impact on the electrical properties of a cell than a partial loss of function. Therefore, the kinetics of many A-type currents is subject to a tight control by the cellular redox milieu.
ROS regulation of N-type inactivation
ROS sensitivity of N-type inactivation can be observed easily when recording current from channels expressed in Xenopus oocytes; in the on-cell configuration of the patch-clamp method, inactivation is fast, but upon patch excision, inactivation gets progressively slower as the cytosolic side of the membrane patch is exposed to ambient oxygen-saturated bath solution (Fig. 5A). Reversibility is obtained by pushing the patch back into the reducing cytosol. Such experiments were first performed by Ruppersberg et al. (106) who demonstrated the ROS sensitivity of rat Kv1.4 and Kv3.4 channels. Both channel types harbor N-terminal inactivation domains and both contain a cysteine residue (Fig. 5B). The mutation C13S in Kv1.4 rendered the inactivation insensitive to changes in the intracellular redox potential. Since Kv1.4 subunits can coassemble with other α-subunits of the Kv1 group and thereby can contribute the N-terminal domains for N-type inactivation, many Kv current components may exist under in-vivo conditions with ROS-dependent inactivation properties.
As simple as this regulation mechanism may seem, it is not unambiguously known what kind of oxidative modification actually occurs and why the inactivation is slowed down or eliminated altogether. A possible scenario is that the normally disordered N-terminal inactivation domain, which is supposed to enter the pore cavity to obstruct K+ current flow, is “immobilized” by forming a disulfide bridge with another protein-based thiol group. Such other group could be a cysteine of one of the other N-terminal domains, a cysteine elsewhere in the α-subunit that is accessible to the N terminus, or an auxiliary protein. Alternatively, covalent attachment of small soluble thiol-containing molecules such as glutathione may change the structure of the N-terminal inactivation domain in a manner sufficient to prevent inactivation. The cysteine itself can be oxidized by hydrogen peroxide (H2O2) to sulfenic (SOH) acid, to sulfinic (SO) acid, and finally to sulfonic (SO2) acid (Fig. 3) (35). Depending on the extent of oxidation, such modified cysteine will then affect inactivation in a reversible or irreversible manner.
ROS-dependent regulation of N-type inactivation may be relevant in the action of Riluzole, which is used in the treatment of amyotropic lateral sclerosis. Riluzole slowed down Kv1.4 N-type inactivation and the effect was diminished by dithiothreitol (DTT) and reduced glutathione, suggesting oxidation of C13 in the inactivation domain might be involved (148). The neuroprotective action of Riluzole could be accounted for controlling glutamate release at the axon terminal via a ROS-mediated increase in Kv1.4 current (148).
A-type currents, primarily mediated by Kv1.4 channels, are also found in small dorsal root ganglia neurons important for pain signaling. Since oxidative challenge removed fast inactivation of K+ channels in these cells, redox modulation of N-type inactivation could be an interesting target for the physiology and pharmacology of pain signaling (53). A plausible scenario of how ROS-dependent removal of N-type inactivation may affect cellular excitability and glutamate release is illustrated in Fig. 4B.
Aging of C. elegans
A link of redox modulation of A-type currents to the process of aging was provided for KvS-1 (a paralog to Kv2.1) channels in C. elegans with a cysteine in the N terminus (C113) (15, 117). Endogenous ROS, which slow down inactivation of KvS-1 because of C113, are responsible for a loss of sensorial capacity during aging. Transgenic worms expressing wild-type and KvS-1-C113S channels revealed that the mutation leads to a preservation of chemotaxis during aging, suggesting that the process of ROS-mediated modulation of N-type inactivation could contribute to the age-associated neuronal degeneration (15, 117).
Kvβ subunits
ROS-regulated N-type inactivation seems to be a potent and widespread phenomenon because several Kvβ subunits also harbor N-terminal inactivation domains with ROS-sensitive cysteine residues (Fig. 5B, C). The first identified was Kvβ1.1, which harbors a cysteine at position 7 (103). Other examples are Kvβ3 [C7 and C22 (44)] and Kvβ1.2 (C8). Since such cytosolic Kvβ subunits can dock to α-subunits of the Kv1 group, many delayed rectifier K+ channels can be transformed to A-type channels with rapid Kvβ-induced inactivation. The occurrence of ROS-dependent and ROS-independent Kvβ subunits suggests that, by means of gene transcription and alternative splicing, a cell can generate K+ current components with degrees of inactivation and sensitivities tailored to changes of the cellular redox status.
In addition to the ability of some Kvβ subunits to induce N-type inactivation, they appear to have an enzymatic function as oxidoreductases, and this enzymatic activity can modulate fast inactivation via oxidation of the cofactor NADPH. Pan et al. (95, 96) showed that either enzymatic conversion of NADPH to NADP+ by a substrate or direct application of NADP+ to excised membrane patches with Kv1.1/Kvβ1.1 channels leads to changes in fast N-type inactivation. Interestingly, such results were also obtained with mutant Kvβ1.1-C7S, suggesting a unique mode of redox modulation without involving this critical cysteine residue (95, 96, 143). Further studies suggested that NADP+ binding to the Kvβ1.1 core domain influences the availability of the Kvβ1.1 N-terminal ball domain (96); likewise, the inactivation may in turn influence the catalytic activity.
Methionine oxidation
A special case of ROS-dependent inactivating Kv channels is the C/B splice variant of Shaker K+ channels in Drosophila (137). These channels (Sh C/B), when expressed in Xenopus oocytes, display a very noticeable variability in inactivation time course. Inspection of the N-terminal inactivation domain, however, does not reveal any cysteine residue. Application of the mild oxidant chloramine-T (ChT) potently removed inactivation suggesting that residues other than cysteine might be modified in an oxidative manner. Sh C/B channels contain a methionine at position 3 (Fig. 5C); when mutated to leucine, the variability of inactivation speed disappears (20).
Following cysteine, methionines, which also harbor a sulfur atom, are the residues that are the next most susceptible to oxidation. In a first oxidative reaction, methionine is oxidized to methionine sulfoxide [Met(O)], in a second to methionine sulfone [Met(O2)]. While Met(O2) formation is physiologically irreversible, Met(O) can be reduced back to methionine by the aid of peptide methionine sulfoxide reductases (Msr) (Fig. 3) (48, 141). When mammalian variants of MsrA were coexpressed in Xenopus oocytes, inactivation of Sh C/B channels was fast and did not display a large scatter (20, 67). In addition, inactivation peptides with the Sh C/B sequence but with Met(O) instead of Met at position 3 did not induce inactivation in N-terminal-deficient Kv channels, while incubation of such peptides with MsrA and DTT partially restored their capability of mimicking inactivation (20). Methionine oxidation appears to be a relevant mechanism as even treatment of the oocytes with the antioxidant vitamin C had an influence on the inactivation time course of Sh C/B channels (21).
It is unclear whether oxidation of M3 in the N-terminal ends of Sh C/B channels is reversible under physiological conditions or whether the activity of MsrA in oocytes simply produces enough scavenging methionine groups to keep the cytosolic oxidative challenge low (48). Further, it must be considered that oxidation of methionine leads to two diasteromers of methionine sulfoxide, namely Met-S-(O) and Met-R-(O), which require different types of Msr: MsrA reduces Met-S-(O) while MsrB reduces Met-R-(O) (Fig. 3) (64, 69). In fact, coexpression of human MSRA and human MSRB2 with Sh C/B in Xenopus oocytes had a stronger effect than MSRA alone arguing that complete protection and/or reduction can be obtained when both diasteromers of Met(O) are addressed (62).
The mechanism by which methionine oxidation affects inactivation most likely is related to the change of the side-chain's physicochemical properties when an oxygen atom is attached to the sulfur. The residue will become less hydrophobic than methionine (12) and may thus eliminate a hydrophobic interaction of the critical methionine residue of the inactivation ball domain with the cavity of the channel. Aside from all N-terminal start methionines, mammalian sequences of N-terminal inactivation domains also contain methionine residues (e.g., M5 and M17 in Kv1.4, Fig. 5C; M29 in Kv3.4); methionine oxidation and enzymatically-driven reduction may be a regulatory mechanism existing beyond Sh C/B channels.
KCNA7 transcripts are found in the heart and skeletal muscle where the channels possibly play a role in repolarization of the action potential (36). In mouse, there are two isoforms of Kv1.7, where the long variant appears to be capable of undergoing N-type inactivation. However, in contrast with other N-type inactivating Kv channels, exposure to oxidants such as DTDP or H2O2 accelerated inactivation. Several redox-sensitive residues of the Kv1.7 protein (H21, C41, C43, and C44) were identified to take part in this process (36).
ROS regulation of C/P-type inactivation
While N-type inactivation is restricted to a few members of the Kv subfamily, almost all of them can undergo some sort of C/P-type inactivation. Thus, redox-mediated modulation of C/P-type inactivation may be a powerful tool to regulate the cellular availability of voltage-gated K+ channels. When exposed to oxidative challenge, inactivation of Shaker channels without N-terminal inactivation domains was accelerated, and the mutation M440L in the pore-segment of the α-subunit abolished that effect. M440, strongly conserved among Kv channels, is located at the apex of the pore helix inside the cavity (17). However, it is unlikely that this site can be reduced by Msr and thus this represents an irreversible modification of M440. Likewise, exposure of Shaker channels to extracellular oxidants also accelerates inactivation and this effect was diminished by the mutation M448L (113). Again, this extracellularly accessible methionine at the pore entrance close to the selectivity filter is conserved and, hence, this kind of oxidative influence on P/C-type inactivation may also apply for many other Kv channels.
