

Abstract
Objectives
The purpose of this study was to test the hypothesis that SCN10A variants contribute to the development of Brugada syndrome (BrS).
Background
BrS is an inherited sudden cardiac death syndrome. Fewer than 35% of BrS probands have genetically identified pathogenic variants. Recent evidence has implicated SCN10A, a neuronal sodium channel gene encoding Nav1.8 in the electrical function of the heart.
Methods
Clinical analysis and direct sequencing of BrS-susceptibility genes were performed on 150 probands, family members and >200 healthy controls. Expression and co-immunoprecipitation studies were performed to functionally characterize the putative pathogenic mutations.
Results
We identified 17 SCN10A mutations in 25 probands (20 M/5 F); 23 of the 25 (92.0%) displayed overlapping phenotypes. SCN10A mutations were found in 16.7% of BrS probands, approaching our yield for SCN5A mutations (20.1%). BrS patients with SCN10A mutations were more symptomatic and displayed significantly longer PR and QRS intervals than SCN10A negative BrS probands. The majority of mutations localized to the transmembrane-spanning regions. Heterologous co-expression of wild-type (WT) SCN10A with WT-SCN5A in HEK cells caused a near doubling of sodium channel current (INa) compared with WT-SCN5A alone. In contrast, co-expression of SCN10A mutants (R14L and R1268Q) with WT-SCN5A caused a 79.4% and 84.4% reduction in INa, respectively. Co-immunoprecipitation studies performed provide evidence for co-association of Nav1.8 and Nav1.5 in the plasma membrane.
Conclusions
Our study identifies SCN10A as a major susceptibility gene for BrS, thus greatly enhancing our ability to genotype and risk stratify probands and family members.
Keywords: Electrophysiology, Cardiac Arrhythmias, Brugada syndrome, Cardiac Conduction disease, Sudden Cardiac Death, Genetics
Introduction
The Brugada syndrome (BrS), introduced as a new clinical entity in 1992 (1), is an inherited sudden cardiac death (SCD) syndrome characterized by the appearance of prominent J waves or ST-segment elevation in leads V1-V3of the electrocardiogram (ECG). An outward shift in the balance of ion channel currents flowing during the early phases of the cardiac action potential have been shown to create the substrate for the development of life-threatening arrhythmias in BrS (2). The syndrome has been associated with 13 genotypes (BrS1 to BrS13) displaying autosomal dominant inheritance (3,4). To date, more than 300 BrS-related mutations in SCN5A have been described (5), accounting for the vast majority (>75%) of BrS genotypepositive cases, but only 11-28% of total BrS probands. Approximately 65% of BrS probands remain genetically undetermined. Thus, there is a pressing need to identify new BrS susceptibility genes for the purpose of early diagnosis, risk stratification, and targeted treatments (6,7). A similar situation is encountered in other inherited cardiac arrhythmia syndromes, including early repolarization syndrome (ERS), cardiac conduction disease (CCD), bradycardia, idiopathic ventricular fibrillation (VF), atrial fibrillation (AF), and right bundle branch block (RBBB).
Nav1.8 (encoded by SCN10A), like Nav1.5 (encoded by SCN5A), is a tetrodotoxinresistant voltage-gated sodium channel located adjacent to SCN5A on human chromosome 3p21–22 (8,9). Until recently, Nav1.8 was principally considered a neuronal sodium channel involved in nociception. The amino acid sequences of human Nav1.8 and Nav1.5 are similar (70.4%). Recent evidence has implicated SCN10A in the electrical function of the heart (10-12). Several genome-wide association studies (GWAS) have reported that single nucleotide polymorphisms in SCN10A are associated with CCD and arrhythmogenesis (13-21). The present study examines the hypothesis that variations in SCN10A contribute to BrS by modulating the expression of Nav1.5 current, the principal cardiac sodium channel. Preliminary results have been reported in abstract form (22).
Methods
Detailed methods are provided in the online supplement.
Clinical analysis and participants
The clinical diagnosis of BrS and ERS was based on criteria provided in the 2005 Consensus Conference document (23) in the case of BrS and criteria suggested in our recent review of the J-wave syndromes in the case of ERS (24).Informed consent was obtained from all patients upon referral to the Masonic Medical Research Laboratory for genetic testing, and patients were tracked anonymously. This study was approved by the regional institutional ethics review board and conducted according to Declaration of Helsinki principles. For each patient, we collected age at time of diagnosis, gender, clinical presentation, family history, and therapy.
