

Abstract
Purpose.
To study the retinal degeneration caused by decreased proteasomal activity in β5t transgenic (β5t-Tg) mice, an animal model of senescence acceleration.
Methods.
β5t-Tg mice and age-matched littermate control (WT) mice were used. Proteasomal activities and protein level of poly-ubiquitinated protein in retinal extracts were quantified. Fundus images of β5t-Tg mice were taken and their features were assessed. For histologic evaluation, the thicknesses of inner nuclear layer (INL), outer nuclear layer (ONL), and photoreceptor outer segment (OS) were measured. For functional analysis, ERG was recorded under scotopic and photopic illumination conditions. Immunofluorescence (IF) staining and TUNEL were performed to investigate the mechanism of photoreceptor degeneration.
Results.
Chymotrypsin-like activity was partially suppressed in retinal tissues of β5t-Tg mice. Retinal degenerative changes with arterial attenuation were present in β5t-Tg, but not in WT mice. Inner nuclear layer thickness showed no significant change between β5t-Tg and WT mice at 1, 3, 6, and 9 months of age. By contrast, thicknesses of ONL and OS in β5t-Tg mice were significantly decreased at 3, 6, and 9 months compared with those in WT mice. Electroretinograms showed decrease of scotopic a-wave amplitude in β5t-Tg mice. The number of TUNEL-positive cells in ONL were significantly increased in β5t-Tg mice and colocalized with apoptosis-inducing factor, but not with cleaved caspase-3 and -9, indicating that the photoreceptor cell death was induced via a caspase-independent pathway.
Conclusions.
The current data showed that impaired proteasomal function causes photoreceptor degeneration.
Keywords: proteasomal dysfunction, neurodegeneration, photoreceptor cell death
This is the first report addressing the impact of proteasome dysfunction in the retina in vivo. The current data demonstrate that decreased function of proteasome causes photoreceptor degeneration, predominantly in rod photoreceptor.
Introduction
The ubiquitin proteasome system (UPS), one of the major proteolytic pathways, contributes to protein quality control in eukaryotic cells. Recent studies have revealed that UPS dysfunction is involved in the pathogenesis of systemic neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease.1–3 In ocular diseases, retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal neurodegenerative disorders characterized by progressive degeneration of photoreceptors.4 To date, over 100 point mutations in the rhodopsin gene have been identified in patients with RP (RetNet https://sph.uth.edu/RetNet/home.htm; provided in the public domain) and it has been revealed that mutations in rhodopsin gene result in the aggregation of unfolded rhodopsin protein and, in turn, cause the UPS dysfunction.5,6 In addition, recent studies have shown that mutation of KLHL7 gene, coding the subunit of E3 ubiquitin ligase complex, causes autosomal dominant RP.7,8 Therefore, emerging evidence indicates that UPS dysfunction also plays a role in retinal neurodegeneration.
Proteasome is a multiunit enzyme complex responsible for protein degradation by the UPS pathway. Proteolysis by proteasome is conducted via the enzymatic response with three β subunits, β1, β2, and β5, which have caspase-like, trypsin-like, and chymotrypsin-like enzymatic activities, respectively.9,10 Decreased proteasomal activity has been reported in neurodegenerative diseases such as Alzheimer's disease11 and Parkinson's disease,12,13 and therefore whether impairment of proteasome function also causes RP has been of great interest. However, since genetic ablation of proteasome subunits including β1, β2, and β5 is embryonic lethal, the changes in ocular tissues caused by proteasome dysfunction have not been studied in vivo so far.
Recently, Tomaru et al.14 generated transgenic mice with decreased proteasomal activity by introducing β5t as a transgene. β5t is a recently discovered β5-like subunit of the 20S proteasome expressed exclusively in the thymus.15 Since β5t has only weak chymotrypsin-like activity and is preferentially incorporated into 20S proteasomes over β5,16 overexpression of β5t in vivo leads to decreased proteasomal activity, which causes senescence acceleration and age-related phenotypes such as lordokyphosis, the loss of subcutaneous fat and degeneration of skeletal muscle fibers.14
In this study, we investigated the retinal changes induced by proteasome dysfunction in β5t-transgenic (β5t-Tg) mice.
Methods
Animals
β5t-Tg mice, generated as previously described,14 and age-matched littermate control (WT) mice (C57BL/6 background) were used in this study. The animals were maintained on a 12 hours light/dark cycle and housed in plastic cages in a climate-controlled animal facility. All animal experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Guidelines for the Care and Use of Laboratory Animals at Hokkaido University Graduate School of Medicine (Sapporo, Japan).