KCNQ Channels
Kv7 (KCNQ) channels are voltage-gated K+ channels known for their role in raising the threshold for action potential firing and stabilizing the resting membrane voltage in excitable cells. They are predominantly expressed in brain, heart, epithelia, and vestibular and auditory organs (52). In neurons, KCNQ underlies M-currents; the corresponding channels are mainly composed of Kv7.2/Kv7.3 heteromultimers and Kv7.4 homomultimers. M-currents were first recorded in frog neurons and later in mammalian peripheral and central neurons; stimulation of muscarinic acetylcholine receptors (hence the name “M”) inhibited the currents, depolarized neurons, and increased neuronal excitability (14). The channels activate slowly at around −60 mV and do not inactivate at neuronal resting potentials. These properties enable Kv7 channels to stabilize the neuronal resting potential. They act as a “brake” on action potential firing when the neuron is exposed to an excitatory stimulus (42). In the heart, Kv7.1 contributes to the slowly activating K+ current IKs, which is essential for action potential termination. The underlying channel is a macromolecular complex composed of α- (Kv7.1) and modulatory β- (KCNE1) subunits. Mutations in either of them can give rise to congenital long QT syndrome and, hence, this channel was originally named KvLQT1 (59, 93).
Structurally, Kv7 channels are different from other channels of the Kv family with respect to a short cytoplasmic N terminus (∼100 residues) and lack of a tetramerization (T1) domain. In addition, Kv7 channels harbor a large C-terminal domain (300–500 residues) with four α-helices, in which two proximal helices serve as a scaffold for cytoplasmic regulatory molecules and also facilitate oligomerization (52).
Kv7 channels have been reported to show oxidation sensitivity. H2O2 at physiological concentrations markedly increases M-currents formed by three of five Kv7 channels (40). Application of 5 μM H2O2 to Kv7 channels expressed in Chinese hamster ovary cells increased currents mediated by Kv7.2/3 heterooligomers and Kv7.4 homooligomers while Kv7.1 and Kv7.3 homooligomers were not affected. A cysteine triplet in the S2–S3 linker of Kv7.4 (C112, C175, and C519) was identified as critical for this effect. These three cysteine residues are conserved in all Kv7 channels except for Kv7.1, suggesting that M-currents are specifically modulated by oxidation. Using a hypoxia-induced model of neurodegeneration by recording from hippocampal slice cultures exposed to oxygen/glucose deprivation, it was further found that enhanced M-currents induced by ROS provide protective effects on oxidative stress-related neurodegeneration. The ROS-triggered enhancement of M-currents could possibly provide a mechanism protecting against oxidative stress-induced excitotoxicity caused by overactivation of ionotropic glutamate receptors (40).
In primary sensory (nociceptive) neurons, substance P enhanced M-type K+ channel activity by releasing ROS from the mitochondria and thereby reduced excitability. The underlying mechanism involves activation of neurokinin receptor 1 and Gi/o proteins and provides a ROS-related link between the neuromodulator and hyeralgesia (71).
NO has been shown to influence IKs in guinea-pig cardiomyocytes. Bai et al. (9) showed that the application of Ginsenoside Re, a traditional drug from Panax ginseng, generally known for its antiarrhythmic and vasorelaxing effect, caused an enhancement of IKs in isolated ventricular myocytes from guinea pig. The enhancement of IKs was abolished in the presence of inhibitors of NO synthase and an NO scavenger, suggesting that NO is responsible for the stimulation of IKs. Further, application of NO donor mimicked Ginsenoside Re and the effect was not mediated via the adenylate cyclase, cAMP-dependent protein kinase, and the serine–threonine phosphatase 2A pathway. The stimulatory effect of NO was reversed by DTT suggesting that NO possibly facilitates S-nitrosylation of a cysteine residue in Kv7.1 channels. This finding could explain the cardioprotective effect of Ginsenoside Re, which underlies NO-redox signaling (9).
In sensory neurons from dorsal root ganglia NO inhibited M-current and increased neuronal excitability (92). It further counteracted the stimulatory effect of ROS on Kv7.4 channels involving a triad of cysteine residues in the S2–S3 linker (C156–158 in Kv7.4). M-channels therefore appear to be regulated by NO and ROS in a reciprocal manner and possibly serve as dynamic redox sensors with a vital role in neuronal excitability (92).
EAG Channels
The EAG subfamily of voltage-gated K+ channels was named according to its first member, the EAG channel, first identified in a mutant of D. melanogaster exhibiting leg-shaking behavior under ether anesthesia (63). Later, mammalian homologs were discovered and formed the EAG group within the EAG subfamily (10, 41). In humans, the EAG subfamily consists of three groups and comprises a total of eight channels (Fig. 2): (i) EAG—EAG1-2 (Kv10.1-2); (ii) EAG-related gene (ERG)—ERG1-3 (Kv11.1-3); (iii) EAG-like K+ channel (ELK)—ELK1-3 (Kv12.1-3) (32, 80, 107, 110, 118, 119). The overall structure of EAG subfamily members is different from other voltage-gated K+ channels in that they possess long N-terminal intracellular domains comprising a Per-Arnt-Sim (PAS) domain, a C-terminal cyclic nucleotide-binding homology domain (cNBHD), and a C-terminal C-linker (e.g., Fig. 6). The functions of PAS and cNBHD domains are not yet understood (10), but PAS domains are often involved in oxygen sensing (132). The C-linker seems to play an important role for EAG channels, and for Kv10.1 and Kv11.1 it has been shown that its function is subject to oxidative modulation (66, 108). It is noteworthy that the C-linker region has been postulated to be a critical component for gating of cyclic nucleotide-modulated channels such as HCN (156) and CNG (164), whose amino-acid sequences are similar to that of the EAG family of channels.
FIG. 6.

Topological model of a human Kv10.1 (EAG1) subunit. Cysteine residues that are implicated in ROS-dependent changes of channel function (108) are indicated by number. CaM-N and CaM-C, N- and C-terminal-binding sites for calmodulin; PAS, Per-Arnt-Sim domain; cNBHD, cyclic nucleotide-binding homology domain.
The EAG group
EAG1 (Kv10.1) was the founding member of the EAG subfamily (142). It is characterized by (i) the dependence of its activation kinetics on the prepulse potential and extracellular Mg2+ ions (80, 134), and (ii) its inhibition by intracellular Ca2+/calmodulin (115, 163). Kv10.1 is expressed in the brain where it presumably regulates neuronal excitability. However, Kv10.1-deficient mice display normal behavior, and no obvious histological or functional changes are observed (139). On the other hand, morpholino-mediated knockdown of Kcnh1 in zebrafish indicates an important role in early development (122). Kv10.1 is abnormally overexpressed in many cancer cells where it facilitates cell cycle progression, proliferation, and migration, and it is considered as a potential cancer biomarker (100, 104).
Kv10.1 expression is linked to oxygen homeostasis as revealed by the following observations: (i) mild hypoxia (5% O2, 4 h) induced a clear increase in HIF1 abundance in Kv10.1-expressing cells while no increase in HIF1 was observed under normoxia; (ii) Kv10.1-expressing tumors show increased vascularization and promotes VEGF secretion. This finding suggests that transformative properties of Kv10.1 are dependent on an interference with the cellular oxygen homeostasis system. Increased vascularization would augment oxygen and nutrient supply to tumor cells and that could provide growth advantage for Kv10.1-expressing cells (30).
Our studies have shown that Kv10.1 is exquisitely sensitive to changes in the intracellular redox conditions. Ionic currents through heterologously expressed Kv10.1 channels are progressively inhibited by treatment with cysteine modifiers such as DTNB, MTSES+, MTSET−, and H2O2 (Fig. 7). A systematic study of cysteine mutants and functional assays revealed that modification of Kv10.1 by cysteine-specific agents contains two components. A rapid component resulted in an alteration of gating parameters: (i) a right-shift of half-activation voltage, (ii) slowdown of activation kinetics, (iii) acceleration of deactivation kinetics, and (iv) a reduction of Po. A slow component caused complete loss of function. While the latter component strongly depends on two cysteines in the C-linker structure (C532:C562, Fig. 6), the fast component is mediated by cysteine residues in the equivalent linker region of the N terminus (C145:C214), suggesting that those protein structures connecting the large cytosolic protein domains to the transmembrane voltage-sensor and pore/gate modules are important redox-dependent relay stations.
FIG. 7.

Voltage-activated K+ channels. (A) Whole-cell current recordings from HEK 293 cells expressing Kv10.1 channels with depolarizing voltage steps ranging from −70 to 50 mV before (ctrl) and after application of bath solution containing H2O2. (B) Maximal currents from the data shown in (A) as a function of voltage.
