

Abstract
The blood-brain barrier (BBB) poses a significant impediment for the delivery of therapeutic drugs into the brain. This is particularly problematic for the treatment of malignant gliomas which are characterized by diffuse infiltration of tumor cells into normal brain where they are protected by a patent BBB. Selective disruption of the BBB, followed by administration of anti-cancer agents, represents a promising approach for the elimination of infiltrating glioma cells. A summary of the techniques (focused ultrasound, photodynamic therapy and photochemical internalization) for site-specific opening of the BBB will be discussed in this review. Each approach is capable of causing localized and transient opening of the BBB with minimal damage to surrounding normal brain as evidenced from magnetic resonance images and histology.
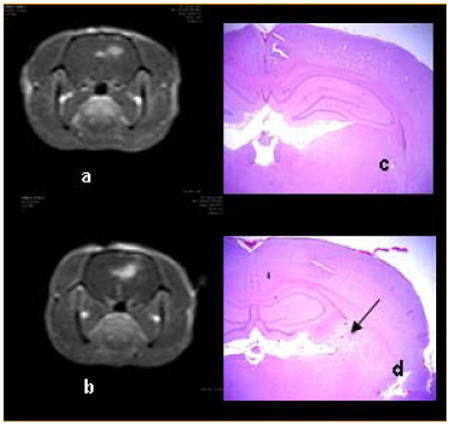
T1-weighted MRI contrast enhanced images (a, b) showing focal enhancement in the area of light treatment. Fluence levels of 9 (a) and 17 J (b) at a fluence rate of 10 mW were performed 4 h following ALA administration (125 mg kg−1 i.p.). Scans were acquired 3–4 h post-PDT and 15 min following i.p. Gd contrast administration. Coronal H&E sections (c, d) taken from the brains of Fischer rats 14 days post-treatment. No significant pathology was observed following delivery of 9 J (c). At the high fluence (17 J), extensive infiltration of lymphocytes and macrophages was apparent as shown by the arrow in (d) [19]. Reprinted with permission.
Keywords: blood-brain barrier, photodynamic therapy, photochemical internalization, focused ultrasound, targeted opening, Clostridium perfringens prototoxin, malignant glioma
1. Introduction
The blood-brain barrier (BBB) controls the passage of blood-borne agents into the central nervous system (CNS) and, as such, it plays a vital role in protecting the brain against pathogens. Although this protective mechanism is essential for normal brain function, it also poses a significant hindrance to the entry of drugs into the brain. In this context, it is hardly surprising that brain diseases account for approximately 30% of the total burden of all diseases [1]. Nearly all large-molecule pharmaceutics (peptides, recombinant proteins, monoclonal antibodies and gene therapeutics) and 98% of small molecules do not cross the BBB [2]. Of the 7000 drugs in the Comprehensive Medicinal Chemistry database, only 5% treat the CNS and these drugs are limited to the treatment of depression, insomnia and schizophrenia [3, 4]. The few drugs that do cross the BBB have a number of features in common including small size (<400 Da), high lipid solubility, and passage across the BBB by passive diffusion. Presently, there are no CNS drugs that effectively address major brain disorders including Alzheimer's, strokes and tumors.
The protective function of the BBB is particularly problematic for the treatment of infiltrating gliomas. Although surgery is used to remove gross tumor, standard adjuvant therapies consisting of radiation and chemotherapy often fail to eliminate infiltrating glioma cells in or beyond the brain-adjacent-to-tumor (BAT) region – a zone that commonly extends several centimeters from the resection margin. This is the reason for the high rate of tumor recurrence (80%) within a 2–3 cm margin of the surgical resection cavity [5]. Infiltrating tumor cells are supplied with nutrients and oxygen by the normal brain vasculature and consequently, protected by the BBB: few anti-cancer drugs are capable of crossing this barrier. Therefore, eradication of gliomas is highly unlikely without addressing the problems posed by the BBB.
In general, three different strategies for the delivery of drugs into the brain have been attempted: widespread opening of the BBB (paracellular approaches), circumventing the BBB and delivery across the BBB (transcellular approaches). In the first approach, intracarotid arterial infusion of either hyperosmolar solutions (e.g. mannitol) or vasoactive drugs (e.g. bradykinin) along with drug administration has been investigated. The utility of this approach is questionable for a number of reasons: (1) the BBB remains open for only a short period of time and therefore the procedure must be repeated in multi-fractionated drug delivery schemes, (2) non-selective opening of the BBB exposes large volumes of normal brain to undesirable substances that may be toxic [6], and (3) the rapid influx of substances produces transient increases in intracranial pressure. A number of techniques for bypassing the BBB have been investigated including direct injection into the cerebrospinal fluid [7], ventricles [8] or tumors [9, 10], implantation of drug-releasing polymers [11, 12], convection-enhanced delivery [13–15] and intranasal drug administration [16]. Due to a number of drawbacks, including limited drug diffusion and the inability to attain therapeutic concentrations, these approaches have failed to demonstrate clinical utility. A number of approaches for drug delivery across the BBB have been attempted including encapsulation into liposomes and nanoparticles. The main drawback with these techniques is the rapid removal of drug from the circulation due to increased uptake in all organs thus reducing the amount of drug reaching the brain [1]. Perhaps the most promising technique for trans-BBB delivery is the conjugation of therapeutic drugs to proteins (e.g. insulin, ApoE and transferrin) that are known to traverse the BBB by receptor-mediated endocytosis [17]. Although this so-called molecular Trojan horse approach has been used for the delivery of a number of therapeutic proteins, there are still some limitations including rapid removal from the circulation, low delivery yields and the need for repeated injections [1].
Site-specific disruption of the BBB represents an alternative approach for drug delivery into the brain. This has been accomplished using either highly focused ultrasound (reviewed in [18]) or laser-based approaches such as photodynamic therapy (PDT) [19] and photochemical internalization (PCI) [20]. These techniques are appealing for a number of reasons including the highly localized nature of the BBB disruption: unlike the use of hyperosmolar solutions, the BBB is only disrupted at sites subjected to sufficient beam power densities which can be controlled by the user to coincide with the location of the pathology. Through judicious choice of beam parameters, the affected volume can be as small as a few mm3. Equally important are observations showing that these highly focused approaches do not cause permanent damage to the BBB, as long as incident power densities remain below threshold levels. Under these conditions the BBB may remain open for relatively long periods of time thus facilitating multi-fractionated drug delivery. In contrast, repeated injections of hyperosmotic compounds are required for extended treatment regimens since the BBB remains open for only a few minutes following bradykinin administration [21].
A summary of the methods (focused ultrasound, PDT and PCI) for site-specific disruption of the BBB in rodents and rabbits will be presented in this review. The utility of these techniques in neuro-oncology will be emphasized especially with regards to their ability to facilitate drug delivery to infiltrating glioma cells protected by a patent BBB. It is shown that each approach is capable of causing localized and transient opening of the BBB with minimal damage to normal structures as evidenced from contrast enhanced magnetic resonance imaging (MRI) and histology.
2. The blood-brain barrier
The first evidence for the existence of the BBB was provided by Ehrlich in 1885 [22]. The primary function of the barrier is to maintain homeostasis of the brain – it is the most important global influx barrier preventing solutes from entering the brain [23]. The human BBB has a total blood vessel length of approximately 600 km occupying a surface area of around 20 m2 [24]. The degree to which the brain is vascularized can be appreciated by the fact that every cubic centimeter of cortex contains 1 km of blood vessels [24].