Western Blotting
Animals were euthanized with overdose administration of anesthesia and eyes were enucleated. Retinal tissues were isolated microsurgically, placed into 120 μL radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, Danvers, MA, USA) with protease inhibitor cocktail (Roche, Basel, Switzerland) and sonicated. The lysate was centrifuged (14,000g for 20 minutes at 4°C) and the supernatant was collected. Samples were separated by SDS-PAGE and blotted to polyvinylidene fluoride membranes (GE Healthcare, Buckinghamshire, UK). To block the nonspecific binding, membranes were washed with solution which consisted of 5% skim milk powder in Tris buffered saline (TBS) and subsequently incubated at 4°C overnight with the following primary antibodies: a rabbit polyclonal antibody against α7 (Abnova, Taipei, Taiwan), a rabbit polyclonal antibody against β5i (Enzo Life Sciences, Farmingdale, NY, USA), a goat polyclonal antibody against β5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), a rabbit monoclonal antibody against actin (Abcam, Tokyo, Japan), a mouse monoclonal antibody against ubiquitin (Santa Cruz Biotechnology) and a rabbit polyclonal anti-β5t antisera, which were raised against 6xHis-tagged recombinant proteins encompassing residues 234 to 302 of mouse β5t.15 Thereafter, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (1:4000 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). The images were obtained with chemiluminescence (Western Lightning Ultra; PerkinElmer, Waltham, MA, USA) using a luminescent image analyzer (LAS-4000; Fujifilm, Tokyo, Japan).
Immunofluorescence (IF) Staining
Eyes fixed in 4% paraformaldehyde (PFA) were embedded in paraffin and sectioned. Dewaxed paraffin sections were prepared and microwave-based antigen retrieval was performed in 10 mM citrate buffer (pH 6.0). Sections were blocked and permeabilized with PBS containing 5% normal goat serum and 0.05% Triton X-100 at room temperature for 1 hour. Thereafter, the sections were probed with the following primary antibodies: a rabbit monoclonal antibody against proteasome 20S α6 subunit (1:500 dilution; Abcam); a rabbit polyclonal antibody against β5t (1:250 dilution; Medical & Biological Laboratories, Nagoya, Japan); a rabbit polyclonal antibody against cone arrestin (1:1000 dilution; Merck Millipore, Billerica, MA, USA); a mouse monoclonal antibody against rhodopsin (4D2, 1:1000 dilution; Merck Millipore); a rabbit polyclonal antibody against cleaved caspase-3 (1:100 dilution; Cell Signaling Technology), cleaved caspase-9 (1:50 dilution; Cell Signaling Technology); and apoptosis-inducing factor (AIF; 1:100 dilution; R&D Systems, Minneapolis, MN, USA). The secondary antibody Alexa Fluor 488 or 546 (Life Technologies, Carlsbad, CA, USA) was used for fluorescent detection, and 4′,6-diamino-2-phenylindole (DAPI) for nuclear staining. Sections were visualized under a BZ-9000 fluorescence microscope (Keyence, Osaka, Japan) or a FluoView 1000 confocal microscope (Olympus, Tokyo, Japan).
Proteasomal Activity Measurement
Sensory retinas were extracted and treated with collagenase. The cells were harvested into 96-well plates and analyzed using proteasome activity assay kits (Proteasome-Glo cell-based assay; Promega, Madison, WI, USA), according to the manufacturer's instructions. Briefly, cells were incubated with specific luminogenic proteasome substrates, Suc-LLVY-aminoluciferin for chymotrypsin-like activity, and substrate luminescence was measured by a luminometer. All data were corrected by the number of cells and expressed as a substrate luminescence/the number of cells (×100).
Enzyme-Linked Immunosorbent Assay (ELISA)
Protein levels of poly-ubiquitinated protein in retinal extracts were determined using ELISA kits (CY-7053, Caltag Medsystems, Buckingham, UK) and normalized to total protein (BCA Protein Assay Kit; Thermo Scientific, Rockford, IL, USA), according to the manufacturer's protocols.
Fundus Photography
A fundus camera (CF-60UVi; Canon, Tokyo, Japan) coupled with a 3 charge-coupled device (CCD) color video camera (Power HA-D; SONY, Tokyo, Japan) with 40 diopter (D) lens (Volk, Mentor, OH, USA) was used. Fundus photographs were taken after pupil dilation with one drop of a mixture of 0.5% tropicamide and 0.5% phenylephrine (Santen Pharmaceutical Co., Osaka, Japan).