The ERG group
Kv11.1 (ERG1) is the most prominent member of the EAG subfamily. It is expressed in cardiac tissue, nervous tissues, smooth muscles, and abnormally overexpressed in tumor tissues (11, 43). The best know function is related to its appearance in the heart where it forms the basis for the rapid delayed-rectifier potassium current (IKr). This current component plays a vital role in the repolarization phase of cardiac action potentials (10, 116). This particular function can be achieved because, upon depolarization, Kv11.1 channels only yield a small steady-state outward current because of fast inactivation and slow activation, while, upon repolarization, rapid recovery from inactivation and slow deactivation give rise to large outward currents (114). Hereditary mutations in the KCNH2 gene or drug-mediated interference with Kv11.1 channels therefore can lead to long QT syndrome and cardiac arrhythmia (68). In neurons, Kv11.1 regulates the spike frequency and controls the resting membrane potential (43, 68). The isoforms Kv11.2 (hERG2) and Kv11.3 (hERG3) are primarily expressed in the brain (119).
Taglialatela et al. (127) showed that ROS induced by iron/ascorbate (Fe/asc) increased Kv11.1-mediated outward current in Xenopus oocytes while the inward current was not affected. This augmentation of Kv11.1 current by ROS was caused by a 12-mV right-shift of the steady-state inactivation. ROS scavengers such as catalase, superoxide dismutase, and mannitol abolished the stimulatory effect of Fe/asc. Molecular analysis identified histidine residues at positions 578 and 587 within the S5–S6 linker to be responsible for the ROS-induced activation of Kv11.1 channels (Fig. 8) (97). This finding suggested that the increased liberation of Fe2+ from hearts during global ischemia followed by reperfusion might promote the Fenton reaction to yield high amounts of ROS; the subsequent oxidation of two histidine residues, presumably to 2-oxo-histidine, then may facilitate the opening of Kv11.1 channels. The Fe/asc-induced Kv11.1 channel activation may give rise to an anti-arrhythmic influence by repolarizing the membrane potential and shortening the action potential duration. Further, the stimulatory effect of Fe/asc on the Kv11.1 channel was counteracted by NO· produced by L-arginine (128). Of note, primarily neuronal Kv11.2 and Kv11.3 do not have the critical Fe/asc-sensitive histidines, suggesting that the histidine-directed redox regulation of IKr might be tissue specific.
FIG. 8.

Topological model of a human Kv11.1 (ERG1) subunit. Cysteine, methionine, and histidine residues that are implicated in ROS-dependent changes of channel function (66, 97) are indicated by number.
Studying Kv11.1 channels expressed in HEK 293 cells, Kolbe et al. (66) demonstrated that Kv11.1 current was inhibited by the cysteine modifier MTSES+, MTSEA, and DTNB when applied from the cytosolic side while the modifiers remained ineffective when applied from the extracellular side. However, extracellular application of either H2O2 or tert-butyl hydroperoxide (tBHP) caused a small transient increase of tail currents at −40 mV, followed by a slower but potent inhibition of the currents. This observation suggested that the transient increase could be due to modification of the two extracellular histidines and the subsequent loss of function due to some intracellular sides of the channel protein. Detailed screening of critical cysteine residues revealed that C723, located in the C-linker region, is most prominently responsible for the channel's oxidation sensitivity; C828 and C740 contribute to a smaller extent (Fig. 8) (66). Mutation of H578:H587 diminished the transient current increase observed with H2O2/tBHP application (66).
Interestingly, oxidative inhibition of Kv11.1 currents was strongly dependent on the deactivation kinetics. The Kv11.1 splice variant “Kv11.1b (hERG1b),” where the N terminus is truncated and deactivation is much faster than in Kv11.1a, was less sensitive toward thiol-directed modification. This finding suggests that the overall oxidation sensitivity of IKr in human cardiac myocytes, comprised of Kv11.1a and Kv11.1b (61), may depend on the composition of both Kv11.1 variants (66).
Methionines of Kv11.1 channels are also sensitive to oxidation. Su et al. (123) have shown that ChT, which preferentially oxidizes methionine, significantly decreased Kv11.1 current. The activation was slowed during depolarization to 30 mV but accelerated during depolarization to 0 or −10 mV and the deactivation was also accelerated upon repolarization. In the presence of recombinant bovine MsrA a diminished effect of ChT was observed (123). The result indicated that methionine oxidation is at least partially responsible for the Kv11.1 current inhibition. This finding was indeed confirmed by a mutagenesis study of Kolbe et al. (66) who observed that three methionine residues located in the C terminus (M554, M651, and M713) contribute to the inhibitory effect of ChT. The influence of ChT was significantly reduced by either mutation of the critical methionine triplet (M554:M651:M713) to leucines or by mutation (C723S), suggesting that the redox sensitivity of Kv11.1 channels is largely cysteine and methionine dependent (66). Collectively, the studies on the redox regulation of the Kv10.1 and Kv11.1 channels underscore the importance of the C-linker and cNBHD segment in gating of the channel and its coupling to oxidative processes (66, 108).
Abnormal QT prolongations were often observed in diabetic patients including type-1 insulin-dependent and type-2 noninsulin-dependent populations and the prolongations also are one of the major causes of sudden cardiac death in cardiac patients (105, 140, 158, 160). Under glucose stress, either by hyperglycemia or hypoglycemia, the Kv11.1 current was reduced, which is a likely cause of IKr dysfunction and QT prolongation. Moreover, under hyperglycemia Kv11.1 was downregulated at the protein level and insulin prevented QT prolongation and rescued IKr function (158). It was also shown for Kv11.1-expressing HEK 293 cells that treatment with the superoxide anion generating system xanthine/xanthine oxidase significantly diminished Kv11.1 current; co-treatment with vitamin E ameliorated that effect suggesting that the antioxidant vitamin E protects Kv11.1 from oxidative damage in diabetic conditions (161). To confirm that the observed Kv11.1 current reduction under hyperglycemia was due to modification of critical oxidation-sensitive residues, Kolbe et al. (66) compared the effect of low and high glucose conditions on the current densities obtained with wild-type and Kv11.1-C723S channels in HEK 293 cells; while the current density for wild-type was significantly smaller in high-glucose condition, Kv11.1-C723S channels were unaffected, strongly suggesting that C723 is a target of ROS produced during conditions of hyperglycemia.
Changes in the oxygen level also affects Kv11.1 channels. Long-term exposure of neuroblastoma (SH-SY5Y) cells to hypoxia (0.1% O2) dramatically altered Kv11.1 currents by causing a 20-mV shift in the voltage dependence of the time constants of recovery from inactivation and deactivation kinetics, and a −19 mV left-shift in steady-state activation (37). Another study by Nanduri et al. (88) found that hypoxia (1% and 5% O2) decreased the synthesis of recombinant Kv11.1 in HEK 293 cells in a proteosomal degradation independent fashion. Moreover, the antioxidant N-acetyl cysteine (NAC) prevented the downregulation of Kv11.1 protein under hypoxia, suggesting that hypoxia enhances the ROS production in the mitochondria and, as a result, Kv11.1 translation was downregulated (88). Carotid bodies have been known to possess O2-sensitive K+ channels that contribute to depolarization upon hypoxia. The O2 sensitivity of dissociated type-I cells from rat carotid body changes with age until 14–16 days (102). Kim et al. (65) found an E-4031-sensitive K+ current (E-4031 is a specific inhibitor of Kv11.1 channels) in 1-day-old rat carotid chemoreceptor cells, and that component was absent in 16-day-old rats. Moreover, E-4031 induced an increase in the cytosolic Ca2+ concentration in response to hypoxia in young cells but not in cells from 16-day-old rats, suggesting that the presence of Kv11.1 currents in the carotid body may dampen the ability of hypoxia to induce depolarization in an age-dependent manner.
Large-Conductance Voltage- and Ca2+-Dependent K+ Channels
Each Slo1 subunit in a tetrameric Slo1 voltage- and Ca2+-dependent BK channel, depending on the alternative splicing process, has ∼1100 residues (residue numbering according to human Slo1 AAB65837). The transmembrane domain of the Slo1 subunit is organized in a manner similar to that of other voltage-gated K+ channels (Fig. 1C) except for the additional transmembrane domain S0 (Fig. 9A). The Slo1 subunit possesses a large cytoplasmic area containing two domains called RCK1 and RCK2 (60). Four sets of the RCK1-RCK2 pairs form the intracellular “gating ring” (145, 153, 154), which undergoes conformational changes on Ca2+ binding (57, 85) leading to opening of the ion conduction gate. Each of the four subunits has two distinct Ca2+ sensors with different functional properties: the RCK1 Ca2+ sensor and the RCK2 Ca2+ sensor (“Ca2+ bowl”) [summarized in Hoshi et al. (49)].
FIG. 9.

Slo1 BK channels. (A) Topological model of a Slo1 BK α-subunit. Cysteine and methionine residues presumably important for the channel's oxidation sensitivity are indicated by residue number. RCK, regulator of conductance for K+ domains. (B) Plausible structure of a Slo1 BK channel complex. The transmembrane domain is a homology model based on the Kv1.2/2.1 structure (PDB 2R9R). The cytoplasmic domain is according to the structure of Wu et al. (PDB 3NAF) obtained in the absence of added Ca2+. S0 is not shown because Kv1.2/2.1 does not contain S0. Sulfur atoms of cysteines in the cytosolic domain are shown in yellow, methionines in green. Side view (top), bottom view (bottom). Numbers indicate important residues. BK, big conductance K+. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Slo1 BK channels, made of four pore-forming Slo1 subunits and often with auxiliary β- or γ-subunits, are dualistically activated by membrane depolarization and an increase in intracellular Ca2+ concentration ([Ca2+]i) (46, 49). The ion conduction gate in the PGD near the selectivity filter (135, 144, 162) typically has a finite but extremely low Po in the absence of activation of the VSDs and of the Ca2+ sensors (Fig. 9A). However, operating in an allosteric manner, activation of all four VSDs by depolarization can increase Po by ∼160,000-fold and binding of Ca2+ to all Ca2+ sensors in the channel can increase Po by ∼4000-fold (46).