The BBB is formed by tightly connected brain capillary endothelial cells (Figure 1). Substantially different than those found in peripheral microvessels, the endothelial cells lining the brain vessels are connected by much tighter junctional complexes in order to completely seal the paracellular spaces and form a continuous physical barrier between the CNS and blood circulation [25]. Both the lumen-facing (luminal) and the brain-facing (abluminal) membranes of the endothelium consist of phospholipid bilayers lacking fenestrations. The luminal and abluminal membranes of the capillary endothelium are separated by approximately 200 nm of endothelial cytoplasm [26]. Molecules crossing the BBB must traverse these two limiting membranes of the endothelium. The brain side of the capillary endothelial cells is completely covered by a basement membrane with the end-foot processes of the astrocytes closely attached to it. Pericytes are embedded in the basement membrane between the endothelial cell and astrocyte process, making particularly close contact with endothelial cells. They are thought to provide structural support to the microvasculature and are important in BBB stability [27]. Both the basement membrane and astrocyte foot processes allow diffusion of molecules [26] which are free to diffuse through the brain extravascular space once successfully across the limiting membranes.
Figure 1.
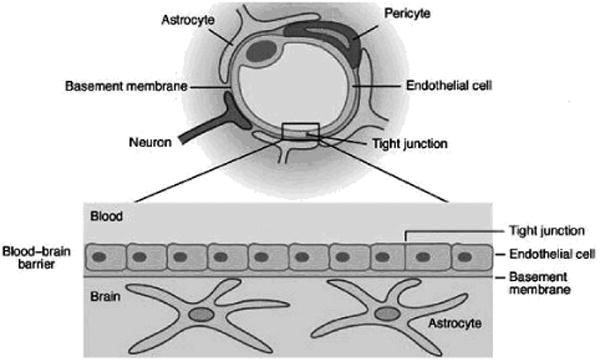
Cross section through a brain microcapillary (upper panel) and the overall structure of the BBB (lower panel). (Modified from: http://www-ermm.cbcu.cam.ac.uk/03006264h.htm.)
The impermeability of the BBB is the result of a number of unique features. Firstly, the physical restriction imposed by tight junctions between endothelial cells greatly reduces paracellular permeability. Additionally, the transport system regulation of endothelial cells limits the number and types of molecules that undergo transcellular transport. Lastly, the metabolic activity of endothelial cells, with powerful enzymes metabolizing many potentially harmful substances, adds to the difficulties faced by molecules trying to penetrate the BBB.
Paracellular transport of substances into the brain is regulated by the tight junctional complex consisting of transmembrane proteins that form beltlike strands between adjacent endothelial cells (Figure 2). Perhaps the most important of these are the occludins and claudins which are transmembrane tight junction (TJ) proteins that play a key role in maintaining the structure and function of the BBB [29]. Other key proteins include the zonula occludens (ZO-1, ZO-2 and ZO-3) which are submembraneous tight junction-associated proteins that anchor the transmembrane TJ proteins to the cytoskeleton and participate in signal transduction [30]. It is hypothesized that impairment of the TJ proteins causes BBB dysfunction and, as such, these proteins would appear to be appealing targets for therapeutic interventions focused on drug delivery to the brain [29].
Figure 2.
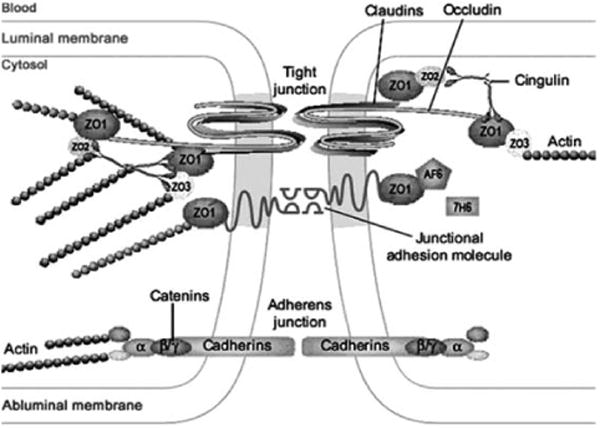
Structure of the tight junctional complex at the BBB. (Modified from [28] and reprinted with permission.)
3. Methods for selective disruption of the blood-brain barrier
3.1 Focused ultrasound
Focused ultrasound (FUS)-induced effects in the brain have been investigated since the early 1940s [31]. By using focused ultrasound, acoustic energy can be concentrated into a focal spot with a diameter of a few mm [32]. Although the technique is capable of producing selective disruption of the BBB, studies have failed to identify sonication parameters resulting in reliable opening of the BBB without concomitant damage to normal brain and therefore the technique appears to have limited clinical utility [33–39]. To address this concern, commercially available contrast agents (e.g. Optison®) consisting of albumin coated microbubbles, have been used in FUS approaches [40]. The presence of these preformed microbubbles circulating in the vasculature confines the ultrasound effects to the blood vessel walls resulting in BBB disruption with minimal damage to surrounding brain tissue [41]. Typically, the contrast agent is injected intravenously a few seconds prior to sonication. The introduction of contrast agents has allowed selective disruption of the BBB at much lower acoustic power levels than previously employed, making this approach substantially easier to apply through the intact skull which is highly absorbing of ultrasound causing heating during sonication [31].
Studies have been conducted in a number of small animal models (mice, rats and rabbits) in order to determine the optimal acoustic parameters for selective opening of the BBB. Typically, the effects of FUS on the BBB are evaluated from contrast-enhanced MR images using commercially available gadolinium (Gd) agents. Since Gd contrast does not traverse the intact BBB, the presence of the compound in the brain parenchyma is suggestive of BBB breakdown. MRI has also been used as a localization tool to guide FUS-induced BBB disruption in targeted regions of the brain.
A major limitation of FUS is the strong phase and amplitude aberrations introduced by the skull [42]. The resultant attenuation and distortion of the ultrasound field increases with acoustic frequency thus preventing the delivery of sufficient acoustic power for BBB disruption at diagnostic frequencies (>3 MHz) through the thin skulls of rodents. Potential solutions to this problem include the use of lower frequencies and sophisticated adaptive techniques, such as large surface area phased arrays [42]. A number of animal studies have shown that trans-cranial focusing can be achieved at frequencies below 1 MHz [43], however, the clinical implications are difficult to evaluate due to the thickness of the human skull. Another reason for using lower frequencies is that they are less likely to produce damage to normal vasculature as indicated by lower levels of red blood cell extravasation [44].
In addition to frequency, there are a number of other parameters of importance for FUS optimization including, acoustic power, pressure amplitude, pulse repetition frequency and burst length [45]. Perhaps the most meaningful metric for FUS-induced BBB disruption is the mechanical index defined as the peak negative pressure amplitude estimated in situ divided by the square root of frequency [44]. The mechanical index was developed from theoretical formulations of inertial cavitation thresholds in water and blood and is commonly used as a standard for setting limits on nonthermal bioeffects produced by ultrasound [46]. Data from a number of animal studies suggest a mechanical index BBB disruption threshold of 0.46 [44].
The exact mechanisms by which the BBB is disrupted by contrast-enhanced FUS remain to be elucidated. The effect is likely due to a combination of cavitation and acoustic radiation forces [43]. Cavitation is defined as the acoustically induced activities of microscopic gas bubbles within the medium and is believed to be the most important of the non-thermal bio-effects of ultrasound [47]. The generation of microbubbles requires high acoustic power densities which may result in damage to the vasculature [37]. With the introduction of microbubble-based contrast agents, high powers are no longer required and therefore the risk of normal tissue damage has decreased significantly.