Polymerase Chain Reaction (PCR)
Deoxyribonucleic acid was isolated from mouse-tail biopsy samples. The negative control DNA was isolated from a normal C57BL/6 mouse. The positive control DNA was purchased from the Jackson Laboratory (STOCK Crb1rd8/J; Bar Harbor, ME, USA). These samples were amplified separately for wild type (Wt) allele and mutant rd8 allele using primers specified by Mehalow et al.17 Primer sequences included mCrb1 mF1: GTGAAGACAGCTACAGTTCTGATC; mCrb1 mF2: GCCCCTGTTTGCATGGAGGAAACTTGGAAGACAGCTACAGTTCTTCTG; and mCrb1 mR: GCCCCATTTGCACACTGATGAC. For PCR amplification approximately 20 ng DNA were used in a 20 μL reaction volume using GoTaq DNA Polymerase (Promega, Madison, WI, USA), 5× Green GoTaq Reaction Buffer (Promega), 200 μM of each dNTP, 1.6 μM each of forward and reverse primer for Wt allele, and 0.8 μM of forward and 1.6 μM of reverse primer for rd8 mutant allele. Reactions initially were denatured at 94°C for 5 minutes followed by 35 cycles at 94°C for 30 seconds, 65°C for 30 seconds, 72°C for 30 seconds, and a final extension at 72°C for 7 minutes.18 Amplicons were separated using 3% agarose gel and visualized using an image analyzer (LAS-4000; Fujifilm). Amplicon sizes for Wt allele and rd8 allele are 220 and 244 bp, respectively.
Histologic Analysis
For histologic analysis, eyes were fixed in 4% glutaraldehyde for 60 minutes at room temperature followed by 4% PFA for at least 24 hours at 4°C, then embedded in paraffin, and sectioned. The sections were stained with hematoxylin and eosin (HE). A BZ-9000 microscope (Keyence) was used for histologic evaluation of the sections, and color micrographs were obtained using a BZ-II Analyzer (Keyence). The thicknesses of outer nuclear layer (ONL) and inner nuclear layer (INL) were measured on sections parallel to the vertical meridian of the eye at a distance of 400 to 2000 μm from both sides of the optic disc, as previously described.19,20 The thickness of photoreceptor outer segment (OS) was also measured at approximately 600 μm from the optic disc and corrected by INL thickness.
For transmission electron microscopy, the eyes were enucleated, and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer at 4°C. The eyes were postfixed for 1.5 hours in 2% aqueous OsO4, dehydrated in ethanol and water, and embedded in epoxy resin. Ultrathin sections were cut from blocks and stained with saturated, aqueous uranyl acetate and Sato's lead stain. The specimens were observed with a Hitachi H-7700 electron microscope (Hitachi High-Technologies Corporation, Tokyo, Japan).
Electroretinogram (ERG)
Mice were dark adapted for at least 12 hours and prepared under dim red illumination, anesthetized with pentobarbital (70 mg/kg), and placed on a heating pad throughout the experiment. The pupils were dilated with one drop of a mixture of 0.5% tropicamide and 0.5% of phenylephrine (Santen Pharmaceutical Co.). The ground electrode was a needle placed subcutaneously in the tail, and the reference electrode was placed in the mouth. The active gold electrodes were placed in contact with the cornea. Recordings were performed (PowerLab System 2/25; AD Instruments, New South Wales, Australia). Full-field scotopic ERGs were measured in response to flash at an intensity ranging from −0.54 to 2.89 log cd·s/m2. One to four responses were averaged with interstimulus intervals of 5 to 180 seconds. Responses were differentially amplified and filtered through a digital bandpass filter ranging from 0.3 to 1000 Hz. The stimuli were delivered through a commercial stimulator (Ganzfeld System SG-2002; LKC Technologies, Inc., Gaithersburg, MD, USA). Subsequently, photopic ERGs were recorded in response to 0.89 to 2.89 log cd·s/m2 flash and 30 cd/m2 background light after 10 minutes of light adaptation. Twenty trials were averaged for single-flash responses. The amplitude of the a-wave was measured from the baseline to the trough of the a-wave, and the amplitude of the b-wave was measured from the trough of the a-wave to the peak of the b-wave.
TdT-Mediated dUTP Nick End Labeling (TUNEL)
TdT-dUTP terminal nick-end labeling assay was performed (Fluorescein Direct In Situ Apoptosis Detection Kit; Merck Millipore) to detect cleaved DNA in the paraffin-embedded sections, according to the manufacturer's instruction.