Genetic screening and analysis
Genomic DNA was extracted from peripheral blood leukocytes and amplified. All known BrS genes and SCN10A were amplified and analyzed by direct sequencing, as previously described (25). The primer sequences for SCN10A are shown in Table S1 (Reference Sequence: NM_006514). More than 200 ethnically matched, healthy controls, plus all available online databases for allele frequency, conservation score, and in silico pathogenic prediction tools, were probed for prediction of pathogenicity of the variants found.
Co-expression of NaV1.5 and NaV1.8 for co-immunoprecipitation (Co-IP) analysis and electrophysiological investigations
Site-directed mutagenesis was performed on full-length human wild-type (WT) and mutant SCN10A-3XFLAG cDNA cloned in pCMV2 vector, the WT SCN3B cloned in pCMV6-XL6 vector, and the WT SCN5A cloned in pcDNA3.1. Co-immunoprecipitation studies were performed using HEK293 cells transfected with SCN5A, SCN10A and SCN3B plasmids were also used for studies. Total protein was isolated 24 hours after transfection with Lysis buffer supplemented with protease inhibitors for Co-IP experiment. Membrane currents were measured using whole-cell patch-clamp techniques using TSA201 cells, as previously described (25).
Statistical analysis
Data are presented as mean±SD, unless otherwise noted. For statistical analysis, two-tailed Student's t-test and ANOVA coupled with Student-Newman-Keuls test, were used to compare two groups and more than three groups of continuous variables separately. Chi-square test was used for compare of categorical variables (SigmaStat, Systat Scientific Inc., San Jose, CA). Differences were considered statistically significant at a value of P<0.05.
Results
Study population
We systematically evaluated 150 unrelated BrS patients and 17 family members using genetic screening (Table 1). Most patients were male (n=101, 67.3%) with a mean age at diagnosis of 44.5±16.1 years. One hundred and sixteen patients (77.3%) were symptomatic, including 39 (26.0%) who suffered from syncope and 20 (13.3%) who experienced cardiac events, documented as aborted cardiac arrest or SCD. Twenty-nine (19.3%) had a family history of cardiac events or SCD. A Type 1 Brugada ECG pattern, characterized by a prominent J-wave appearing as a coved type ST-segment elevation, was observed spontaneously in 57 patients (38.0%, Figure 1A), appeared after sodium channel blockers in 76 patients (50.7%, Figure 1B) or during fever in the remaining 17 patients (11.3%, Fig. 1C). Some BrS patients also displayed ERS (Fig. 1D), CCD (Fig. 1E), RBBB (Fig. 1F), ventricular tachycardia/ventricular fibrillation (VT/VF), or AF.
Table 1. Demographics of Patients with Brugada Syndrome (BrS).
| Probands with BrS | Probands with BrS and CCD | |||||
|---|---|---|---|---|---|---|
| Overall | SCN10A+ | SCN10A- | Overall | SCN10A+ | SCN10A- | |
| Patient Demographics | ||||||
| Number of probands | 150 | 25 (Yield 16.67%) | 125 | 36 | 12 (Yield 33.33%) | 24 |