Electron microscopy of animal brains following FUS suggests that sonication results in transendothelial transport by both transcellular and paracellular pathways [48–50]. These include: (1) transcytosis; (2) endothelial cell cytoplasmic openings (both fenestrations and channel formation); (3) passage through leaking tight junctions and; (4) free passage through injured endothelium (usually only observed at high powers). There is compelling evidence suggesting that FUS causes disassembly of the tight junctional molecular structures leading to loss of barrier functions in brain microvessels [29]. For example, immunoelectron microscopy for the tight junction-specific proteins shows a loss of immunosignals for occludins, claudin-5 and ZO-1 as early as 1 h following sonication [29]. The barrier function of the tight junction appears completely restored 4–5 h following sonication.
Data from animal studies suggest that FUS-induced BBB disruption does not result in permanent damage to the brain as evidenced by the lack of ischemic or apoptotic areas indicative of compromised vasculature or regions of neuronal damage due to extravasated red blood cells [31]. The effects that have been observed (small extravasations and very mild inflammatory reactions) do not appear to affect neurons up to four weeks following sonication [31].
Based on MR imaging studies in small animals, BBB opening is observed over a relatively short time span ranging from approximately 10 min. to 5 h following sonication [43]. Although this time window is sufficient for administering therapeutic agents in single fractionated treatment regimens, it is unsuitable for the type of repeated drug administrations required to maintain high titers of chemotherapeutic agents for the treatment of brain tumors. This can only be achieved through repeated sonication exposure which may be difficult to implement from a practical point of view.
The feasibility of MR image-guided FUS for the delivery of antibodies and chemotherapeutic agents to the brain has been demonstrated in a number of rodent models [32, 50, 51]. These studies show that anti-dopamine D4 receptor antibody [32], Herceptin [50] and the chemotherapeutic agent, doxorubicin [51] can be delivered in sonicated regions of the brain. The size of the FUS-induced opening is not known, however, it must be relatively large to allow passage of Herceptin (mol. wt. = 150 kDa). Although interesting, the clinical relevance of these results is not entirely clear since all studies were performed in normal brain. The use of disease models will be required to determine whether sufficient drug concentrations can be delivered to impact quality of life and/or survival.
3.2 Photodynamic therapy
PDT is a local form of treatment involving the administration of a tumor-localizing photosensitizer that is subsequently activated by light of a wavelength matching a major absorption resonance of the drug [52]. The resultant photochemical and photobiological events cause irreversible damage to tissues. Several studies have shown that PDT may prove useful in prolonging survival and/or improving the quality of life of glioma patients [53–55]. The aim of PDT in neuro-oncology is to eliminate the nests of tumor cells remaining in the BAT region following surgical resection while minimizing damage to surrounding normal brain. This may be difficult to achieve since glioma cells in the BAT are protected by a relatively intact BBB that prevents the passage of sufficient levels of photosensitizers and prodrugs required to elicit a PDT response [56, 57].
The prodrug, 5-aminolevulinic acid (ALA), appears to be particularly well suited for the treatment of gliomas due to its excellent tumor specificity [58] and rapid systemic clearance [60]. This latter characteristic makes it particularly appealing for repetitive PDT regimens which have been shown to have clear advantages over single treatments in a number of experimental and clinical studies [55, 60, 61].
In ALA-induced endogenous photosensitization, the heme biosynthetic pathway is used to produce protoporphyrin IX (PpIX) – a photosensitizer that has been shown to accumulate in high concentrations in glial tumors [62–64]. Not surprisingly, animal studies have demonstrated the efficacy of ALA in PDT of bulk tumor [65] while its excellent tumor specificity has made it useful in fluorescence-guided resection of high-grade gliomas (64, 66). The high drug levels observed in gliomas are likely due to passive diffusion across a compromised BBB. The low PpIX levels (<1% of that found in bulk tumor) observed in normal brain following ALA administration, suggest that only trace amounts of ALA traverse the intact BBB [56].
PDT using either hematoporphyrin derivatives or ALA has been reported to induce brain edema [67–70]. The edema region surrounding the site of light treatment suggests a local degradation of the BBB. PDT therefore appears to have a two-fold effect: a direct antineoplastic effect on the remaining tumor cells as well as an effect that causes localized opening of the BBB.
A systematic study of the effects of ALA-PDT on the BBB in normal rat brain was recently completed [19]. Intracranial light delivery from a 632 nm diode laser was accomplished by stereotactic placement of a flat end optical fiber (numerical aperture = 0.22) directly into the brain to a depth of 5 mm below the dura. Contrast enhanced T1-weighted MRI was used to monitor the degree of BBB disruption which was inferred from the intensity and volume of the Gd contrast agent visualized. The results show that the degree of BBB disruption was sensitively dependent on both light fluence and fluence rate. The exact fluence rates (and fluences) in the rat brain are unknown since direct measurements of light distributions were not attempted. The stated fluence rates represent the incident irradiances measured from the flat end fiber prior to insertion into the brain. The intracranial fluence rate will be many times higher near the fiber due to multiple scattering [71]. At a relatively low fluence rate of 10 mW, ALA-PDT at increasing fluence levels resulted in increased contrast flow rates (Figure 3). Histological sections taken 14 days post-PDT showed local tissue damage (including necrosis and hemorrhage) only at the higher fluences (17 and 26 J) suggesting an upper limit to the maximum fluence that can be employed. Higher fluence rates had a profound effect on BBB patency as evidenced from both increased contrast volumes and signal intensities (Figure 4). At all light fluences investigated (9–26 J), fluence rates exceeding 10 mW induced pronounced brain edema which often led to increased morbidity, and even death (Figure 5). Analysis of T2-weighted MR images showed an exponential decrease in edema volume as a function of time post-PDT treatment (Figure 6). At the highest fluence investigated (26 J), edema volume decreased by approximately 50% within three days of treatment. This time period was found to be a significant prognostic indicator: any animal surviving past day three was highly likely to be a long-term survivor.
Figure 3.
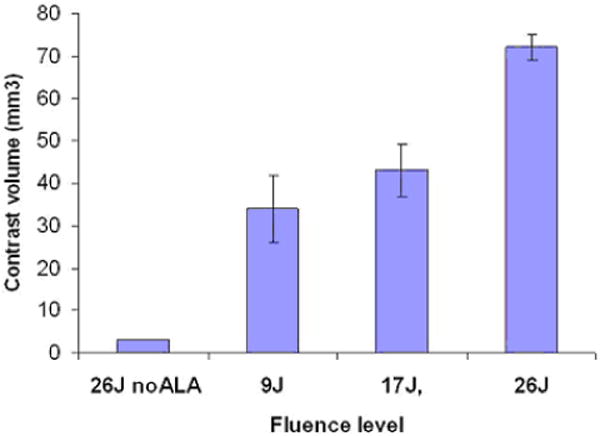
(online color at: www.biophotonics-journal.org) Mean contrast volume measured from MRI scans performed 24 h post-PDT using a fluence rate of 10 mW. ALA (125 mg kg−1 i.p.) was administered 4–5 h prior to light delivery. A significant light dose response was apparent with increasing light fluence resulting in increased contrast. A fluence of 26 J, in the absence of ALA (light-only control), resulted in minimal contrast enhancement suggesting an intact BBB [19]. (Reprinted with permission.)