Caspase Activity Measurement
The retinas were isolated and used for the assay of caspase-3 or caspase-9 activity. The enzymatic activities of caspase-3 and caspase-9 were measured using caspase-3 and caspase-9 colorimetric protease assay kits (Medical & Biological Laboratories), respectively. The light emission of chromophore p-nitroanilide (pNA) cleaved from labeled DEVD-pNA or LEHD-pNA was measured at 415 nm using a 96-well microplate reader (Tecan Japan, Kawasaki, Japan).
Statistical Analysis
All the results are expressed as the mean ± SEM as indicated. Mann-Whitney U test was used for statistical comparison between groups. Differences between the means were considered statistically significant when the probability values were less than 0.05.
Results
Suppression of Proteasomal Activity in Retinal Tissues of β5t-Tg Mice
To verify the overexpression of β5t protein in retinal tissues of β5t-Tg mice, Western blotting and IF were performed. β5t protein was detected in retinal lysates obtained from β5t-Tg mice, but not from WT mice, while β5 protein was not detected in retinal tissues of β5t-Tg mice (Fig. 1A). β5i and α7 proteins were detected in both β5t-Tg mice and WT mice (Fig. 1A). In accord with Western blotting data, IF staining showed that β5t was ubiquitously present in retina of β5t-Tg mice, but not in those of WT mice (Fig. 1B). By contrast, α6 subunit, proteasome 20S component,21 was found throughout the retina in both β5t-Tg mice and WT mice (Fig. 1C). The present data suggested that β5t protein incorporated into 20S proteasomes in β5t-Tg mice, similar to the other organs such as liver, muscle, and brain, as previously described.14 Furthermore, chymotrypsin-like activity was significantly decreased in sensory retina of β5t-Tg mice compared with that of WT mice (10.87 ± 0.86 vs. 16.24 ± 1.08 arbitrary unit/cells (×100), n = 5 each, P < 0.05), whereas both trypsin-like activity (3.60 ± 0.10 vs. 4.60 ± 0.61, n = 5 each, P = 0.14) and caspase-like activity (4.09 ± 0.32 vs. 5.22 ± 0.68, n = 5 each, P = 0.21) were not statistically different between β5t-Tg mice and WT mice (Fig. 1D). In addition, poly-ubiquitinated protein was increased in the retinal extraction of β5t-Tg mice compared with that of WT mice (0.36 ± 0.02 units/mg vs. 0.23 ± 0.01 units/mg, n = 5–7, P < 0.001, Figs. 1E, 1F).
Figure 1.

Expression of β5t and decreased proteasomal activity in retinal tissues. (A) Immunoblotting of β5t and other proteasome subunits using retinal extracts from mice at 3 months of age (10 μg protein/lane). β5t protein was detected only in the retinal extracts from β5t-Tg mice, but not WT mice. (B, C) Representative fluorescent micrographs of retinal tissues from WT mice and β5t-Tg mice immunostained for β5t protein (B), and proteasome 20S α6 subunit protein (C) showing the distribution of all proteasome subtypes (n = 3–5). Scale bar: 20 μm. (D) Chymotrypsin-like activity, trypsin-like activity and caspase-like activity in the retina from WT mice and β5t-Tg mice at 1 month of age (n = 5 each). Data are expressed as the substrate luminescence/the number of cells (×100). (E) Protein levels of poly-ubiquitinated protein in the retina from mice at 3 months of age (n = 5–7). (F) Western blotting for ubiquitin (10 μg protein/lane). Values are mean ± SEM. *P < 0.05; **P < 0.01. N.S., not significant.
Fundus Features in β5t-Tg Mice
To assess the fundus features of β5t-Tg mice, we compared the fundus images of β5t-Tg mice with those of age-matched WT mice at 6 months of age. Comparison of the images highlighted characteristic features in the retina of β5t-Tg mice (e.g., disseminated retinal white deposits, atrophic changes and attenuation of retinal arteries; n = 8–10, Fig. 2). Deoxyribonucleic acid analysis revealed the presence of rd8 mutation in homozygous form in both WT and β5t-Tg mice (data not shown).
Figure 2.