Any one of the allosteric gating components of the Slo1 BK channel, the PGDs, VSDs, and Ca2+ sensors, and the strengths of allosteric interactions among them could be subject to oxidative regulation. Numerous examples of changes in functional properties of native and heterologously expressed Slo1 BK channels by various experimental manipulations of the redox status have been reported. In some of the cases, the biophysical mechanisms and the molecular components responsible for the phenomena have been elucidated but the atomic structural and physicochemical bases remain largely unknown. The reported cases of oxidative modifications of Slo1 BK channels appear to involve modification of cysteine and/or methionine residues (Fig. 9). A typical human Slo1 protein (e.g., AAB65837) has 29 cysteine and 30 methionine residues, that is, 116 and 120, respectively, in one tetrameric complex. Oxidation of other amino acids, such as histidine and tryptophan, has not been explicitly implicated. Depending on their locations in the protein (e.g., extracellular, transmembrane, intracellular) and reactivity (e.g., adjacent residues and pH), any and many of the cysteine and methionine residues could be oxidatively modified. However, oxidative modifications of only a subset of the residues may lead to observable functional changes.
Oxidative status of cysteine in Slo1
Consistent with the oxidizing nature of the extracellular compartment, the two cysteine residues of Slo1 facing the extracellular side (C14 and C141) are disulfide linked under ambient conditions when heterologously expressed (73) (Fig. 9A). The remaining cysteine residues are more likely to remain reduced. Consequently, the side chains should contain a free SH or S− group depending on the local environment, and they retain potential to be oxidized or modified. The atomic structures of the cytoplasmic domain (145, 153, 154) show that many cysteine residues reside near the protein surface (e.g., C430, C612, C615, C695, C995, C1001, and C1028). Further, some cysteine residues are found in the close vicinity of each other; their Sγ atoms are within 2.4 to 5.5 Å of each other (C348-C422, C722-C800, C797-C800, and C1011-C105). In comparison, the typical disulfide bond length is about ∼2 Å. Assuming that the atomic structures are physiologically relevant, the aforementioned cysteine pairs have potential to be oxidized to form disulfide bonds. The side chains of other cysteine residues may be modified to form sulfenic, sulfinic, or sulfonic groups (Fig. 3). It is possible that the sulfenic groups may react with glutathione under physiological conditions to form mix disulfides (35).
Functional consequences of cysteine oxidation in Slo1
Currents through heterologously expressed human Slo1 channels progressively decrease in size after patch excision with 1 or 100 μM [Ca2+]i (28). During this rundown process, the activation kinetics slows, and the macroscopic conductance-voltage curve (GV curve) becomes shallower and shifts to the positive direction without altering the maximal conductance (but see below) or the underlying unitary current size. These functional changes are readily reversed by the reducing agent DTT and mimicked by the oxidizing agent H2O2, leading to the idea that oxidation of cysteine caused the gating changes observed. The extent of rundown of Slo1 channels may depend on [Ca2+]i. The rundown is less noticeable in the virtual absence of Ca2+ (129, 130, 159) and most prominent at intermediate concentrations of Ca2+ (159), indicating that the Ca2+-dependent activation mechanism of the channel is subject to oxidative regulation. Note that rundown may be observed in some patches without Ca2+, suggesting that multiple mechanisms and multiple targets may be operative (129). While one study (28) reported no change in the maximal macroscopic conductance, other studies documented a clear decrease in the maximal conductance and the number of active channels during rundown, which is reversed by the reducing agent DTT (121, 159).
Cysteine 911 in the RCK2 Ca2+ sensor
Of the two Ca2+ sensors, the RCK2 Ca2+ sensor (“Ca2+ bowl”) is influenced more readily by cysteine oxidation (130). In particular, C911 is located in close proximity of the RCK2 Ca2+ sensor; the distances between the Ca2+-ligating carbonyl oxygen atoms in the RCK2 Ca2+ sensor and the Sγ atom of C911 are ∼7 to 13 Å. The oxidant H2O2 or the thiol modifier MTSEA impairs the contribution of the RCK2 Ca2+ bowl sensor, but not of the RCK1 sensor, to the overall Ca2+-dependent activation process of the channel. Mutation of C911 noticeably diminishes the propensity of Slo1 channels to rundown on patch excision (159) and impairs the effects of H2O2 and MTSEA at moderate [Ca2+]i (127). The absence of a free SH/S− group at position 911 thus disrupts the normal function of the RCK2 Ca2+ sensor and the Ca2+-dependent gating of the Slo1 channel.
Cysteine 430
Oxidation/modification of cysteine residues other than C911 may also lead to noticeable functional changes. Cysteine modifiers such as DTNB, NEM, MTSES−, and MTSET+ stimulate or inhibit currents through Slo1 channels (127, 158). A detailed biophysical analysis suggests that these thiol modifiers alter multiple components of the allosteric gating mechanism such as the intrinsic stability of the ion conduction gate and its coupling to the Ca2+ sensors and VSDs (158). One of the residues critical in mediating the channel's sensitivity to the cysteine modifiers is C430. This residue faces the inner central opening of the gating ring domain and the mutation C430A abolishes many of the effects observed (158). C430 is 25 to 30 Å away from the RCK1 Ca2+ sensor and the RCK2 Ca2+ sensor, and it is unlikely to participate directly in Ca2+ sensing of the channel. Comparison of the gating ring structures obtained under different conditions (PDB IDs 3NAF vs. 3U6N) suggests that the region containing C430 may exhibit a moderate conformational change (∼4 Å) (49). This gating ring movement may be altered by modification of the side chain, affecting multiple gating characteristics. Mutation C430A, however, does not appreciably antagonize the inhibitory effect of H2O2 at 5 μM [Ca2+]i (159), illustrating the distinct functional consequences of oxidation of different cysteine residues.
Cysteines 612 and 615
Free heme is thought to bind to the sequence CKACH located in the unstructured RCK1-RCK2 linker segment with nM affinities (56, 131, 151). The cysteine residues in this sequence, C612 and C615, modulate the affinity to the heme binding domain peptide to heme; the reduced peptide binds heme more tightly than the oxidized form (151). Electrophysiologically, the mutation C615S eliminates the effect of MTSET on the apparent Ca2+ affinity of the channel, suggesting that modification of the C615 side chain may occur. Because C615 is near the periphery of the gating ring domain and ≥30 Å away from the Ca2+ sensor sites, the effect is probably indirect or allosteric.
STREX insert
The SLO1 transcript is extensively spliced to create many variants (18, 109). Some native Slo1 channels contain an insertion of 58 residues, of which 6 are cysteine, termed STREX (coded by the “stress regulated exon”) between the RCK1 and RCK2 domains (136, 147). Select cysteine residues in the STREX insert are palmitoylated and mediate the interaction of the STREX-containing channel with the plasma membrane in a phosphorylation-dependent manner (136). Application of the thiol modifier thimerosal, which is capable of creating ethylmercury adducts, decreases Po of the STREX-containing Slo1 channel at moderate [Ca2+] and this inhibition is reversed by the reducing agent DTT (34). Inclusion of STREX may offer another mechanism of regulation of the Slo1 channel by cysteine oxidation. The STREX insert is also implicated in hypoxia sensing by the Slo1 channel but the mechanism is redox independent (84).
Other cysteine residues?
While mutagenesis of C911, C430, and C615 diminishes many effects of application of H2O2 and select thiol modifiers, oxidation/modification of other cysteine residues could also contribute. The behavior of the ion conduction gate is intimately associated with the conformation changes of the gating ring domain (57, 85). Any change in the gating ring domain motion has potential to alter the channel gating and oxidative modification of the cysteine side chain at multiple positions may exert such an effect.
Functional consequences of methionine oxidation in Slo1
Of 30 methionine residues in Slo1, 11 of them are located in the transmembrane domain (N through S6; Fig. 9A) and the remainder in the cytoplasmic domain. As with cysteine, those facing the extracellular domain (M1, M10, and M21) are exposed to a more oxidizing condition. However, the exact oxidation status of these residues, whether their side chains exist as Met, Met(O), or Met(O2) is unknown. The methionine residues within the membrane and the cytoplasmic domain are more likely to be reduced under basal conditions and retain potential to be oxidized. Some of them are clearly solvent exposed in the available structures (M442, M513, M663, M679, M712, and M1053). The remaining methionine residues face the protein interior, consistent with the notion that methionine is nonpolar. Since Met(O) is much more hydrophilic and comparable to aspartate, glutamate, and lysine (12), oxidation of buried methionine residues to Met(O), if it occurs, may have a greater impact on the channel function.
