

Abstract
Eudistomin U is a member of a subclass of naturally occurring indole alkaloids known as β-carbolines. These molecules are reported to have diverse biological activity and high binding affinity to DNA, which make them attractive targets for total synthesis. We describe an efficient, five-step synthesis of eudistomin U by employing two key reactions: a Bischler-Napieralski cyclization and a Suzuki cross coupling. We also describe the cytotoxicity of eudistomin U against various cancer cell lines and human pathogens, in which we observed potent antibacterial activity against Gram-positive bacteria.
Keywords: β-carboline, Suzuki cross coupling, eudistomin U, antibacterial, palladium catalysis
β-Carbolines are indole alkaloids that exist as secondary metabolites in many living organisms.1 One particular family of β-carboline natural products, known as the eudistomins, has been isolated from several species of marine ascidians.2 The eudistomins are structurally diverse and show wide-ranging biological properties, making them attractive targets for synthetic chemists (Figure 1). For example, nearly a third of the eudistomin alkaloids have antimicrobial activities.1 Other eudistomins have been shown to exhibit antiviral activities against herpes simplex virus-1 and polio vaccine type-1 virus.2c,2d,3 Antitumor activity has also been observed for many family members in both mouse and human cancer cells.2b,2d,4 Furthermore, a synthetic derivative of eudistomin K was found to be incredibly potent (as low as 0.005 μg/mL) against L1210, Molt-4F, MT-4, and P-388 leukemic cells,5 demonstrating that the eudistomins are privileged lead structures in drug discovery.
Figure 1.
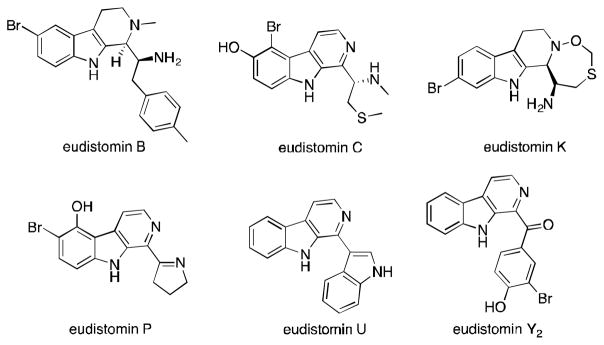
Structures of several biologically active eudistomins
While structural variants of this class of natural products have been prepared and evaluated, no such study has been undertaken for eudistomin U. This is striking since eudistomin U is the only natural product in its class with an aromatic group at the 1-position of the β-carboline. Furthermore, only a brief mention of its biological activity has appeared in the literature.2a,4 Given these gaps, we wondered whether we could devise a synthesis of eudistomin U that would enable a more comprehensive evaluation of its cytotoxicity while also laying the foundation for the formation of derivatives. In this paper, we report a new synthesis of eudistomin U that employs a late-stage Suzuki cross coupling to form the key aryl-aryl C-C bond. Preliminary cytotoxicity measurements of eudistomin U against selected bacteria, fungi, and human cancer cells are also reported.
Eudistomin U has succumbed to total synthesis five times (Figure 2) and was first reported by Molina using an iminophosphorane intermediate to prepare the β-carboline ring.6 This was followed shortly thereafter by Quéginer,7 who prepared the natural product via sequential metalations. More recently, Waters8 reported an efficient IBX oxidation of a Pictet-Spengler intermediate to generate eudistomin U while Witulski and coworkers9 used a novel Rh- or Ru-catalyzed [2+2+2] cyclization to generate the natural product. Finally, Yamaguchi and Itami reported a novel oxidative C–H/C–H coupling in their total synthesis.10 Given that none of these procedures allowed for investigation of the 1-position of the β-carboline very late in the synthesis, we devised a synthetic strategy that would allow us to modify this position rapidly from a common precursor. We thought that preparation of a 1-halo-β-carboline would allow us to attach various groups to the 1-position using cross coupling conditions. In our synthesis of eudistomin U reported herein, we modified the approach of Bracher and Hilderbrand, who successfully employed this strategy in the synthesis of naturally occurring 1-aryl-β-carbolines.11
Figure 2.

Previous synthetic strategies for the total synthesis of eudistomin U
We began our synthesis with commercially available starting material tryptamine (1), which underwent a Bischler-Napieralski12 cyclization to provide the lactam 3 (Scheme 1).11 Attempts to oxidize lactam 3 to the unsaturated pyridone 4 were unsuccessful using Pd/C or Ce(NH4)2(NO3)6. DDQ oxidation, however, provided 4 in a crude yield of 94% and was taken directly to the next step without further purification.13 We first attempted to convert pyridone 4 into the aryl chloride or bromide, but found that the harsh reaction conditions (refluxing POCl3 or POBr3) led to low yields of the corresponding product.14 Instead, formation of triflate 5 proceeded smoothly in 76% yield using trifluoromethanesulfonic anhydride under basic conditions.
Scheme 1.