Figure 4.
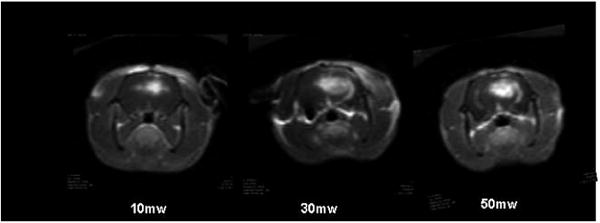
T1-weighted MR contrast enhanced images showing focal contrast enhancement as evidence of BBB disruption. Increased fluence rates resulted not only in increased contrast volume but increased signal intensity as well, indicating an increased contrast concentration. In all cases, Fischer rats were irradiated to a total fluence of 26 J 4–5 h following ALA administration (125 mg kg−1 i.p.) Images were acquired 15 min following i.p. Gd contrast administration [19]. (Reprinted with permission.)
Figure 5.
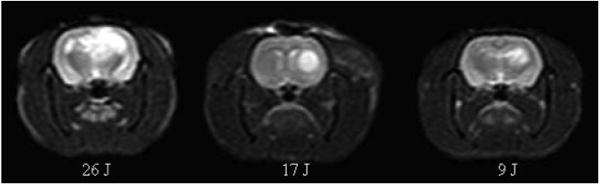
(online color at: www.bio-photonics-journal.org) T2-weighted MR images showing increased signal intensities as evidence of light dose dependent edema formation. Light was delivered at a fluence rate of 10 mW and images were acquired 24 h following PDT. ALA was administered (125 mg kg−1 i.p.) 4-5 h prior to light administration.
Figure 6.
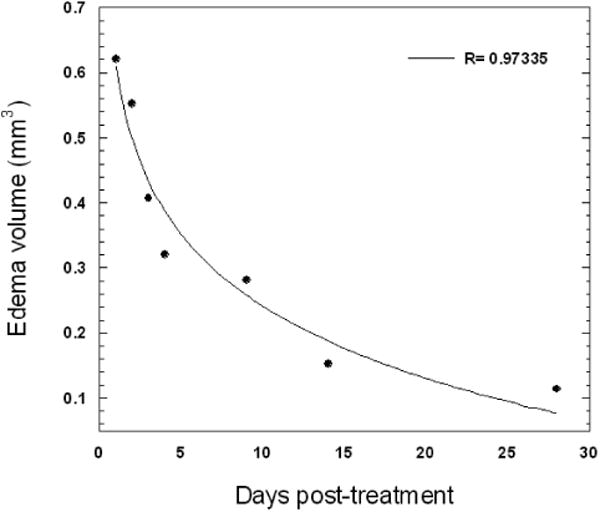
Temporal dependence of edema volume in ALA-PDT-treated Fischer rats. Light fluence and fluence rates were 17 J and 10 mW, respectively. Edema volumes were estimated from T2-weighted MR images. The curve represents the best exponential fit to the data.
ALA-PDT-mediated disruption of the BBB was found to be temporary in nature opening rapidly following treatment and significantly restored 72 h later (Figure 7). The BBB was found to be disrupted as early as 2 h following PDT and approximately 90% restored 72 h later. Although significantly longer in duration compared to FUS-induced BBB opening, this window may be insufficient for the application of anti-cancer agents in extended multi-fractionated treatments.
Figure 7.
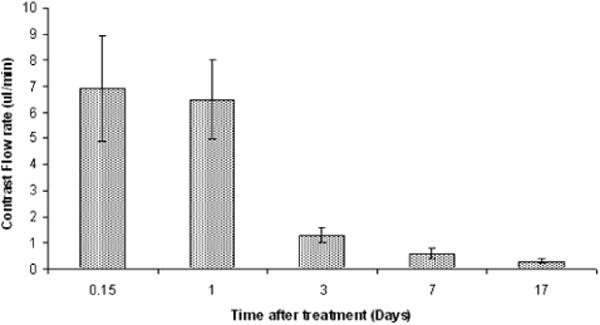
Time course of BBB closing. MRI scans were acquired 4 h and 1, 3, 7 and 17 days post-PDT treatment. At each time point, contrast flow rate (based on increasing volume of T1 signal intensity), was estimated by rescanning the rats at several intervals following contrast injection. The flow rate decreased rapidly (80%) between days 1 and 3. PDT was performed 4 h following i.p. injection of 125 mg kg−1 ALA. All animals were subjected to light fluence and fluence rates of 17 J and 10 mW, respectively [19]. (Reprinted with permission.)
The mechanisms of PDT-mediated BBB opening are unknown but they likely include direct PDT effects on the endothelial cytoskeleton that lead to cell rounding and contraction, probably mediated by PDT-induced microtubule depolarization [72]. Furthermore, the formation and/or enlargement of endothelial gaps has been observed in response to PDT [73]. Additional studies in a rat brain tumor model have found extensive edema formation in response to ALA-PDT which is likely due to a breakdown of the blood-tumor barrier (BTB) [65, 69]. In these cases, the BTB was significantly altered by PDT most probably in a manner similar to the one proposed for the BBB, i.e., by enlarging endothelial gaps. Electron microscopy studies in a rat C6 glioma model demonstrated stretching of the tight junctions, with an enlargement of the gaps between endothelial cells following PDT [74]. The treatment was found to have only a minimal impact on the normal subcellular structures of the BBB suggesting that the endothelial cells were not permanently damaged by PDT.
3.3 Photochemical internalization
Photochemical internalization (PCI) is a novel technology that can enhance the delivery of macromolecules in a site-specific manner [75]. The concept is based on the use of specially designed photosensitizers, which localize preferentially in the membranes of endocytic vesicles. Upon light activation, the photosensitizer interacts with ambient oxygen causing vesicular membrane damage resulting in the release of encapsulated macromolecules into the cell cytosol instead of being transported and degraded in the lysosomes. PCI has been shown to potentiate the biological activity of a large variety of macromolecules and other molecules that do not readily penetrate the plasma membrane, including proteins (e.g. protein toxins and immunotoxins), peptides, DNA delivered as a complex with cationic polymers or incorporated in adenovirus or adeno-associated virus, peptide-nucleic acids (PNA) and chemotherapeutic agents [76–79].
Localized BBB opening via PCI-mediated delivery of Clostridium perfringens epsilon prototoxin (ETXp) was recently investigated in Fischer rats [20]. The rationale for using ETXp is due to the ability of active toxin (ETX) to cause widespread but reversible opening of the BBB [80–82]. Following systemic administration, ETXp is converted to fully active toxin by proteolytic cleavage. Administration of ETXp in rats has also been shown to result in a reduction of the endothelium barrier antigen in rat brain endothelial cells accompanied by a reversible opening of the BBB [83]. Since the overall objective of the study was to evaluate the efficacy of PCI for localized BBB disruption, only low concentrations of ETXp were administered, i.e., concentrations sufficiently below the threshold for BBB opening. Disruption of the BBB was accomplished by combining sub-threshold doses of ETXp with sub-threshold PDT light fluences. In all cases, a membrane localizing photosensitizer (aluminium phthalocyanine disulfonate; AlPcS2a) was used and contrast-enhanced MRI was employed to track BBB disruption following each procedure. Light illumination was accomplished by coupling the 670 nm light output from a diode laser into a flat end optical fiber placed on the surface of the rat's skull. As with the ALA-PDT studies described in the previous section, direct intracranial measurements of fluence rates were not attempted: the stated fluence rates represent the incident irradiances measured from the flat end fiber and fluences were calculated based on these values.