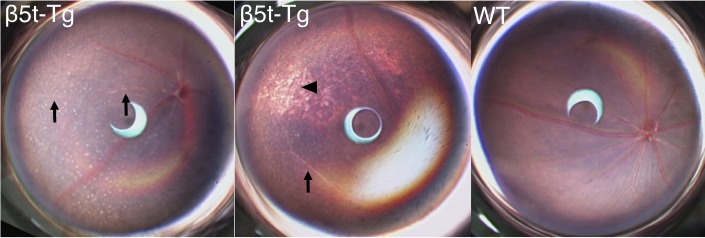
Fundus features of β5t-Tg mice. Fundus images of β5t-Tg mice at 6 months of age showed disseminated retinal white deposits (left), peripheral retinal degeneration (middle, arrowhead), and attenuation of retinal arteries (left and middle, arrows), none of which were found in age-matched WT mice (right). Since DNA analysis revealed the presence of rd8 mutation in homozygous form in both WT and β5t-Tg mice, it is possible that the deposits are originated from interference between β5t incorporation and the rd8 mutation.
Photoreceptor Degeneration in β5t-Tg Mice
To investigate the time course of retinal degeneration, retinal thickness was measured in β5t-Tg and WT mice at 1, 3, 6, and 9 months of age (Fig. 3A). Whereas INL thickness showed no difference between β5t-Tg and WT mice, ONL thickness remarkably decreased in β5t-Tg mice, compared with WT mice (Fig. 3B). There was significant difference in ONL/INL ratio between WT and β5t-Tg mice at 3 months of age (1.58 ± 0.03 vs. 1.32 ± 0.06, n = 3–6, P < 0.05), 6 months of age (1.66 ± 0.03 vs. 0.86 ± 0.02, n = 5–7, P < 0.01) and at 9 months of age (1.49 ± 0.02 vs. 0.63 ± 0.04, n = 4–8, P < 0.01), but not at 1 month of age (1.56 ± 0.04 vs. 1.57 ± 0.004, n = 3–4, P = 0.60, Fig. 3C). Similarly, the thickness of OS was shortened in β5t-Tg mice compared with WT mice at 3, 6, and 9 months (n = 3–7, Fig. 3D). However, the ultrastructure of disc membrane in OS was relatively maintained in β5t-Tg mice compared with those in WT mice (Fig. 3E).
Figure 3.

Photoreceptor cell degeneration caused by proteasome dysfunction. (A) Representative images of retinal sections stained with HE from WT mice and β5t-Tg mice at 1, 3, 6, and 9 months of age. Scale bar: 10 μm. (B) Quantification analysis of INL and ONL thickness (n = 3–8). Closed circles indicate WT mice, and open circles indicate β5t-Tg mice. (C) Outer nuclear layer/INL ratio (n = 3–8). (D) Outer segment/INL ratio (n = 3–7). (E) Ultrastructure of OS at 6 months of age. Scale bar: 2 μm. Values are mean ± SEM. *P < 0.05; **P < 0.01.
Predominant Impairment of Rod Function in β5t-Tg Mice
To elucidate whether the proteasome dysfunction causes photoreceptor degeneration in rod photoreceptor and/or cone photoreceptor, ERG was recorded under full-field scotopic and photopic illumination conditions. At 6 months of age, scotopic a-wave and b-wave were detectable (Fig. 4A); however, the amplitude of scotopic a-wave was, except at −0.54 log cd·s/m2, significantly decreased in β5t-Tg mice compared with WT in an intensity-dependent manner (−0.54 log cd·s/m2, P < 0.05, 1.89 log cd·s/m2, P < 0.01, and 2.89 log cd·s/m2, P < 0.001, n = 7–8, Fig. 4B). By contrast, whereas photopic ERG responses showed tendency toward the amplitude of a-wave was reduced in β5t-Tg mice compared with WT mice (Fig. 4C); however, it was not statistically significant (P = 0.60, 0.77, and 0.52, respectively, n = 7–8, Fig. 4D). The functional analysis indicates that photoreceptor degeneration is predominantly caused in rod photoreceptors. In support with the ERG data, IF staining for cone arrestin showed no differences between WT and β5t-Tg mice, whereas ONL was thinner in β5t-Tg mice (Figs. 4E, 4F).
Figure 4.

Impairment of retinal function caused by proteasome dysfunction. (A, C) Representative wave responses of scotopic ERG (A) and photopic ERG (C) from WT mice and β5t-Tg mice at 6 months of age. (B, D) Quantification of scotopic a-wave amplitude (B) and photopic a-wave amplitude (D) at 6 months of age (n = 7–8). (E) Representative fluorescent micrographs of retinal tissues from WT mice and β5t-Tg mice at 6 months of age immunostained for cone arrestin. (F) Quantification analysis of cone arrestin–positive cells within 600 μm from both sides of the optic disc. Scale bar: 20 μm. Values are mean ± SEM. *P < 0.05; **P < 0.01.