Synthesis of eudistomin U and analogs
Since intermediate 5 contains a reactive aryl triflate functionality, we chose to attach the remaining indole ring to the β-carboline core via a Suzuki cross coupling reaction.15 Following Bracher and Hildebrand’s conditions for the synthesis of komaroine and perlolyrine,11 we obtained an initial yield of 38%. Seeking to optimize this reaction yield, we tested conditions that varied the solvent, palladium precatalyst, and exogenous base (Table 1). Toluene or tetrahydrofuran in combination with absolute ethanol proved to be the best reaction solvent (entry 1), with ethanol required for the solubility of 6. Several palladium(0) and palladium(II) complexes were tested under otherwise identical conditions (entries 4–7), with the highest yields obtained using palladium(0) complexes. Using tris(dibenzylideneacetone) dipalladium(0) as precatalyst, we also investigated the effect of base strength. We found that stronger bases reduced the reaction yield (entries 8 and 9), in one case significantly. Replacing aqueous sodium carbonate with an organic base also resulted in a slightly less efficient synthesis (entry 10). With the optimal Suzuki cross coupling conditions in hand (entry 7, 73% yield), efficient removal of the benzenesulfonyl group under basic conditions gave the natural product in an overall yield of 19% over five steps (Scheme 1). To underscore the brevity of this sequence, we also prepared 9 (54%) and 10 (94%) via identical Suzuki cross coupling conditions, demonstrating the generality of this scheme and the facility with which close analogs can be made.
Table 1.
Optimization of conditions for the Suzuki cross coupling reaction (5 7)
| Entry | Conditionsa | Yield (%) |
|---|---|---|
| 1 | 5% Pd(PPh3)4, aq. Na2CO3, EtOH, toluene | 38 |
| 2 | 5% Pd(PPh3)4, aq. Na2CO3, EtOH, THF | 38 |
| 3 | 5% Pd(PPh3)4, K2CO3, DMF | 20 |
| 4 | 5% Pd(PPh3)2Cl2, aq. Na2CO3, PPh3, EtOH, toluene | 52 |
| 5 | 5% Pd(OAc)2, aq. Na2CO3, PPh3, EtOH, toluene | 7 |
| 6 | 5% Pd(dba)2, aq. Na2CO3, PPh3, EtOH, toluene | 68 |
| 7 | 5% Pd2(dba)3, aq. Na2CO3, PPh3, EtOH, toluene | 73 |
| 8 | 5% Pd2(dba)3, KOtBu, PPh3, EtOH, toluene | 22 |
| 9 | 5% Pd2(dba)3, K2CO3, PPh3, EtOH, toluene | 61 |
| 10 | 5% Pd2(dba)3, Et3N, PPh3, EtOH, toluene | 69 |
All reactions were run under a N2 atmosphere at 80 °C for 1 h using 1.0 equivalents of triflate 5 and 1.4 equivalents of boronic acid 6.
With multimilligram quantities available, we expanded the cytotoxicity profile for eudistomin U beyond the limited data currently published. The isolation chemists reported a strong antibacterial response against Agrobacterium tumefaciens, but provided no quantitative results or experimental procedures.2a In one other study, eudistomin U was found to inhibit the growth of mouse P388 cells (38% inhibition) at 10 μM.4 To further evaluate its activity, we determined the cytotoxicity of eudistomin U in three classes of organisms: bacteria, fungi, and human cancer cells (Table 2).
Table 2.
Cytotoxicity of eudistomin U (8) in model organisms
| Cell Type | Organism | IC50 (μg/mL)a |
|---|---|---|
| bacteria | Streptococcus pyogenes | 3.4 |
| Staphylococcus aureus | 6.4 | |
| Mycobacterium smegmatis | 3.6 | |
| Pseudomonas aeruginosa | 27.7 | |
| Escherichia coli | 12.3 | |
| fungi | Candida albicans | >30 |
| Saccharomyces cerevisiae | >300 | |
| human cancer | C19 leukemia | 15.6 |
| CaOV3 ovarian | 24.9 | |
| WM266-4 melanoma | 27.5 |
Cytotoxicity (IC50) is the concentration of 8 that causes 50% growth inhibition relative to an untreated control; see supporting information.
We tested the antibacterial activity of eudistomin U against five different bacteria. Using an optical density based assay,16 percent survivorship was calculated as a function of increasing concentration (μg/mL). Table 2 shows that the Gram-positive bacteria (S. pyogenes, S. aureus, and M. smegmatis) were most susceptible to treatment with eudistomin U (IC50 = 3.4–6.4 μg/mL), with IC50 values roughly two-fold more potent than Gram-negative bacteria. Surprisingly, no inhibition was observed for either fungal strain under similar conditions. Cancer cell cytotoxicity, which was measured using an MTT cell viability assay,17 showed moderate activity across three different cell lines. The lowest IC50 was observed in C19 leukemia cells (15.6 μg/mL). Together, these results indicate that the development of eudistomin U or its derivatives as antibacterial agents holds the most promise. The underlying causes of this activity are currently under investigation.