The results show that ETXp-PCI is capable of causing localized BBB disruption at very low light fluences (0.5 and 1 J) (Figures 8 and 9). In comparison, the BBB remained relatively intact when exposed to AlPcS2a-PDT at these light levels. At higher fluences (2.5 J), the PDT effect was so pronounced that the addition of ETXp had no apparent effect on BBB disruption. Based on histological data, no significant damage was noted in rat brains subjected to ETXp-PCI at light fluences of 0.5 and 1 J. In contrast, at fluence levels of 2.5 J, even in the absence of ETXp, a focally extensive area of necrosis and degeneration of brain parenchyma and infiltration of lymphocytes, plasma cells and foamy macrophages were evident. These observations are consistent with treatment-induced cerebral ischemia.
Figure 8.
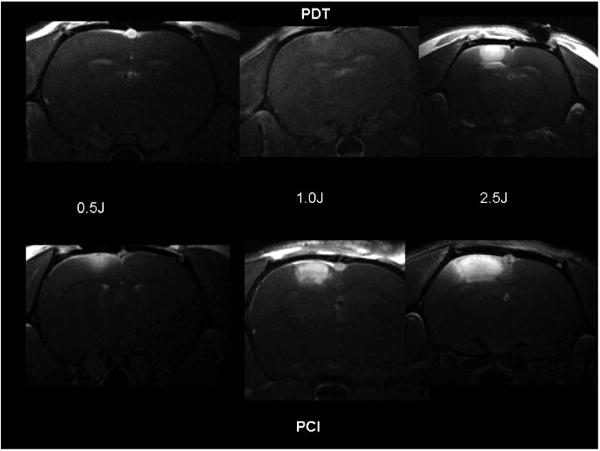
T1-weighted contrast enhanced MR images showing focal enhancement directly below the light source. Forty-eight hours following AlPcS2a administration (1 mg kg−1 i.p.), six groups of Fischer rats (n = 4 per group) were irradiated to 3 different fluences using a fluence rate of 10 mW. The images were acquired 3 days following PDT (AlPcS2a + light) or PCI (ETXp + AlPcS2a + light). ETXp was administered (20 μg kg−1 i.p.) 1 h prior to light treatment. In all cases, images were acquired 15 min following i.p. Gd contrast administration [20]. (Reprinted with permission.)
Figure 9.
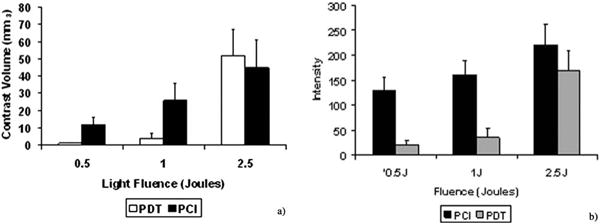
(a) Average contrast volume and (b) signal intensity measured from MR images performed 3 days post-PDT or ETXp-PCI treatment for three light fluences (fluence rate = 10 mW). Each data point represents the mean (± standard error) of four animals [20]. (Reprinted with permission.)
Of particular interest is the time duration and evolution of the ETXp-PCI BBB disruption since this represents the therapeutic window for drug delivery. Based on an analysis of MR images, enhancement volumes were observed to peak three days following ETXp-PCI suggestive of maximum BBB opening at that time (Figure 10). Thereafter, contrast volumes were observed to decrease and by day 11, only trace amounts of contrast were observed in the 0.5 and 1 J animals. Based on these results, the optimum therapeutic drug delivery window appeared to be between days 2 and 5 following ETXp-PCI BBB opening. The data in Figure 10 also illustrate the ineffectiveness of AlPcS2a-PDT: it is doubtful that this technique can be used to open the BBB for effective delivery of therapeutic agents at the low light fluences required to avoid causing permanent brain damage.
Figure 10.
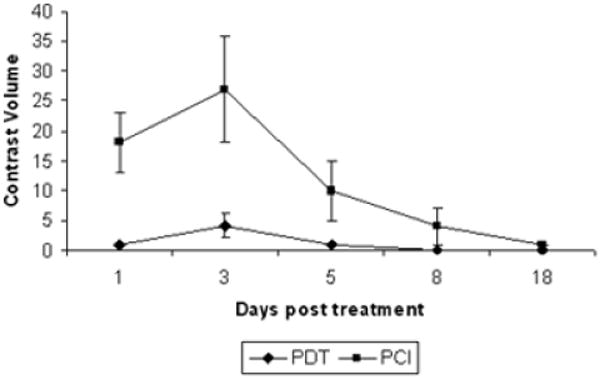
Time course of BBB opening induced by PDT or ETXp-PCI. Fischer rats (n = 4 per group) received 1 mg kg−1 AlPcS2a (i.p.) and were irradiated (1 J; 10 mW) 48 h later. The PCI group received an i.p. injection (20 μg kg−1) of ETXp 1 h prior to light irradiation. T1-weighted images were acquired 15 min after i.p. Gd contrast administration [20]. (Reprinted with permission.)
The ability of a chemotherapeutic agent (bleomycin; BLM) to eradicate infiltrative glioma cells following ETXp-PCI-mediated BBB disruption was investigated in an orthotopic brain tumor model consisting of F98 glioma cells in Fischer rats [20]. This model exhibits many of the hallmarks of glioblastoma multiforme and, as such, is considered to be highly relevant in therapeutic studies of malignant gliomas [84]. Since BBB disruption (and subsequent BLM administration) was initiated within 24 h of cell inoculation, the model was considered to be an accurate representation of infiltrating tumor cells. The 24 h time period is insufficient for the development of bulk tumor and subsequent BBB breakdown, but long enough for the cells to form small sequestered micro-clusters protected by an intact BBB. Compared to controls, animal survival was significantly extended following ETXp-PCI BBB opening and BLM therapy (Figure 11).
Figure 11.

Kaplan-Meier survival of tumor cell implanted Fischer rats. All animals received 1 × 104 F98 glioma cells and were subjected to treatment 24 h later. Three groups were followed: non-treated controls, PDT-BLM controls (AlPcS2a + 1 J), ETXp-PCI BLM experimental group (AlPcS2a+ ETXp + 1 J); BLM (4 mg kg−1) was injected i.p. twice daily for 3 days [20]. Reprinted with permission.)
Collectively, the results show that ETXp-PCI can be used to open the BBB in a highly localized and transient manner without causing permanent damage to the brain or BBB. As evidenced from survival studies in tumor cell implanted animals, the extent and time course of disruption were sufficient to allow the passage of therapeutic doses of a high molecular weight (ca. 1.4 kDa) chemotherapeutic agent into the brain.
Although a detailed understanding of the reversible low light fluence BBB disruption is presently unknown, a direct effect on the capillary endothelial cells is the most probable cause. This would include direct interaction of ETX on the endothelial cell cytoskeleton in the illuminated regions that could lead to cell rounding and contraction, with the formation and/or enlargement of endothelial gaps that have previously been described in response to PDT [73].
ETX has recently been shown to cause rapid destruction of certain types of cancer cells expressing the ETX receptors claudin-3 and claudin-4. Unfortunately, the utility of this toxin is limited by systemic toxicity because its receptors are expressed in numerous organs as evidenced from the relatively low levels of systemically (i.p.) administered ETXp resulting in animal death (LD50 ≈ 40 μg kg−1) [20]. Nevertheless, under the appropriate conditions, ETXp-PCI appears to be a safe and efficient method for localized BBB opening in rats.