Photoreceptor Cell Death in β5t-Tg Mice
To further explore the mechanism of photoreceptor cell death in β5t-Tg mice, TUNEL assay and IF study were performed. Immunofluorescence study showed the presence of rhodopsin exclusively in OS, but not in inner segment, indicating that no ectopic accumulation of rhodopsin was induced by proteasome dysfunction (Fig. 5A). The number of TUNEL-positive cells in ONL significantly increased in β5t-Tg mice in comparison with WT mice (n = 5 each, P < 0.05, Figs. 5B, 5C). Activities of cleaved caspase-3 and -9 were not increased in β5t-Tg mice compared with WT mice (Fig. 5D). In addition, IF study showed no staining of cleaved caspase-3 and -9 in TUNEL-positive cells (data not shown). By contrast, TUNEL-positive photoreceptor nuclei in β5t-Tg mice were stained with antibody against AIF (Fig. 5E). The data indicate that the photoreceptor cell death in β5t-Tg mice is induced via a caspase-independent pathway.
Figure 5.

Apoptotic changes of photoreceptor cells caused by proteasome dysfunction. (A) Representative fluorescent micrographs of rhodopsin, showing the exclusive localization of rhodopsin protein in OS, but not in inner segment of photoreceptors or ONL in WT mice and β5t-Tg mice at 3, 6, and 9 months of age. Scale bar: 10 μm. (B) Representative micrographs of TUNEL staining. Arrows indicate TUNEL-positive cells. Nuclei were counterstained with DAPI. Scale bar: 10 μm. (C) Quantification analysis of TUNEL-positive cells in ONL from WT mice and β5t-Tg mice at 3 months of age (n = 5 each). (D) Activities of caspase-3 and -9 in retinal tissues from WT mice and β5t-Tg mice at 3 months of age (n = 10 each). Values are mean ± SEM. (E) Representative fluorescent micrographs of AIF protein, showing the costaining of AIF protein (arrows) in TUNEL-positive cells (n = 4 each). Nuclei were counterstained with DAPI. These sections were obtained from mice at 3 months of age. Scale bar: 10 μm. Values are mean ± SEM. *P < 0.05.
Discussion
The present study demonstrated that (1) decreased function of proteasome causes retinal degeneration in vivo, largely attributed to photoreceptor cell death, (2) photoreceptor cell degeneration occurs even in the absence of gene mutation of photoreceptor-related proteins, and (3) proteasomal dysfunction induces apoptosis of photoreceptor cells via a caspase-independent pathway. In addition, the current data suggest that photoreceptor cells are vulnerable to the functional disturbance of proteasome in the retina.
Functional impairment of proteasome is known to trigger systemic neurodegenerative diseases. For instance, proteasomal function was shown to be impaired in substantia nigra of patients of Parkinson's disease.13 In addition, proteasomal activity was significantly decreased in the hippocampus and parahippocampal gyrus of patients with Alzheimer's disease.11 Therefore, we hypothesized that proteasomal dysfunction was also relevant to retinal neurodegeneration. In β5t-Tg mice used in this study, an animal model of senescence acceleration, β5t protein was incorporated into 20S proteasomes over β5 protein, chymotrypsin-like activity was decreased and subsequently poly-ubiquitinated protein was increased in the retina of β5t-Tg mice. The data demonstrated that incorporation of β5t protein induced partial suppression of proteasomal activity in the retinal tissues of β5t-Tg mice.
Histologic analysis revealed that degenerative changes exclusively occurred in ONL and OS of photoreceptor cells, while inner retinal tissue was not changed. The present data may indicate that photoreceptors are vulnerable to proteasomal dysfunction. In addition, the number of cone photoreceptor was not reduced in β5t-Tg mice with age, suggesting that photoreceptor cells affected by proteasomal dysfunction were rod. In line with the histologic findings, scotopic ERGs showed that amplitude of a-wave was significantly reduced in β5t-Tg mice. By contrast, the amplitudes of photopic a-wave were reduced in β5t-Tg mice compared with WT mice, while statistical significance was not achieved in the same number of experiments that were performed with rods. Further evaluation is required to elucidate the impact of proteasomal dysfunction in cone photoreceptors.