In summary, we have developed a short, five-step synthesis of eudistomin U and have provided a preliminary characterization of its biological activity. Eudistomin U shows no antifungal activity but modest anticancer and strong antibacterial properties. Our approach is unique since intermediate 5 contains functionality that can be replaced by other groups in the final step of the sequence, thereby facilitating structure-activity investigations. Work in this area is currently being explored.
Supplementary Material
Acknowledgments
This research was supported by Providence College, the Rhode Island Foundation Medical Research Funds, and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under the grant number 5 P20 GM103430-13. The authors would also like to acknowledge Dr. Tun-Li Shen at Brown University for HR-MS measurements. We also thank Dr. Charles Toth (leukemia), Dr. Brett Pellock (bacteria), Dr. Nicanor Austriaco (fungi), and Dr. Yinsheng Wan (melanoma and ovarian) of the Providence College Department of Biology for various cell lines.
Footnotes
Supplementary data associated with this article, including all experimental details, cytotoxicity assay protocols, and the full characterization of new compounds can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cao R, Peng W, Wang Z, Xu A. Curr Med Chem. 2007;14 (4):479–500. doi: 10.2174/092986707779940998. [DOI] [PubMed] [Google Scholar]
- 2.(a) Badre A, Boulanger A, Abou-Mansour E, Banaigs B, Combaut G, Francisco C. J Nat Prod. 1994;57 (4):528–33. doi: 10.1021/np50106a016. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi J, Cheng J, Ohta T, Nozoe S, Ohizumi Y, Sasaki T. Journal of Organic Chemistry. 1990;55:3666. [Google Scholar]; (c) Kobayashi J, Harbour GC, Gilmore J, Rinehard JKL. Journal of the American Chemical Society. 1984;106:1526–1528. [Google Scholar]; (d) Lake RJ, Blunt JW, Munro MHG. Australian Journal of Chemistry. 1989;42 (7):1201–1206. [Google Scholar]; (e) Schupp P, Poehner T, Edrada R, Ebel R, Berg A, Wray V, Proksch P. J Nat Prod. 2003;66 (2):272–5. doi: 10.1021/np020315n. [DOI] [PubMed] [Google Scholar]; (f) Davis RA, Carroll AR, Quinn RJ. Journal of Natural Products. 1998;61:959–960. doi: 10.1021/np9800452. [DOI] [PubMed] [Google Scholar]
- 3.(a) Rinehart KL, Jr, Kobayashi J, Harbour GC, Hughes RG, Jr, Mizask SA, Scahill TA. Journal of the American Chemical Society. 1984;106:1524. [Google Scholar]; (b) Lake RJ, Brennan MM, Blunt JW, Munro MHG, Panell LK. Tetrahedron Letters. 1988;29:2255. [Google Scholar]
- 4.Dong XC, Wen R, Zheng J. Acta Pharmaceutica Sinica. 2004;39:259. [PubMed] [Google Scholar]
- 5.Van Maarseveen JH, Hermkens PHH, DeClerq E, Balzarini J, Schereen HW, Kruse CG. Journal of Medicinal Chemistry. 1992;35 (17):3223–3230. doi: 10.1021/jm00095a019. [DOI] [PubMed] [Google Scholar]
- 6.Molina P, Fresneda PMS, G-Z Tetrahedron Letters. 1995;36 (20):3581–3582. [Google Scholar]
- 7.Rocca P, Marsais F, Godard A, Queguiner G, Adams L, Alo B. Tetrahedron Letters. 1995;36 (39):7085–7088. [Google Scholar]
- 8.Panarese JD, Waters SP. Org Lett. 2010;12 (18):4086–9. doi: 10.1021/ol101688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nissen F, Richard V, Alayrac C, Witulski B. Chem Commun (Camb) 2011;47 (23):6656–8. doi: 10.1039/c1cc11298h. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi AD, Mandal D, Yamaguchi J, Itami K. Chemistry Letters. 2011;40:555–557. [Google Scholar]
- 11.Bracher F, Hildebrand D. Liebigs Annalen der Chemie. 1992;1992 (12):1315–1319. [Google Scholar]
- 12.Bischler A, Napieralski B. Berichte. 1893;26:1903–1908. [Google Scholar]
- 13.Hawkins A, Jakubec P, Ironmonger A, Dixon DJ. Tetrahedron Letters. 2013;54 (5):365–369. [Google Scholar]
- 14.Bracher F, Hildebrand D. Tetrahedron. 1994;50 (43):12329–12336. [Google Scholar]
- 15.Miyaura N, Yanagi T, Suzuki A. Synthetic Communications. 1981;11:513–519. [Google Scholar]
- 16.De La Fuente R, Sonawane ND, Arumainayagam D, Verkman AS. Br J Pharmacol. 2006;149 (5):551–9. doi: 10.1038/sj.bjp.0706873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mossman T. Journal of Immunological Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.