ChT increases Slo1 currents without any change in the single-channel current-voltage property (129). The current-enhancing effect was partially but not completely reversed by treatment with the enzyme MsrA and DTT together, implicating oxidation of methionine. In contrast with the current-enhancing effect of ChT, the cysteine modifiers DTNB, MTSEA, and PCMB all decreased currents. Biophysically, treatment with ChT moves the overall voltage dependence of activation to the negative direction by 30 mV, and this shift has been suggested to involve a change in the VSD function (129). An exhaustive mutagenesis study showed that contemporaneous substitution of three methionine residues (M536, M712, and M739) in the cytoplasmic gating ring domain eliminated the functional changes caused by ChT (112). M536 and M739 are located in nonpolar areas while M712 is solvent exposed toward the “bottom” of the gating ring domain (Fig. 9B). Presumably oxidation of the side chains of these methionine residues to Met(O) [or possibly to Met(O2)] changes the conformation of the cytoplasmic gating ring domain, which is then transmitted to the transmembrane VSDs. It is unknown whether treatment with ChT selectively oxidizes M536, M712, and M739 or that oxidation of other methionine residues has no functional sequence; biochemical or proteomic information is unavailable. The partial reversal of the effect of ChT by MsrA (129) may be mediated by its action on the solvent exposed M712. While rundown is attributed to cysteine oxidation, run-up is considered to be a consequence of methionine oxidation in Slo1, and both types of oxidation can occur so that the current size is sometimes unstable after patch excision (129).
Oxidation of β-subunits
Auxiliary β-subunits also contain multiple cysteine residues, many of which are well conserved among different β-variants and face the extracellular side (four to seven extracellular cysteine residues in human β-subunits). These cysteine residues are most probably disulfide linked. In particular, in Slo1+β3 channels, the reducing agent DTT markedly alters ion permeation properties of the channel complex (157). Whether these disulfide links are subject to physiological regulation remains unknown because the extracellular compartment is oxidizing.
Variable numbers of methionine residues are found in human β-subunits (from 3 to 11). Treatment of Slo1+β1 channels with ChT induces a larger shift in the voltage dependence (−75 mV) than that observed with Slo1 without any β-subunits (−30 mV) (108). The augmented response to ChT, however, does not require any methionine or cysteine within β1 and it is thought that β1 amplifies the functional consequence of oxidation of the aforementioned three methionine residues within Slo1 (111).
Physiological relevance
The physiological importance of Slo1 regulation by ROS remains to be elucidated. However, given the involvement of this channel in many physiological processes, fine-tuned adjustment of BK channel activity via reactive species is a likely scenario. It might be particularly relevant at sites of high ROS concentration. For example, H2O2 is generated from dismutation of superoxide anion (O2−), which is in turn created by the NADPH oxidase complex located in plasma membranes and organelles and also by the mitochondria. Thus, cellular metabolism via the NADPH oxidases and mitochondria could oxidatively influence Slo1 BK channels. Interestingly, some BK channels have been reported to exist in the inner mitochondria membrane (120, 149), where the channels may face greater concentrations of reactive species.
Oxidative modification of Slo1 channels is likely to be pathophysiologically important. One such example is found in dysfunction of Slo1 BK channels in type-2 diabetes. In Zucker diabetic fatty rats, a model system for type-2 diabetes, dilation of coronary artery vessels is impaired (79). Because Slo1 BK channels provide an important vasodilatory influence (13, 89), Lu et al. tested the hypothesis that dysfunction of Slo1 BK channels may contribute to the vascular dysfunction associated with type-2 diabetes by manipulating glucose levels in cell culture (78). A high level of glucose in the cell culture medium increased the overall ROS level and decreased Po at 0.5 μM [Ca2+]i. The high glucose medium failed to alter the C911A channel, suggesting that high glucose leads to oxidative modification of C911 and impairs the Ca2+-dependent activation of the Slo1 channel (78).
Conclusion and Perspectives
ROS regulation of voltage-gated K+ channels is a multifaceted process ranging from protein degradation to “real” regulation in the sense that oxidative events are reversible. Thus, also with respect to the channels discussed here, ROS challenge is a double-edged sword, but several cases are reported in which oxidative modification of K+ channels results in increased activity and, hence, a potential beneficial or protective influence.
Most of the studies examining oxidative regulation of Kv channels thus far utilized exogenous oxidants and modifiers, often in high concentrations. Some of these oxidants such as H2O2 are in fact found under physiological conditions, but the time- and spatially resolved quantification in cells remains a challenging task. Further, one ROS source alone may not be sufficient in many cases as, for example, the concurrent presence of transition metal ions often is required to catalyze ROS-related chemical reactions. Thus, a limiting factor of ROS-related research on ion channels is ROS monitoring in the appropriate physiological background.
Nevertheless, great progress was made in the understanding of how ion channels are regulated by reactive species. In many cases very detailed insight could be gained because the function of ion channels can be studied with very high precision—in many cases in inside-out and outside-out mode—providing access to both sides of the membrane. Voltage-gated K+ channels bear the additional advantage of allowing automated leak correction and, by means of voltage-step protocols, the exact timing of channel activation and deactivation, such that detailed quantitative data sets with high time resolution can be generated. Using recombinant channel proteins and exogenous ROS sources together with site-directed mutagenesis enables identification of potential molecular targets (e.g., side chains) and the biophysical mechanisms underlying the consequent functional changes. A technical limitation that still needs to be overcome is the ROS-related proteomics of membrane proteins—ideally in subcellular and temporal resolution—to better understand the chemical nature of oxidative protein modification. In addition, given the advent of many new 3D structures of ion channels, structural variants subjected to oxidative modulation may prove very insightful.
Abbreviations Used
- ΔVm
change in membrane potential
- BK
big conductance K+
- Ca2+
calcium ion
- [Ca2+]i
cytosolic free Ca2+ concentrations
- Cat
catalase
- CaV
voltage-gated Ca2+
- ChT
chloramine-T
- cNBHD
cyclic nucleotide-binding homology domain
- CO
carbon monoxide
- DPPX
dipeptidyl aminopeptidase-like protein
- DTDP
2,2′- or 4,4′- dithiodipyridine
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- DTT
dithiothreitol
- EAG
ether à go-go
- Fe/asc
iron/ascorbate
- Fe2+/Fe3+
iron(II) ion/iron(III) ion
- Glu
glutamate
- GPx
glutathione peroxidase
- GRed
glutathione reductase
- GSH-red/GSH-ox
glutathione, reduced and oxidized
- GV curve
conductance-voltage curve
- H2O2
hydrogen peroxide
- HIF-1α
hypoxia-inducible factor-1α
- HPV
hypoxic pulmonary vasoconstriction
- HUGO
nomenclature of human genes
- IKr
rapid delayed rectifier current
- IKs
slow delayed rectifier current
- IKur
repolarizing ultra-rapid delayed-rectifier current
- K+
potassium ion
- KChiP
K channel interacting protein
- Kv
voltage-gated potassium (channel)
- Kvβ
β-subunit of voltage-gated potassium channels
- Lck
lymphocyte-specific protein tyrosine kinase
- Met
methionine
- Met(O)
methionine sulfoxide
- Met(O2)
methionine sulfone
- Msr
methionine sulfoxide reductase
- MTSEA
2-aminoethyl methanethiosulfonate hydrobromide
- MTSES+
2-sulfonatoethyl methanethiosulfonate
- MTSET−
2-(trimethylammonium)ethyl methanethiosulfonate
- NAC
N-acetyl cysteine
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NEM
N-ethylmaleimide
- NFT
nuclear factor-activating T
- NO
nitric oxide
- O2
molecular oxygen
- ONOO−
peroxynitrite
- PAH
pulmonary arterial hypertension
- PAS
Per-Arnt-Sim
- PC-12
pheochromocytoma 12
- PCMB
p-chloromercuribenzoic acid
- PGD
pore/gate domain
- Po
open probability
- RCK
regulator of conductance for K+
- ROS
reactive oxygen species
- src
family of nonreceptor tyrosine kinases
- SOD
superoxide dismutase
- STREX
stress regulated exon
- tBHP
tert-butyl hydroperoxide
- TM
transmembrane domain
- VSD
voltage-sensor domain
- WT
wildtype
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft FOR 1738 (S.H.H., T.H.) and National Institutes of Health (T.H.).