Recently, lines of evidence have demonstrated that rhodopsin gene mutation causes ectopic accumulation of rhodopsin protein and rhodopsin aggregation have been thought to be crucial for photoreceptor cell death.5 Rhodopsin mutations have been classified into two groups, class I and II. In class I category, mutations at the C-terminus of rhodopsin affect the post-Golgi trafficking and impair its normal targeting to photoreceptor OS.22,23 Subsequent ectopic rhodopsin accumulation has been shown to cause cell death in vitro setting and in vivo setting.22–24 In class II, mutations in the transmembrane, intradiscal or cytoplasmic domains of rhodopsin lead to protein misfolding, followed by the ectopic accumulation of rhodopsin protein in ONL and inner segment25–27 and by the retention of rhodopsin protein in the endoplasmic reticulum.28,29 By contrast, the present data demonstrated that decrease of proteolytic activity induced photoreceptor cell degeneration without mutation of photoreceptor-associated protein including rhodopsin. Furthermore, ectopic accumulation of rhodopsin was not detected and structure of OS was unexpectedly maintained, that is, ultrastructural alterations reported in human RP, for example, disoriented disc formation30 and electron-dense inclusions31 were not observed in animals with proteasomal dysfunction. These data indicate the possible and novel mechanism(s) to induce photoreceptor cell degeneration without mutant protein accumulation.
Apoptosis of photoreceptor cells is caused through both a caspase-dependent and a caspase-independent pathway.32–34 The current study showed the AIF protein was present in TUNEL-positive photoreceptor nuclei in β5t-Tg mice. Translocation of AIF from the mitochondrial intermembrane space to the nucleus participates in apoptosis characterized by caspase-independent peripheral chromatin condensation and large-scale DNA fragmentation.35 Therefore, it is likely that a caspase-independent pathway, but not a caspase-dependent pathway, is implicated in photoreceptor cell death in β5t-Tg mice. In line with the current study, it was previously shown that proteasome inhibition led to neuronal cell death through caspase-independent pathways.36 Alternatively, Lobanova et al.37 recently described that insufficient capacity of proteasomes to process abnormally large amounts of misfolded protein causes photoreceptor cell death. The previous and current data may indicate that imbalance between proteasomal activity and amount of its substrate protein causes retinal degeneration. The mechanism by which functional impairment of proteasome causes photoreceptor degeneration is of interest and needs to be elucidated by further studies.
The limitation of this study is that the strain C57BL/6 used in this study carries a homozygous rd8 mutation. The rd8 mutation is a single nucleotide deletion in the Crb1 gene, which results in exhibiting retinal white deposits covering the inferior nasal quadrant of the retina and a slow progressive retinal degeneration.38 Whereas retinal white deposits were seen even in superior quadrant of the retina in β5t-Tg mice, it is undeniable that the white deposits are caused by the presence of rd8 mutation. By contrast, morphologic and functional photoreceptor degeneration was exclusively found in β5t-Tg mice, but not in its littermate mice.
In summary, the current data showed that decreased proteasomal activity induces apoptosis of photoreceptor cells in vivo. Retinitis pigmentosa is one of the representative neurodegenerative disorder in the eye and the causative gene of RP has been defined only in 50% and 30% of autosomal dominant and recessive RP, respectively.4 The current data raise an interesting question regarding the impact of proteasome dysfunction in the pathogenesis of RP.
Acknowledgments
The authors thank Ikuyo Hirose, Shiho Namba, and Erdal Tan Ishizuka for their skillful technical assistance.
Supported by Grant-in-Aid for Scientific Research (C) (25462702) from the Japan Society for the Promotion of Science (Tokyo, Japan).