4. Summary and future work
Effective drug delivery to the brain is of vital importance for the treatment of a wide variety of neurological conditions. It is clear that the BBB plays a central role in preventing the effective management of these diseases. Although a number of strategies have been proposed to address the protective function of this barrier, relatively few investigations have focused on localized opening of the BBB. Focussed ultrasound with microbubble contrast agents has been the most widely investigated technique for site-specific disruption of the BBB. This approach has shown promise in a number of small animal models however, there remain a number of hurdles which could hamper its clinical utility. Perhaps the most vexing problem is the requirement for sub MHz frequencies for effective penetration through the skull. Although frequencies in the range of 0.26 to 2.1 MHz have been shown to cause selective and transient opening of the BBB, it's not clear whether these frequencies can penetrate the human skull which is approximately 20 times thicker than the rat skull. FUS-induced BBB disruption is highly transient in nature, lasting at most 5 h. Although this is significantly longer than can be achieved with intra-arterial injection of vasoactive compounds (a few minutes), it is too short for the type of cancer therapies requiring multi-fractionated treatments necessary for maintaining sufficient drug concentrations over extended time periods. Although repeated FUS treatments is a potential solution, it is not clear what effects such treatments would have on the endothelial cells comprising the BBB: even at relatively low acoustic powers, extravasation of red blood cells has been observed. The complex geometry of the resection cavity resulting from surgical debulking poses another challenge for FUS in neuro-oncology applications. Although the beam can be steered, it may be more difficult to treat the entire resection margin compared to laser-based approaches in which an optical fiber can be inserted into an applicator which can be placed into the resection cavity resulting in uniform irradiation of the entire margin [85]. For example spherical balloon type applicators, similar to those currently used in breast brachytherapy, may be ideal for reducing hot spots near the light irradiation source. The MRI results presented in this review suggest that it is not necessary to irradiate the entire BAT in order to achieve therapeutic drug concentrations throughout this region. MR images show that Gd contrast is capable of diffusing 0.5–1.0 cm from the region of BBB disruption. Theoretical calculations have shown that the fluence rate of 630 nm light in brain tissue decreases by less than an order of magnitude within 0.5 cm from the surface of a 2 cm diameter balloon [86]. Therefore it should be possible to open the BBB at distances of up to 0.5 cm into the BAT without causing significant damage to brain tissue immediately adjacent to the applicator. Due to drug diffusion, the effective treatment volume would then extend 1–1.5 cm from the resection margin which represents a significant fraction of the BAT.
As with FUS, ALA-PDT is also capable of producing localized and transient BBB opening with no apparent long term effects. PDT-induced opening of the BBB is sensitively dependent on both light fluence and fluence rate. Both parameters must be chosen carefully since local tissue damage has been observed at fluence levels just above those required for significant BBB disruption. In addition, relatively high fluence rates are required for significant BBB disruption. Significant edema, and resultant morbidity, are often observed at the light fluence/fluence rate combinations required for non-destructive BBB opening thus limiting the usefulness of this approach. From a drug delivery perspective, the BBB remains open for a relatively long period of time (72 h) compared to FUS approaches (5 h) thus making ALA-PDT potentially useful for some neuro-oncology applications.
ETXp-PCI is the newest and least understood of the BBB opening techniques reviewed in this work. In spite of its underlying complexity, the approach has a number of appealing characteristics that may make it a useful tool for localized drug delivery in the brain. ETXp-PCI can induce opening of the BBB at lower light fluences than PDT thus reducing the risk of permanent damage to the BBB and the brain. Since the BBB remains open for extended time periods (up to 14 days), the PCI technique would appear to be the most useful of the three approaches for treatments requiring extended delivery of therapeutic agents. Unlike PDT and FUS, ETXp-PCI has demonstrated its clinical utility in an infiltrative glioma model. These results show that the technique causes sufficient opening of the BBB to allow the passage of a high molecular weight chemotherapeutic agent into glioma infiltrated brain. Due to ETX or ETXp toxicity, it is doubtful that ETXp-based PCI approaches can be used in human clinical protocols. Nevertheless, the findings provide the basis for further PCI studies using non-toxic vasoactive compounds such as bradykinin and its analogs.
A common unifying thread linking the therapeutic approaches considered in this review is the relative lack of understanding of the BBB disruption mechanisms. A rudimentary understanding of FUS-induced effects at the cellular level exists, however, further studies are required for a more complete elucidation. Additional studies are also necessary to address the limitations posed by the human skull: it's not clear whether the present FUS techniques developed in small animals can be translated to humans. Few studies have addressed the mechanisms of the effects of PDT on the BBB, and to our knowledge, there have been no mechanistic studies of PCI-induced BBB opening. In the case of PCI, this will have to be addressed in order for it to progress from a curiosity to a serious treatment option.
Biographies

Steen J. Madsen received his Ph.D. in Applied Nuclear Physics from McMaster University in 1993. He completed a Postdoctoral Fellowship at the Beckman Laser Institute, University of California, Irvine in 1995. From 1995–1997 he was a Medical Physics Resident at the Mallinckrodt Institute of Radiology, Washington University Medical School in St. Louis. He is currently an Associate Professor of Health Physics and Diagnostic Sciences at the University of Nevada, Las Vegas. His research is focused on the use of laser-based approaches for the treatment of brain tumors.

Henry Hirschberg received his M.D. and Ph.D. degrees (specializing in Immunology) from the University in Oslo. He received his board certification in medicine and neurosurgery in Norway in 1978 and 1984, respectively. From 1990–2006 Dr. Hirschberg served as Consultant in Neurosurgery and Chief of Image Guided and Stereotactic Surgery and Neurosurgical Oncology in the Department of Neurosurgery at the University Hospital Rikhospitalet, Oslo, Norway. He is currently research professor at the Beckman Laser Institute UCI and adjunct professor at the University of Nevada, Las Vegas.