References
- 1.Abbott GM. Molecular mechanisms of cardiac voltage-gated potassium channelopathies. Curr Pharm Des 12: 3631–3644, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Ammon HP. and Wahl MA. Islet redox ratios: their role in insulin release. In: Frontiers of Insulin Secretion and Pancreatic β-cell Research, edited by Flatt PR. and Lenzen S. London: Smith-Gordon, 1994, pp. 112–113 [Google Scholar]
- 3.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, and Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature 403: 553–556, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Annunziato L, Pannacaccione A, Cataldi M, Secondo A, Castaldo P, Di Renzo G, and Taglialatela M. Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol Aging 23: 819–834, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Antz C. and Fakler B. Fast inactivation of voltage-gated K+ channels: from cartoon to structure. News Physiol Sci 13: 177–182, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Archer S. and Michelakis E. The mechanism(s) of hypoxic pulmonary vasoconstriction: potassium channels, redox O2 sensors, and controversies. News Physiol Sci 17: 131–137, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, and Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Archer SL, Weir EK, and Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121: 2045–2066, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai CX, Takahashi K, Masumiya H, Sawanobori T, and Furukawa T. Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br J Pharmacol 142: 567–575, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer CK. and Schwarz JR. Physiology of EAG K+ channels. J Membr Biol 182: 1–15, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Faravelli L, Olivotto M, and Wanke E. herg encodes a K+ current highly conserved in tumours of different histogenesis: a selective advantage for cancer cells? Cancer Res 58: 815–822, 1998 [PubMed] [Google Scholar]
- 12.Black SD. and Mould DR. Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal Biochem 193: 72–82, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, and Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Brown DA. and Adams PR. Muscarinic suppression of a novel voltage-sensitive K+-current in a vertebrate neurone. Nature 283: 673–676, 1980 [DOI] [PubMed] [Google Scholar]
- 15.Cai SQ. and Sesti F. Oxidation of potassium channels causes progressive sensory function loss during aging. Nat Neurosci 12: 611–617, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, and Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 25: 280–289, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Avdonin V, Ciorba MA, Heinemann SH, and Hoshi T. Acceleration of P/C-type inactivation in voltage-gated K+ channels by methionine oxidation. Biophys J 78: 174–187, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Tian L, MacDonald SH, McClafferty H, Hammond MS, Huibant JM, Ruth P, Knaus HG, and Shipston MJ. Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) α-subunits generated from a single site of splicing. J Biol Chem 280: 33599–33609, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Chimote AA, Kuras Z, and Conforti L. Disruption of Kv1.3 channel forward vesicular trafficking by hypoxia in human T lymphocytes. J Biol Chem 287: 2055–2067, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciorba MA, Heinemann SH, Weissbach H, Brot N, and Hoshi T. Modulation of potassium channel function by methionine oxidation and reduction. Proc Natl Acad Sci U S A 94: 9932–9937, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciorba MA, Heinemann SH, Weissbach H, Brot N, and Hoshi T. Regulation of voltage-dependent K+ channels by methionine oxidation: effect of nitric oxide and vitamin C. FEBS Lett 442: 48–52, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Conforti L. and Millhorn DE. Selective inhibition of a slow-inactivating voltage-dependent K+ channel in rat PC12 cells by hypoxia. J Physiol 502: 293–305, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conforti L, Bodi I, Nisbet JW, and Millhorn DE. O2-sensitive K+ channels: role of the Kv1.2-subunit in mediating the hypoxic response. J Physiol 524: 783–793, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conforti L, Petrovic M, Mohammad D, Lee S, Ma Q, Barone S, and Filipovich AH. Hypoxia regulates expression and activity of Kv1.3 channels in T lymphocytes: a possible role in T cell proliferation. J Immunol 170: 695–702, 2003b [DOI] [PubMed] [Google Scholar]
- 25.Conforti L, Takimoto K, Petrovic M, Pongs O, and Millhorn D. The pore region of the Kv1.2α subunit is an important component of recombinant Kv1.2 channel oxygen sensitivity. Biochem Biophys Res Commun 306: 450–456, 2003a [DOI] [PubMed] [Google Scholar]
- 26.Cotella D, Hernandez-Enriquez B, Wu X, Li R, Pan Z, Leveille J, Link CD, Oddo S, and Sesti F. Toxic role of K+ channel oxidation in mammalian brain. J Neurosci 32: 4133–4144, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dallas ML, Boyle JP, Milligan CJ, Sayer R, Kerrigan TL, McKinstry C, Lu P, Mankouri J, Harris M, Scragg JL, Pearson HA, and Peers C. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J 25: 1519–1530, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dichiara TJ. and Reinhart PH. Redox modulation of hslo Ca2+-activated K+ channels. J Neurosci 17: 4942–4955, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dodson PD, Billups B, Rusznak Z, Szucs G, Barker MC, and Forsythe ID. Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol 550: 27–33, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Downie BR, Sánchez A, Knötgen H, Contreras-Jurado C, Gymnopoulos M, Weber C, Stühmer W, and Pardo LA. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem 283: 36234–36240, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbins JM, Cohen SL, Chait BT, and MacKinnon R. The structure of the potassium channels: molecular basis of K+ conduction and selectivity. Science 280: 69–77, 1998 [DOI] [PubMed] [Google Scholar]
- 32.Engeland B, Neu A, Ludwig J, Roeper J, and Pongs O. Cloning and functional expression of rat ether-à-go-go like K+ channel genes. J Physiol 513: 647–654, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erkinjuntti T, Roman G, and Gauthier S. Treatment of vascular dementia-evidence from clinical trials with cholinesterase inhibitors. J Neurol Sci 226: 63–66, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Erxleben C, Everhart AL, Romeo C, Florance H, Bauer MB, Alcorta DA, Rossie S, Shipston MJ, and Armstrong DL. Interacting effects of N-terminal variation and strex exon splicing on slo potassium channel regulation by calcium, phosphorylation, and oxidation. J Biol Chem 277: 27045–27052, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Finkel T. Redox-dependent signal transduction. FEBS Lett 476: 52–54, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Finol-Urdaneta RK, Strüver N, and Terlau H. Molecular and functional differences between heart mKv1.7 channel isoforms. J Gen Physiol 128: 133–145, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fontana L, D'Amico M, Crociani O, Biagiotti T, Solazzo M, Rosati B, Arcangeli A, Wanke E, and Olivotto M. Long-term modulation of HERG channel gating in hypoxia. Biochem Biophys Res Commun 286: 857–862, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Franco-Obregon A. and Lopez-Barneo J. Differential oxygen sensitivity of calcium channels in rabbit smooth muscle cells of conduit and resistance pulmonary arteries. J Physiol 491: 511–518, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchs B, Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, and Weissmann N. Redox signaling and reactive oxygen species in hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 174: 282–291, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, and Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J 25: 4996–5004, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganetzky B. and Wu CF. Neurogenetic analysis of potassium currents in Drosophila: synergistic effects on neuromuscular transmission in double mutants. J Neurogenet 1: 17–28, 1983 [DOI] [PubMed] [Google Scholar]
- 42.Hansen HH, Waroux O, Seutin V, Jentsch TJ, Aznar S, and Mikkelsen JD. Kv7 channels: interaction with dopaminergic and serotonergic neurotransmission in the CNS. J Physiol 586: 1823–1832, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He FZ, McLeod HL, and Zhang W. Current pharmacogenomic studies on hERG potassium channels. Trends Mol Med 19: 227–238, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Heinemann SH, Rettig J, Graack HR, and Pongs O. Functional characterization of Kv channel β-subunits from rat brain. J Physiol 493: 625–633, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinemann SH, Rettig J, Wunder F, and Pongs O. Molecular and functional characterization of a rat brain Kvβ3 potassium channel subunit. FEBS Lett 377: 383–389, 1995 [DOI] [PubMed] [Google Scholar]
- 46.Horrigan FT. and Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol 120: 267–305, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoshi T. and Armstrong CM. C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J Gen Physiol 141: 151–160, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoshi T. and Heinemann SH. Regulation of cell function by methionine oxidation and reduction. J Physiol 531: 1–11, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoshi T, Pantazis A, and Olcese R. Transduction of voltage and Ca2+ signals by Slo1 BK channels. Physiology 28: 172–189, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoshi T, Zagotta WN, and Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250: 533–538, 1990 [DOI] [PubMed] [Google Scholar]
- 51.Hoshi T, Zagotta WN, and Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7: 547–556, 1991 [DOI] [PubMed] [Google Scholar]
- 52.Howard RJ, Clark KA, Holton JM, and Minor DL., Jr Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53: 663–675, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsieh CP. Redox modulation of A-type K+ currents in pain-sensing dorsal root ganglion neurons. Biochm Biophys Res Comm 370: 445–449, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, and Takahashi T. Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci 23: 10445–10453, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iverson LE, Tanouye MA, Lester HA, Davidson N, and Rudy B. A-type potassium channels expressed from Shaker locus cDNA. Proc Natl Acad Sci U S A 85: 5723–5727, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, and Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res 97: 805–812, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Javaherian AD, Yusifov T, Pantazis A, Franklin S, Gandhi CS, and Olcese R. Metal-driven operation of the human large-conductance voltage- and Ca2+-dependent potassium channel (BK) gating ring apparatus. J Biol Chem 286: 20701–20709, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen MØ, Jogini V, Borhani DW, Leffler AE, Dror RO, and Shaw DE. Mechanism of voltage gating in potassium channels. Science 336: 229–233, 2012 [DOI] [PubMed] [Google Scholar]