Disclosure: R. Ando, None; K. Noda, None; U. Tomaru, None; M. Kamoshita, None; Y. Ozawa, None; S. Notomi, None; T. Hisatomi, None; M. Noda, None; A. Kanda, None; T. Ishibashi, None; M. Kasahara, None; S. Ishida, None
References
- 1. Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012; 181: 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cook C, Stetler C, Petrucelli L. Disruption of protein quality control in Parkinson's disease. Cold Spring Harb Perspect Med. 2012; 2: a009423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000; 9: 2009–2018 [DOI] [PubMed] [Google Scholar]
- 4. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809 [DOI] [PubMed] [Google Scholar]
- 5. Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002; 277: 34150–34160 [DOI] [PubMed] [Google Scholar]
- 6. Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002; 115: 2907–2918 [DOI] [PubMed] [Google Scholar]
- 7. Friedman JS, Ray JW, Waseem N, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009; 84: 792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kigoshi Y, Tsuruta F, Chiba T. Ubiquitin ligase activity of Cul3-KLHL7 protein is attenuated by autosomal dominant retinitis pigmentosa causative mutation. J Biol Chem. 2011; 286: 33613–33621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998; 92: 367–380 [DOI] [PubMed] [Google Scholar]
- 10. Groll M, Bochtler M, Brandstetter H, Clausen T, Huber R. Molecular machines for protein degradation. Chembiochem. 2005; 6: 222–256 [DOI] [PubMed] [Google Scholar]
- 11. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000; 75: 436–439 [DOI] [PubMed] [Google Scholar]
- 12. McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003; 179: 38–46 [DOI] [PubMed] [Google Scholar]
- 13. McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001; 297: 191–194 [DOI] [PubMed] [Google Scholar]
- 14. Tomaru U, Takahashi S, Ishizu A, et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol. 2012; 180: 963–972 [DOI] [PubMed] [Google Scholar]
- 15. Murata S, Sasaki K, Kishimoto T, et al. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science. 2007; 316: 1349–1353 [DOI] [PubMed] [Google Scholar]
- 16. Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009; 10: 104–115 [DOI] [PubMed] [Google Scholar]
- 17. Mehalow AK, Kameya S, Smith RS, et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003; 12: 2179–2189 [DOI] [PubMed] [Google Scholar]
- 18. Mattapallil MJ, Wawrousek EF, Chan CC, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakao T, Tsujikawa M, Notomi S, Ikeda Y, Nishida K. The role of mislocalized phototransduction in photoreceptor cell death of retinitis pigmentosa. PLoS One. 2012; 7: e32472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsuruma K, Yamauchi M, Inokuchi Y, Sugitani S, Shimazawa M, Hara H. Role of oxidative stress in retinal photoreceptor cell death in N-methyl-N-nitrosourea-treated mice. J Pharmacol Sci. 2012; 118: 351–362 [DOI] [PubMed] [Google Scholar]
- 21. Ferrington DA, Hussong SA, Roehrich H, et al. Immunoproteasome responds to injury in the retina and brain. J Neurochem. 2008; 106: 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung CH, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994; 14: 5818–5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tam BM, Moritz OL, Hurd LB, Papermaster DS. Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J Cell Biol. 2000; 151: 1369–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Green ES, Menz MD, LaVail MM, Flannery JG. Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000; 41: 1546–1553 [PubMed] [Google Scholar]
- 25. Surgucheva I, Ninkina N, Buchman VL, Grasing K, Surguchov A. Protein aggregation in retinal cells and approaches to cell protection. Cell Mol Neurobiol. 2005; 25: 1051–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roof DJ, Adamian M, Hayes A. Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest Ophthalmol Vis Sci. 1994; 35: 4049–4062 [PubMed] [Google Scholar]
- 27. Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992; 9: 815–830 [DOI] [PubMed] [Google Scholar]
- 28. Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994; 33: 6121–6128 [DOI] [PubMed] [Google Scholar]
- 29. Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 8840–8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolb HJ. Biological and immunological activity of fructose 1,6-bisphosphatase. Effect of structural changes on the quantitative displacement radioimmunoassay of fructose 1,6-bisphosphatase. Eur J Biochem. 1974; 43: 145–153 [DOI] [PubMed] [Google Scholar]
- 31. To K, Adamian M, Dryja TP, Berson EL. Retinal histopathology of an autopsy eye with advanced retinitis pigmentosa in a family with rhodopsin Glu181Lys. Am J Ophthalmol. 2000; 130: 790–792 [DOI] [PubMed] [Google Scholar]
- 32. Liu C, Li Y, Peng M, Laties AM, Wen R. Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation s334ter during photoreceptor degeneration. J Neurosci. 1999; 19: 4778–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ. 2002; 9: 1220–1231 [DOI] [PubMed] [Google Scholar]
- 34. Doonan F, Groeger G, Cotter TG. Preventing retinal apoptosis–is there a common therapeutic theme? Exp Cell Res. 2012; 318: 1278–1284 [DOI] [PubMed] [Google Scholar]
- 35. Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999; 397: 441–446 [DOI] [PubMed] [Google Scholar]
- 36. Papa L, Gomes E, Rockwell P. Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis. 2007; 12: 1389–1405 [DOI] [PubMed] [Google Scholar]
- 37. Lobanova ES, Finkelstein S, Skiba NP, Arshavsky VY. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci U S A. 2013; 110: 9986–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002; 42: 517–525 [DOI] [PubMed] [Google Scholar]