| Family history | 29 (19.33%) | 4 (16.00%) | 25 (20.00%) | 8 (22.22 %) | 3 (25.00%) | 5 (20.83%) |
| Age for diagnosis | 44.48±16.10 | 42.50±13.56 | 44.83±16.61 | 44.75±19.92 | 43.25±15.15 | 45.50±22.18 |
| Male | 101 (67.33%) | 20 (80.00%) | 81 (64.80%) | 27 (75.00%) | 9 (75.00%) | 18 (75.00%) |
| Female | 49 (32.67%) | 5 (20.00%) | 44 (35.20 %) | 9 (25.00%) | 3 (25.00%) | 6 (25.00 %) |
| with CCD | 36 (24.00%) | 12 (48.00%) | 24 (19.20%) | 36 (100.00%) | 12 (100.00%) | 24 (100.00%) |
| ER† | 24 (16.00%) | 8 (32.00%) | 16 (12.80%) | 8 (22.22%) | 3 (25.00%) | 5 (20.83%) |
| bradycardia | 24 (16.00%) | 5 (20.00%) | 19 (15.20%) | 10 (27.78%) | 3 (25.00%) | 7 (29.17%) |
| VT/VF | 33 (22.00%) | 12 (48.00%) | 21 (16.80%) | 12 (33.33%) | 7 (58.33%) | 5 (20.83%) |
| AF | 13 (8.67%) | 2 (8.00%) | 11 (8.80%) | 1 (2.78%) | 0 (0.00%) | 1 (4.17%) |
| Symptom | ||||||
| Asymptomatic | 34 (22.67%) | 1 (4.00%) | 33 (26.40%) | 9 (25.00%) | 1 (8.33%) | 8 (33.33%) |
| Syncope | 39 (26.00%) | 10 (40.00%) | 29 (23.20%) | 10 (27.78%) | 4 (33.33%) | 6 (25.00%) |
| SCD | 20 (13.33%) | 6 (24.00%) | 14 (11.20%) | 3 (16.67%) | 3 (25.00%) | 3 (12.50%) |
| chest pain | 11 (7.33%) | 5 (20.00%) | 6 (4.80%) | 3 (8.33%) | 3 (25.00%) | 0 (0.00%) |
| other symptoms‡ | 54 (36.00%) | 12 (48.00%) | 42 (33.60%) | 11 (33.33%) | 4 (33.33%) | 8 (33.33%) |
| Electrocardiograms | ||||||
| HR (bpm) | 73.82±16.58 | 72.58±16.35 | 74.10±16.69 | 68.07±17.77 | 72.00±21.39 | 65.11±15.31 |
| PR (ms) | 175.97 ± 38.23 | 193.44±31.79* | 171.47±38.42 | 218.29±34.59 | 217.42±24.20 | 218.77±39.66 |
| QRS (ms) | 98.84±17.82 | 105.72±18.85* | 97.26±17.28 | 103.47±19.34 | 107.67±24.01 | 101.38±16.72 |
| QT (ms) | 378.94±42.43 | 384.44±38.67 | 377.70±43.30 | 398.28±44.45 | 388.67±48.18 | 403.08±42.71 |
| QTc (ms) | 414.40±36.22 | 416.05±37.42 | 414.04±36.11 | 416.29±42.24 | 420.40±49.82 | 414.23±38.92 |
P<0.05 compared between SCN10A+ and SCN10A-groups.
Early repolarization (ER) pattern other than V1-V3
Other symptoms include palpitation, dizziness, sleep apnea, coma, et al.
AF: atrial fibrillation; CCD: cardiac conduction disease; HR: heart rate; SCD: sudden cardiac death; VT/VF: ventricular tachycardia/ventricular fibrillation.
Figure 1. Representative Cases of the Different Brugada Syndrome (BrS) Phenotypes Associated with the SCN10A Mutations/Rare Variants Identified.
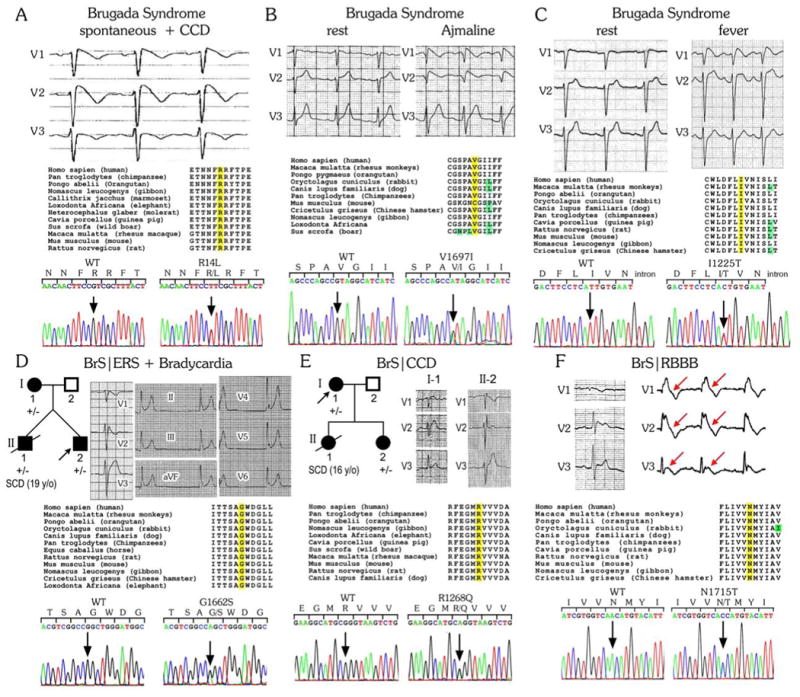
Each panel shows the ECG phenotype, amino acid alignments of the mutated residue position in a number of mammalian species, and DNA chromatogram of wild-type (WT) and mutant SCN10A. For the pedigrees in panels D&E, +/- denotes heterozygous for the mutation; circles represent female subjects and squares represent male subjects. The arrow denotes the proband. Clinically affected and unaffected subjects are labeled as black and white, respectively. CCD: cardiac conduction disease; ERS= early repolarization syndrome; RBBB= right bundle branch block; SCD= sudden cardiac disease.