- 59.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30, 2000 [DOI] [PubMed] [Google Scholar]
- 60.Jiang Y, Pico A, Cadene M, Chait BT, and MacKinnon R. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron 29: 593–601, 2001 [DOI] [PubMed] [Google Scholar]
- 61.Jones EMC, Roti Roti EC, Wang J, Delfosse SA, and Robertson GA. Cardiac IKr channels minimally comprise hERG 1α and 1β subunits. J Biol Chem 279: 44690–44694, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Jung S, Hansel A, Kasperczyk H, Hoshi T, and Heinemann SH. Activity, tissue distribution and site-directed mutagenesis of a human peptide methionine sulfoxide reductase of type B: hCBS1. FEBS Lett 527: 91–94, 2002 [DOI] [PubMed] [Google Scholar]
- 63.Kaplan WD. and Trout WE. The behavior of four neurological mutants of Drosophila. Genetics 61: 399–409, 1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim HY. and Gladychev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J 407: 321–329, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Kim I, Boyle KM, and Carroll JL. Postnatal development of E-4031-sensitive potassium current in rat carotid chemoreceptor cells. J Appl Physiol 98: 1469–1477, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Kolbe K, Schönherr R, Gessner G, Sahoo N, Hoshi T, and Heinemann SH. Cysteine 723 in the C-linker segment confers oxidation inhibition of hERG1 potassium channels. J Physiol 588: 2999–3009, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuschel L, Hansel A, Schönherr R, Weissbach H, Brot N, Hoshi T, and Heinemann SH. Molecular cloning and functional expression of a human peptide methionine sulfoxide reductase (hMsrA). FEBS Lett 456: 17–21, 1999 [DOI] [PubMed] [Google Scholar]
- 68.Larsen AP. Role of ERG1 isoforms in modulation of ERG1 channel trafficking and function. Pflügers Arch 460: 803–812, 2010 [DOI] [PubMed] [Google Scholar]
- 69.Lee BC. and Gladychev VN. The biological significance of methionine sulfoxide stereochemistry. Free Radic Biol Med 50: 221–227, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Gutterman DD, Rusch NJ, Bubolz A, and Liu Y. Nitration and functional loss of voltage-gated K+ channels in rat coronary microvessels exposed to high glucose. Diabetes 53: 2436–2442, 2004 [DOI] [PubMed] [Google Scholar]
- 71.Linley JE, Ooi L, Pettinger L, Kirton H, Boyle JP, Peers C, and Gamper N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc Natl Acad Sci U S A 109: E1578–E1586, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipton S. and Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330: 613–622, 1994 [DOI] [PubMed] [Google Scholar]
- 73.Liu G, Zakharov SI, Yang L, Deng SX, Landry DW, Karlin A, and Marx SO. Position and role of the BK channel α subunit S0 helix inferred from disulfide crosslinking. J Gen Physiol 131: 537–548, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long SB, Campbell EB, and MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309: 897–903, 2005 [DOI] [PubMed] [Google Scholar]
- 75.Long SB, Tao X, Campbell EB, and MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450: 376–382, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Loots E. and Isacoff EY. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J Gen Physiol 112: 346–389, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lorincz A. and Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci 28: 14329–14340, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu T, He T, Katusic ZS, and Lee H-C. Molecular mechanisms mediating inhibition of human large conductance Ca2+-activated K+ channels by high glucose. Circ Res 99: 607–616, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Lu T, Wang XL, He T, Zhou W, Kaduce TL, Katusic ZS, Spector AA, and Lee HC. Impaired arachidonic acid-mediated activation of large-conductance Ca2+-activated K+ channels in coronary arterial smooth muscle cells in Zucker diabetic fatty rats. Diabetes 54: 2155–2163, 2005 [DOI] [PubMed] [Google Scholar]
- 80.Ludwig J, Terlau H, Wunder F, Brüggemann A, Pardo LA, Marquardt A, Stühmer W, and Pongs O. Functional expression of a rat homologue of the voltage gated ether-à-go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J 13: 4451–4458, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AMF, Backx PH, and Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol Endocrinol 15: 1423–1435, 2001 [DOI] [PubMed] [Google Scholar]
- 82.MacDonald PE, Salapatek AM, and Wheeler MB. Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic β-cells. J Physiol 546: 647–653, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, Wang J, Saleh MC, Chan CB, Tsushima RG, Salapatek AM, and Wheeler MB. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic β-cells enhances glucose-dependent insulin secretion. J Biol Chem 277: 44938–44945, 2002 [DOI] [PubMed] [Google Scholar]
- 84.McCartney CE, McClafferty H, Huibant JM, Rowan EG, Shipston MJ, and Rowe IC. A cysteine-rich motif confers hypoxia sensitivity to mammalian large conductance voltage- and Ca-activated K (BK) channel α-subunits. Proc Natl Acad Sci U S A 102: 17870–17876, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miranda P, Contreras JE, Plested AJ, Sigworth FJ, Holmgren M, and Giraldez T. State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc Natl Acad Sci U S A 110: 5217–5222, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moudgil R, Michelakis ED, and Archer SL. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13: 615–632, 2006 [DOI] [PubMed] [Google Scholar]
- 87.Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, and Rudy B. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 37: 449–461, 2003 [DOI] [PubMed] [Google Scholar]
- 88.Nanduri J, Wang N, Bergson P, Yuan G, Ficker E, and Prabhakar NR. Mitochondrial reactive oxygen species mediate hypoxic down-regulation of hERG channel protein. Biochem Biophys Res Commun 373: 309–314, 2008 [DOI] [PubMed] [Google Scholar]
- 89.Nelson MT. and Bonev AD. The β1 subunit of the Ca2+-sensitive K+ channel protects against hypertension. J Clin Invest 113: 955–957, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nielsen NH, Winkel BG, Kanters JK, Schmitt N, Hofman-Bang J, Jensen HS, Bentzen BH, Sigurd B, Larsen LA, Andersen PS, Haunsø S, Kjeldsen K, Grunnet M, Christiansen M, and Olesen SP. Mutations in the Kv1.5 channel gene KCNA5 in cardiac arrest patients. Biochem Biophys Res Commun 354: 776–782, 2007 [DOI] [PubMed] [Google Scholar]
- 91.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, and Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet 15: 2185–2191, 2006 [DOI] [PubMed] [Google Scholar]
- 92.Ooi L, Gigout S, Pettinger L, and Gamper N. Triple cysteine module within M-type K+ channels mediates reciprocal channel modulation by nitric oxide and reactive oxygen species. J Neurosci 33: 6041–6046, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Osteen JD, Sampson KJ, and Kass RS. The cardiac IKs channel, complex indeed. Proc Natl Acad Sci U S A 107: 18751–18752, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pal S, Hartnett KA, Nerbonne JM, Levitan ES, and Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 23: 4798–4802, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pan Y, Weng J, Cao Y, Bhosle RC, and Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvβ1. J Biol Chem 283: 8634–8642, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pan Y, Weng J, Levin EJ, and Zhou M. Oxidation of NADPH on Kvβ1 inhibits ball-and-chain type inactivation by restraining the chain. Proc Natl Acad Sci U S A 108: 5885–5890, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pannaccione A, Castaldo P, Ficker E, Annunziato L, and Taglialatela M. Histidines 578 and 587 in the S5–S6 linker of the human ether-à-gogo related gene-1 K+ channels confer sensitivity to reactive oxygen species. J Biol Chem 277: 8912–8919, 2002 [DOI] [PubMed] [Google Scholar]
- 98.Panyi G, Sheng Z, and Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys J 69: 896–903, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Papazian DM, Schwarz TL, Temple BL, Jan YN, and Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science 237: 749–753, 1987 [DOI] [PubMed] [Google Scholar]
- 100.Pardo LA. and Sühmer W. Eag1 as a cancer target. Expert Opin Ther Targets 12: 837–843, 2008 [DOI] [PubMed] [Google Scholar]
- 101.Pongs O, Kecskemethy N, Müller R, Krah-Jentgens I, Baumann A, Kiltz HH, Canal I, Llamazares S, and Ferrus A. Shaker encodes a family of putative potassium channel proteins in the nervous system of Drosophila. EMBO J 7: 1087–1096, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol 88: 2287–2295, 2000 [DOI] [PubMed] [Google Scholar]
- 103.Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, and Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature 369: 289–294, 1994 [DOI] [PubMed] [Google Scholar]
- 104.Rodríguez-Rasgado JA, Acuña-Macías I, and Camacho J. Eag1 channels as potential cancer biomarkers. Sensors (Basel) 12: 5986–5995, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rossing P, Breum L, Major-Peteresen A, Sato A, Winding H, Pietersen A, Kastrup J, and Parving HH. Prolonged QTc interval predicts mortality in patients with type 1 diabetes mellitus. Diabet Med 18: 199–205, 2001 [DOI] [PubMed] [Google Scholar]
- 106.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, and Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 352: 711–714, 1991 [DOI] [PubMed] [Google Scholar]
- 107.Saganich MJ, Vega-Saenz de Miera E, Nadal MS, Baker H, Coetzee WA, and Rudy B. Cloning of components of a novel subthreshold activating K+ channel with a unique pattern of expression in the cerebral cortex. J Neurosci 19: 10789–10802, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sahoo N, Schönherr R, Hoshi T, and Heinemann SH. Cysteines control the N- and C-linker-dependent gating of KCNH1 potassium channels. Biochim Biophys Acta 1818: 1187–1195, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sakai Y, Harvey M, and Sokolowski B. Identification and quantification of full-length BK channel variants in the developing mouse cochlea. J Neurosci Res 89: 1747–1760, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanguinetti MC, Jiang C, Curran ME, and Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81: 299–307, 1995 [DOI] [PubMed] [Google Scholar]