References
- 1.Teichberg VI. P Natl Acad Sci USA. 2007;104:7315–7316. doi: 10.1073/pnas.0702450104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardridge WM. Drug Discov Today. 2007;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Pardridge WM. Brain drug targeting: the future of brain drug development. Cambridge University Press; Cambridge: 2001. p. 111. [Google Scholar]
- 4.Ghose AK, Viswanadhan VN, Wendoloski JJ. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 5.Wallner KE, Galicich JH, Krol G, Arbit E, Malkin MG. Int J Radiat Oncol Biol Phys. 1989;16:1405–1409. doi: 10.1016/0360-3016(89)90941-3. [DOI] [PubMed] [Google Scholar]
- 6.Kroll RA, Neuwelt EA. Neurosurgery. 1998;42:1083–1099. doi: 10.1097/00006123-199805000-00082. [DOI] [PubMed] [Google Scholar]
- 7.Fenstermacher J, Kaye K. Ann NY Acad Sci. 1988;531:29–39. doi: 10.1111/j.1749-6632.1988.tb31809.x. [DOI] [PubMed] [Google Scholar]
- 8.Nagaraja TN, Patel P, Gorski M, Gorevic PD, Patlak CS, Fenstermacher JD. Cerebrospinal Fluid Res. 2005;2:5. doi: 10.1186/1743-8454-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Clin Cancer Res. 2000;6:2157–2165. [PubMed] [Google Scholar]
- 10.Sampson JH, Akabani G, Archer GE, Bigner DD, Berger MS, Friedman AH, Friedman HS, Herndon JE, Kunwar S, Marcus S, McLendon RE, Paolino A, Penne K, Provenzale J, Quinn J, Reardon DA, Rich J, Stenzel T, Tourt-Uhlig S, Wikstrand C, Wong T, Williams R, Yuan F, Zalutsky MR, Pastan I. J Neurooncol. 2003;65:27–35. doi: 10.1023/a:1026290315809. [DOI] [PubMed] [Google Scholar]
- 11.Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black KL, Sisti M, Brem S, Mohr G, Muller P, Morawetz R, Schold SC. Lancet. 1995;345:1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 12.Lawson HC, Sampath P, Bohan E, Park MC, Hussain N, Olivi A, Weingart J, Kleinberg L, Brem H. J Neurooncol. 2007;83:61–70. doi: 10.1007/s11060-006-9303-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. P Natl Acad Sci USA. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman DM, Laske DW, Morrison PF, Bankiewicz KS, Oldfield EH. J Neurosurg. 1995;82:1021–1029. doi: 10.3171/jns.1995.82.6.1021. [DOI] [PubMed] [Google Scholar]
- 15.Lidar Z, Mardor Y, Jonas T, Pfeffer R, Faibel M, Hadani M, Ram Z. J Neurosurg. 2004;100:472–479. doi: 10.3171/jns.2004.100.3.0472. [DOI] [PubMed] [Google Scholar]
- 16.Graff CL, Pollack GM. J Pharm Sci. 2005;94:1187–1195. doi: 10.1002/jps.20318. [DOI] [PubMed] [Google Scholar]
- 17.Pardridge WM. Discovery Med. 2006;6:139–143. [PubMed] [Google Scholar]
- 18.Vykhodtseva N, McDannold N, Hynynen K. Ultrasonics. 2008;48:279–296. doi: 10.1016/j.ultras.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirschberg H, Uzal FA, Chighvinadze D, Zhang MJ, Peng Q, Madsen SJ. Laser Surg Med. 2008;40:535–542. doi: 10.1002/lsm.20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirschberg H, Zhang MJ, Gach HM, Uzal FA, Peng Q, Sun CH, Chighvinadze D, Madsen SJ. J Neurooncol. 2009;95:317–329. doi: 10.1007/s11060-009-9930-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy LJ, Hachey DL, Oates JA, Morrow JD, Brown NJ. JPET. 2000;294:263–269. [PubMed] [Google Scholar]
- 22.Ehrlich P. Das Sauerstoff-Bedurfnis des Organismus: Eine Farbenanalytische studie. Hirschwald, Berlin: 1885. p. 85. [Google Scholar]
- 23.Begley DJ. Pharmacol Ther. 2004;104:29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Keep RG, Jones HC. Brain Res Dev Brain Res. 1990;56:47–53. doi: 10.1016/0165-3806(90)90163-s. [DOI] [PubMed] [Google Scholar]
- 25.deBoer AG, Gaillard PJ. Annu Rev Pharmacol Toxicol. 2007;47:323–355. doi: 10.1146/annurev.pharmtox.47.120505.105237. [DOI] [PubMed] [Google Scholar]
- 26.Pardridge W. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballabh P, Braun A, Nedergaard M. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Huber J, Egleton R, Davis T. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 29.Sheikov N, McDannold N, Sharma S, Hynynen K. Ultrasound Med Biol. 2008;34:1093–1104. doi: 10.1016/j.ultrasmedbio.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Mariscal L, Chavez de Ramirez B, Avila-Flores A. Cell Dev Biol. 2000;11:315–324. doi: 10.1006/scdb.2000.0178. [DOI] [PubMed] [Google Scholar]
- 31.McDannold N, Vykhodtseva N, Raymond S, Jolesz FA, Hynynen K. Ultrasound Med Biol. 2005;31:1527–1537. doi: 10.1016/j.ultrasmedbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Biochem Biophys Res Commun. 2006;340:1085–1090. doi: 10.1016/j.bbrc.2005.12.112. [DOI] [PubMed] [Google Scholar]
- 33.Bakay L, Hueter TF, Ballantine HT, Sosa D. Arch Neurol. 1956;76:457–467. doi: 10.1001/archneurpsyc.1956.02330290001001. [DOI] [PubMed] [Google Scholar]
- 34.Ballantine HT, Bell E, Manlapaz J. J Neurosurg. 1960;17:858–876. doi: 10.3171/jns.1960.17.5.0858. [DOI] [PubMed] [Google Scholar]
- 35.Vykhodtseva N. Proceedings of the 5th International Symposium on Ultrasound in Biology and Medicine; Puschino, Russia. 1981. pp. 95–97. [Google Scholar]
- 36.Patrick JT, Nolting MN, Goss SA, Dines KA, Clendenon JL, Rea MA, Heimburger RF. Adv Exp Med Biol. 1990;267:369–381. doi: 10.1007/978-1-4684-5766-7_36. [DOI] [PubMed] [Google Scholar]
- 37.Vykhodtseva NI, Hynynen K, Damianou C. Ultrasound Med Biol. 1995;21:969–979. doi: 10.1016/0301-5629(95)00038-s. [DOI] [PubMed] [Google Scholar]
- 38.Mesiwala AH, Mourad PD. Radiology. 2002;224:294–296. doi: 10.1148/radiol.2241011774. [DOI] [PubMed] [Google Scholar]
- 39.McDannold N, Vykhodtseva N, Jolesz FA, Hynynen K. Magn Reson Med. 2004;51:913–923. doi: 10.1002/mrm.20060. [DOI] [PubMed] [Google Scholar]
- 40.Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Radiology. 2001;220:640–646. doi: 10.1148/radiol.2202001804. [DOI] [PubMed] [Google Scholar]
- 41.Hynynen K, McDannold N, Sheikov NA, Jolesz FA, Vykhodtseva N. Neuroimage. 2005;24:12–20. doi: 10.1016/j.neuroimage.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 42.Choi JJ, Pernot M, Small SA, Konofagou EE. Ultrasound Med Biol. 2007;33:95–104. doi: 10.1016/j.ultrasmedbio.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 43.Vykhodtseva N, McDannold N, Hynynen K. Ultrasonics. 2008;48:279–296. doi: 10.1016/j.ultras.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDannold N, Vykhodtseva N, Hynynen K. Ultrasound Med Biol. 2008;34:834–840. doi: 10.1016/j.ultrasmedbio.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDannold N, Vykhodtseva N, Hynynen K. Ultrasound Med Biol. 2008;34:930–937. doi: 10.1016/j.ultrasmedbio.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apfel RE, Holland CK. Ultrasound Med Biol. 2001;17:179–185. doi: 10.1016/0301-5629(91)90125-g. [DOI] [PubMed] [Google Scholar]