Mutation yield and analysis
Overall, 17 putative pathogenic SCN10A rare variants [16 missense and 1 frameshift mutation] were identified in 25 probands (Fig. 2A, Tables 2 and 3). Seven family members were positive for SCN10A variants. Eleven mutations were identified only once (64.7%), while 6 variants were found in multiple unrelated patients (Fig. 2B). The most frequent mutation was R14L (Fig. 1A), which was carried by 4 BrS probands. The other mutations/rare polymorphisms present in the population were V1697I (3 patients), G1662S (3 patients), I206M (2 patients), I1225T (2 patients), and R1869C (2 patients). Most variants localized to the transmembrane-spanning regions (P-loop 42.3%, S1-S4 23.1% for BrS probands, Fig. 2C).
Figure 2. Clinical and Genetic Prevalence of SCN10A Mutations/Rare Variants in probands with Brugada Syndrome (BrS) Identified in the Present Study.
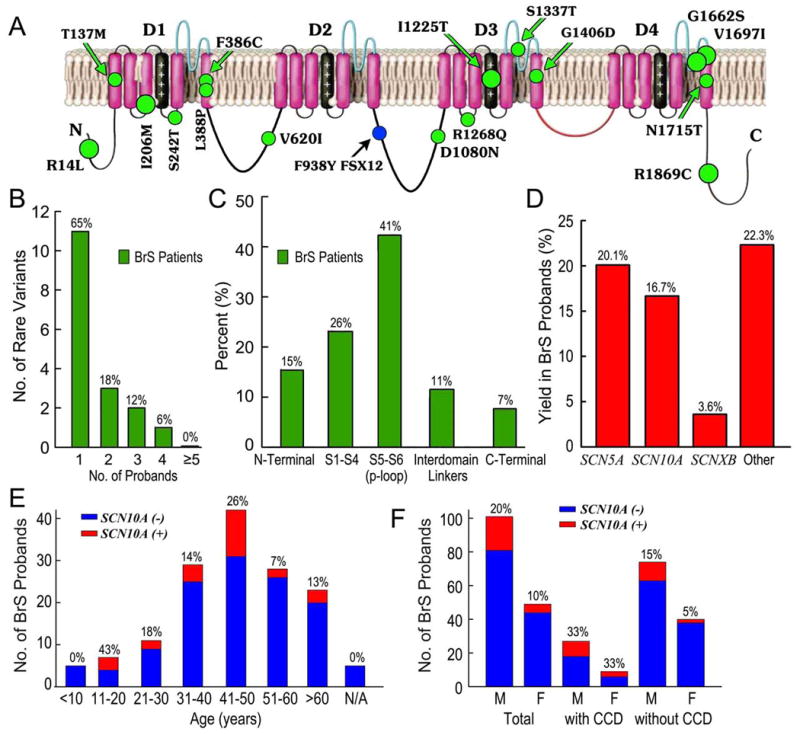
A: Schematic showing topology of Nav1.8, the pore-forming α subunit encoded by SCN10A and location of putative BrS-causing variants. B: Frequency distribution of SCN10A mutations/rare variants in BrS cases (green). C: Percentage of mutations/rare variants in BrS cases (green) by location. D: Mutation detection yield by gene in Masonic Medical Research Laboratory BrS cases. E&F: Bar graph showing age and gender distribution of BrS cases. CCD= cardiac conduction disease.