- 111.Santarelli LC, Chen J, Heinemann SH, and Hoshi T. The β1 subunit enhances oxidative regulation of large-conductance calcium-activated K+ channels. J Gen Physiol 124: 357–370, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Santarelli LC, Wassef R, Heinemann SH, and Hoshi T. Three methionine residues located within the regulator of conductance for K+ (RCK) domains confer oxidative sensitivity to large-conductance Ca2+-activated K+ channels. J Physiol 571: 329–348, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schlief T, Schönherr R, and Heinemann SH. Modification of C-type inactivating Shaker potassium channels by chloramine-T. Pflügers Arch 431: 483–493, 1996 [DOI] [PubMed] [Google Scholar]
- 114.Schönherr R. and Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol 493: 635–642, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schönherr R, Löber K, and Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin. EMBO J 19: 3263–3271, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwarz JR. and Bauer CK. Functions of erg K+ channels in excitable cells. J Cell Mol Med 8: 22–30, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sesti F, Liu S, and Cai S-Q. Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol 20: 45–51, 2009 [DOI] [PubMed] [Google Scholar]
- 118.Shi W, Wang HS, Pan Z, Wymore RS, Cohen IS, McKinnon D, and Dixon JE. Cloning of a mammalian elk potassium channel gene and EAG mRNA distribution in rat sympathetic ganglia. J Physiol 511: 675–682, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi W, Wymore RS, Wang HS, Pan Z, Cohen IS, McKinnon D, and Dixon JE. Identification of two nervous system-specific members of the erg potassium channel gene family. J Neurosci 17: 9423–9432, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Singh H, Stefani E, and Toro L. Intracellular BKCa (iBKCa) channels. J Physiol 590: 5937–5947, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Soto MA, Gonzalez C, Lissi E, Vergara C, and Latorre R. Ca2+-activated K+ channel inhibition by reactive oxygen species. Am J Physiol Cell Physiol 282: C461–C471, 2002 [DOI] [PubMed] [Google Scholar]
- 122.Stengel R, Rivera-Milla E, Sahoo N, Ebert C, Bolling F, Heinemann SH, Schönherr R, and Englert C. Kcnh1 voltage-gated potassium channels are essential for zebrafish early embryonic development. J Biol Chem 287: 35565–35575, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Su Z, Limberis J, Martin RL, Xu R, Kolbe K, Heinemann SH, Hoshi T, Cox BF, and Gintant G. Functional consequences of methionine oxidation of hERG potassium channels. Biochem Pharmacol 74: 702–711, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Svoboda LK, Reddie KG, Zhang L, Vesely ED, Williams ES, Schumacher SM, O'Connell RP, Shaw R, Day SM, Anumonwo JM, Carroll KS, and Martens JR. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res 111: 842–853, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sweeney M. and Yuan JX. Hypoxic pulmonary vasoconstriction: role of voltage-gated potassium channels. Respir Res 1: 40–48, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Szigligeti P, Neumeier L, Duke E, Chougnet C, Takimoto K, Lee SM, Filipovich AH, and Conforti L. Signalling during hypoxia in human T lymphocytes—critical role of the src protein tyrosine kinase p56Lck in the O2 sensitivity of Kv1.3 channels. J Physiol 573: 357–370, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Taglialatela M, Castaldo P, Iossa S, Pannaccione A, Fresi A, Ficker E, and Annunziato L. Regulation of the human ether-a-gogo related gene (HERG) K+ channels by reactive oxygen species. Proc Natl Acad Sci U S A 94: 11698–11703, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Taglialatela M, Pannaccione A, Iossa S, Castaldo P, and Annunziato L. Modulation of the K+ channels encoded by the human ether-à-gogo-related gene-1 (hERG1) by nitric oxide. Mol Pharmacol 56: 1298–1308, 1999 [DOI] [PubMed] [Google Scholar]
- 129.Tang XD, Daggett H, Hanner M, Garcia ML, McManus O, Brot N, Weissbach H, Heinemann SH, and Hoshi T. Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol 117: 253–273, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tang XD, Garcia ML, Heinemann SH, and Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol 11: 171–178, 2004 [DOI] [PubMed] [Google Scholar]
- 131.Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, and Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 425: 531–535, 2003 [DOI] [PubMed] [Google Scholar]
- 132.Taylor BL. and Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev 63: 479–506, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tempel BL, Papazian DM, Schwarz TL, Jan YN, and Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 237: 770–775, 1987 [DOI] [PubMed] [Google Scholar]
- 134.Terlau H, Ludwig J, Steffan R, Pongs O, Stühmer W, and Heinemann SH. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflügers Arch 432: 301–312, 1996 [DOI] [PubMed] [Google Scholar]
- 135.Thompson J. and Begenisich T. Selectivity filter gating in large-conductance Ca2+-activated K+ channels. J Gen Physiol 139: 235–244, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tian L, Jeffries O, McClafferty H, Molyvdas A, Rowe IC, Saleem F, Chen L, Greaves J, Chamberlain LH, Knaus HG, Ruth P, and Shipston MJ. Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc Natl Acad Sci U S A 105: 21006–21011, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Timpe LC, Jan YN, and Jan LY. Four cDNA clones from the Shaker locus of Drosophila induce kinetically distinct A-type potassium currents in Xenopus oocytes. Neuron 1: 659–667, 1988 [DOI] [PubMed] [Google Scholar]
- 138.Tseng-Crank JCL, Tseng GN, Schwartz A, and Tanouye MA. Molecular cloning and functional expression of a potassium channel cDNA isolated from a rat cardiac library. FEBS Lett 268: 63–68, 1990 [DOI] [PubMed] [Google Scholar]
- 139.Ufartes R, Schneider T, Mortensen LS, de Juan Romero C, Hentrich K, Knoetgen H, Beilinson V, Moebius W, Tarabykin V, Alves F, Pardo LA, Rawlins JN, and Stühmer W. Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum Mol Genet 22: 2247–2262, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Veglio M, Chinaglia A, and Cavallo-Perin P. QT interval, cardiovascular risk factors and risk of death in diabetes. J Endocrinol Invest 27: 175–181, 2004 [DOI] [PubMed] [Google Scholar]
- 141.Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med 18: 93–105, 1995 [DOI] [PubMed] [Google Scholar]
- 142.Warmke JW. and Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A 91: 3438–3442, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weng J, Cao Y, Moss N, and Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem 281: 15194–15200, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wilkens CM. and Aldrich RW. State-independent block of BK channels by an intracellular quaternary ammonium. J Gen Physiol 128: 347–364, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu Y, Yang Y, Ye S, and Jiang Y. Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature 466: 393–397, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, and Chandy KG. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest 111: 1703–1713, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xie J. and McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science 280: 443–446, 1998 [DOI] [PubMed] [Google Scholar]
- 148.Xu L, Enyeart JA, and Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage-dependent oxidative mechanism. J Pharmacol Exp Ther 299: 227–237, 2001 [PubMed] [Google Scholar]
- 149.Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, and O'Rourke B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298: 1029–1033, 2002 [DOI] [PubMed] [Google Scholar]
- 150.Yang Q, Chen SR, Li DP, and Pan HL. Kv1.1/1.2 channels are downstream effectors of nitric oxide on synaptic GABA release to preautonomic neurons in the paraventricular nucleus. Neuroscience 149: 315–327, 2007 [DOI] [PubMed] [Google Scholar]
- 151.Yi L, Morgan JT, and Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem 285: 20117–20127, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yuan H, Wang WP, Feng N, Wang L, and Wang XL. Donepezil attenuated oxygen-glucose deprivation insult by blocking Kv2.1 potassium channels. Eur J Pharmacol 657: 76–83, 2011 [DOI] [PubMed] [Google Scholar]
- 153.Yuan P, Leonetti MD, Hsiung Y, and MacKinnon R. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 481: 94–97, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yuan P, Leonetti MD, Pico AR, Hsiung Y, and MacKinnon R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 Å resolution. Science 329: 182–186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zagotta WN, Hoshi T, and Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 250: 568–571, 1990 [DOI] [PubMed] [Google Scholar]
- 156.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, and Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425: 200–205, 2003 [DOI] [PubMed] [Google Scholar]
- 157.Zeng XH, Xia XM, and Lingle CJ. Redox-sensitive extracellular gates formed by auxiliary β subunits of calcium-activated potassium channels. Nat Struct Biol 10: 448–454, 2003 [DOI] [PubMed] [Google Scholar]
- 158.Zhang G. and Horrigan FT. Cysteine modification alters voltage- and Ca2+-dependent gating of large conductance (BK) potassium channels. J Gen Physiol 125: 213–236, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang G, Xu R, Heinemann SH, and Hoshi T. Cysteine oxidation and rundown of large-conductance Ca2+-dependent K+ channels. Biochem Biophys Res Commun 342: 1389–1395, 2006 [DOI] [PubMed] [Google Scholar]
- 160.Zhang Y, Han H, Wang J, Wang H, Yang B, and Wang Z. Impairment of HERG (human ether-a-go-go related gene) K channel function by hypoglycemia and hyperglycemia: similar phenotypes but different mechanisms. J Biol Chem 278: 10417–10426, 2003 [DOI] [PubMed] [Google Scholar]
- 161.Zhang Y, Sun X, Zhang Y, Wang J, Lu Y, Yang B, and Wang Z. Potential therapeutic value of antioxidants for abnormal prolongation of QT interval and the associated arrhythmias in a rabbit model of diabetes. Cell Physiol Biochem 28: 97–102, 2011 [DOI] [PubMed] [Google Scholar]
- 162.Zhou Y, Xia XM, and Lingle CJ. Cysteine scanning and modification reveal major differences between BK channels and Kv channels in the inner pore region. Proc Natl Acad Sci U S A 108: 12161–12166, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ziechner U, Schönherr R, Born AK, Gavrilova-Ruch O, Glaser R, Malesevic M, Küllertz G, and Heinemann SH. Inhibition of human ether-à-go-go potassium channels hEAG1 by Ca2+/calmodulin binding to the cytosolic N- and C-termini. FEBS J 273: 1074–1086, 2006 [DOI] [PubMed] [Google Scholar]
- 164.Zong X, Zucker H, Hofmann F, and Biel M. Three amino acids in the C-linker are major determinants of gating in cyclic nucleotide-gated channels. EMBO J 17: 353–362, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]