- 47.Frizzell LA. In: Ultrasound: its Chemical Physical and Biological Effects. Suslick KS, editor. VCH; New York: 1998. pp. 287–303. [Google Scholar]
- 48.Sheikov N, McDannold N, Vykhodtseva N, Jolesz F, Hynynen K. Ultrasound Med Biol. 2004;30:979–989. doi: 10.1016/j.ultrasmedbio.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 49.Sheikov N, McDannold N, Jolesz F, Zhang YZ, Tam K, Hynynen K. Ultrasound Med Biol. 2006;32:1399–1409. doi: 10.1016/j.ultrasmedbio.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 50.Kinoshita M, McDannold N, Jolesz FA, Hynynen K. P Natl Acad Sci USA. 2006;103:11719–11723. doi: 10.1073/pnas.0604318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treat LH, McDannold N, Vykhodtseva N, Zhang Y, Tam K, Hynynen K. Int J Cancer. 2007;121:901–907. doi: 10.1002/ijc.22732. [DOI] [PubMed] [Google Scholar]
- 52.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. J Nat Can Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng MS, McKean J, Boisvert D. Surg Neurol. 1986;25:423–435. doi: 10.1016/0090-3019(86)90080-7. [DOI] [PubMed] [Google Scholar]
- 54.Muller PJ, Wilson BC, Lilge LD, Yang V, Hetzel FW, Chen Q, Selker R, Abrams J. Proc SPIE. 2001;4248:34–41. [Google Scholar]
- 55.Eljamel MS, Goodman C, Moseley H. Laser Med Sci. 2008;23:361–367. doi: 10.1007/s10103-007-0494-2. [DOI] [PubMed] [Google Scholar]
- 56.Madsen SJ, Angell-Petersen E, Spetalen S, Carper SW, Ziegler SA, Hirschberg H. Laser Surg Med. 2006;38:540–548. doi: 10.1002/lsm.20274. [DOI] [PubMed] [Google Scholar]
- 57.Kostron H, Fiegele T, Akatuna E. Med Laser App. 2006;21:285–290. [Google Scholar]
- 58.Lilge L, Wilson BC. J Clin Laser Med Surg. 1998;16:81–92. doi: 10.1089/clm.1998.16.81. [DOI] [PubMed] [Google Scholar]
- 59.Peng Q, Warloe T, Berg K, Moan J, Kongshaug M, Giercksky KE, Nesland JM. Cancer. 1997;79:2282–2308. doi: 10.1002/(sici)1097-0142(19970615)79:12<2282::aid-cncr2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 60.Madsen SJ, Sun CH, Tromberg BJ, Hirschberg H. J Neurooncol. 2003;62:243–250. doi: 10.1023/a:1023362011705. [DOI] [PubMed] [Google Scholar]
- 61.Hirschberg H, Angell-Petersen E, Peng Q, Tromberg B, Sun CH, Madsen SJ. J Environ Pathol Toxicol Oncol. 2006;25:261–280. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i1-2.170. [DOI] [PubMed] [Google Scholar]
- 62.Hirschberg H, Angell-Petersen E, Peng Q, Madsen SJ, Sioud M, Sørensen D. Proc SPIE. 2004;5312:405–414. [Google Scholar]
- 63.Angell-Petersen E, Sørensen DR, Madsen SJ, Hirschberg H. Proc SPIE. 2004;5312:415–423. [Google Scholar]
- 64.Stummer W, Novotny A, Stepp H, Goetz C, Bise K, Reulen HJ. J Neurosurg. 2000;93:1003–1013. doi: 10.3171/jns.2000.93.6.1003. [DOI] [PubMed] [Google Scholar]
- 65.Angell-Petersen E, Madsen SJ, Spetalen S, Sun CH, Peng Q, Carper SW, Hirschberg H. J Neurosurg. 2006;104:109–117. doi: 10.3171/jns.2006.104.1.109. [DOI] [PubMed] [Google Scholar]
- 66.Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Lancet Oncol. 2006;7:392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 67.Stummer W, Goetz C, Hassan A, Heimann DVM, Kempski O. Neurosurgery. 1993;33:1075–1082. doi: 10.1227/00006123-199312000-00016. [DOI] [PubMed] [Google Scholar]
- 68.Hebeda KM, Saarnak AE, Olivo M. Acta Neurochir. 1998;140:503–513. doi: 10.1007/s007010050132. [DOI] [PubMed] [Google Scholar]
- 69.Hirschberg H, Mathews MS, Angell-Petersen E, Spetalen S, Madsen SJ. Proc SPIE. 2007;6424:64242B1–B8. [Google Scholar]
- 70.Itol S, Rachinger W, Stepp H, Reulen HJ, Stummer W. Acta Neurochir. 2005;147:57–65. doi: 10.1007/s00701-004-0422-1. [DOI] [PubMed] [Google Scholar]
- 71.Angell-Petersen E, Hirschberg H, Madsen SJ. J Biomed Opt. 2007;12:0140031–9. doi: 10.1117/1.2709882. [DOI] [PubMed] [Google Scholar]
- 72.Sporn LA, Foster TH. Cancer Res. 1992;52:3443–3448. [PubMed] [Google Scholar]
- 73.Fingar VH. J Clin Laser Med Surg. 1996;14:323–328. doi: 10.1089/clm.1996.14.323. [DOI] [PubMed] [Google Scholar]
- 74.Hu SS, Cheng HB, Zheng YR, Zhang RY, Yue W, Zhang H. Biomed Environ Sci. 2007;20:269–273. [PubMed] [Google Scholar]
- 75.Berg K, Selbo PK, Prasmickaite L, Tjelle TE, Sandvig K, Moan J, Gaudernack G, Fodstad Ø, Kjølsrud S, Anholt H, Rodal GH, Rodal SK, Høgset A. Cancer Res. 1999;59:1180–1183. [PubMed] [Google Scholar]
- 76.Dietze A, Peng Q, Selbo PK, Kaalhus O, Muller C, Bown S, Berg K. Br J Cancer. 2005;92:2004–2009. doi: 10.1038/sj.bjc.6602600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Selbo PK, Kaalhus O, Sivam G, Berg K. Photochem Photobiol. 2001;74:303–310. doi: 10.1562/0031-8655(2001)074<0303:aabpio>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 78.Selbo PK, Sivam G, Fodstad Ø, Sandvig K, Berg K. Int J Cancer. 2000;87:853–859. doi: 10.1002/1097-0215(20000915)87:6<853::aid-ijc15>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 79.Prasmickaite L, Høgset A, Selbo P, Engesæter B, Hellum M, Berg K. Br J Cancer. 2002;86:652–657. doi: 10.1038/sj.bjc.6600138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Worthington R, Mulders M. Onderstepoort J Vet Res. 1975;42:25–28. [PubMed] [Google Scholar]
- 81.Nagahama M, Sakurai J. Toxicon. 1991;29:211–217. doi: 10.1016/0041-0101(91)90105-z. [DOI] [PubMed] [Google Scholar]
- 82.Dorca-Arevalo J, Soler-Jover A, Gibert M, Popoff M, Martin-Satue M, Blasi J. Vet Microbiol. 2008;131:14–25. doi: 10.1016/j.vetmic.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 83.Zhu C, Ghabriel M, Blumbergs P, Reilly P, Manavis J, Youssef J, Hatami S, Finnie J. Exp Neurol. 2001;169:72–82. doi: 10.1006/exnr.2001.7652. [DOI] [PubMed] [Google Scholar]
- 84.Barth RF. J Neurooncol. 1998;36:91–102. doi: 10.1023/a:1005805203044. [DOI] [PubMed] [Google Scholar]
- 85.Madsen SJ, Sun CH, Tromberg BJ, Hirschberg H. Laser Surg Med. 2001;29:406–412. doi: 10.1002/lsm.10005. [DOI] [PubMed] [Google Scholar]
- 86.Madsen SJ, Svaasand LO, Tromberg BJ, Hirschberg H. Proc SPIE. 2001;4257:41–49. [Google Scholar]