Table 2. Clinical and Genetic Characteristics of Affected Probands.
| No. | General Information |
Symptom | Arrhythmia | Genetic Rare Variants |
ECG | Treatme nt |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age for Dx (y/o ) |
Sex | FH* | Sync ope |
SCD | Ch est pai n |
Other† | BrS/ERS | ER Pattern other than V1-V3 |
CCD | Brady cardia |
VT/VF | AF |
SCN10 A |
Othe r suspe cted gene |
H R (b pm ) |
PR (m s) |
Q RS (m s) |
Q T (m s) |
Q Tc (m s) |
||
| 1 | 65 | M | N | N | N | N | Y | BrS (Spontaneous BrS/RBBB) | N | Y (IAVB) | N | Y (induced) | N | N1715T | N | 71 | 220 | 72 | 426 | 463 | ICD |
| 2 | 50 | F | Y | N | N | Y | Y | BrS (Spontaneous) | I, avL | Y (IAVB) | N | N | N | R14L | N | 73 | 234 | 112 | 402 | 443 | |
| 3 | 42 | F | N | Y | N | Y | Y | BrS (Spontaneous) | N | N | N | Y (induced) | N | F938YFSX12 | N | 68 | 160 | 120 | 400 | 426 | ICD |
| 4 | 44 | M | N | Y | N | N | N | BrS (Spontaneous) | N | N | N | Y (induced) | N | G1662S | N | 79 | 190 | 120 | 380 | 436 | Quinidine |
| 5 | 20 | M | N | Y | Y | N | N | BrS/ERS3 (Spontaneous/Procainamide+) | II, III, avF | N | N | N | N | S242T | N | 67 | 170 | 105 | 376 | 396 | |
| 6 | 19 | M | N | N | N | Y | N | BrS (Procainamide+) | N | N | N | Y (induced) | N | R14L | N | 66 | 185 | 113 | 397 | 416 | |
| 7 | 36 | M | N | N | N | N | Y | BrS (Ajmaline+) | N | N | N | Y (spontaneous) | N | G1406D | N | 73 | 150 | 110 | 400 | 442 | ICD |
| 8 | 19 | M | Y | Y | Y | Y | Y | BrS/ERS3 (Flecainide+) | II, III, avF, V4-V6 | Y (IAVB) | Y | Y (spontaneous) | N | G1662S | N | 44 | 200 | 120 | 410 | 352 | |
| 9 | 39 | M | N | Y | N | N | N | BrS(Ajmaline+) | I, avL | N | N | N | N | I206M/V1697I | N | 78 | 168 | 80 | 340 | 388 | |
| 10 | 46 | M | N | Y | N | N | Y | BrS (Procainamide+) | N | N | N | N | Y | S1337T | N | 69 | 148 | 105 | 418 | 448 | |
| 11 | 37 | M | N | N/A | N/A | N/A | N/A | BrS (Ajmaline+) | N | Y (I AVB) | N | N | N | T137M | N | 59 | 205 | 72 | 350 | 348 | |
| 12 | 49 | F | Y | Y | N | N | Y | BrS (Spontaneous) | N | Y (IAVB) | N | Y (spontaneous) | N | R1268Q | N | 79 | 170-200 | 102 | 380 | 436 | ICD |
| 13 | 44 | M | N/A | N | N | N | Y | BrS (fever+) | N | N | N | N | N | I1225T | N | 80 | 180 | 110 | 350 | 404 | |
| 14 | 27 | M | N | N | Y | N | N | BrS (fever+) | N | Y (IAVB) | N | Y (during fever) | N | G1662S | N | 120 | 220 | 120 | 300 | 387 | ICD, Metoprolol |
| 15 | 44 | M | N | Y | N | N | N | BrS (fever+) | N | Y (IAVB) | N | Y (during fever) | N | I1225T | N | 103 | 200 | 116 | 400 | 525 | ICD |
| 16 | 56 | M | N/A | N | N | N | Y | BrS (fever+) | N | N | N | N | N | L388P | N | 86 | 180 | 112 | 350 | 418 | |
| 17 | 31 | M | Y | N | N | N | N | BrS (Ajmaline+) | N | Y (IAVB) | N | Y (induced) | N | V1697I | N | 75 | 200 | 120 | 360 | 403 | |
| 18 | 65 | M | Y | Y | Y | N | N | BrS/ERS3 (Spontaneous) | II, III, avF | N | Y | Y (spontaneous) | N | R14L | N | 54 | 194 | 94 | 400 | 378 | ICD, Amiodarone |
| 19 | 49 | M | N/A | N | N | N | Y | BrS (fever+) | AvL | N | N | N | N | R14L | N | 88 | 130 | 110 | 340 | 411 | |
| 20 | 58 | M | N | Y | N | N | N | BrS (Ajmaline+) | N | Y (IAVB) | N | N | N | I206M/V1697I | CAC NB2B | 84 | 210 | 100 | 344 | 408 | ICD, Metoprolol |
| 21 | 46 | F | N | N | Y | N | N | BrS/ERS3 (Spontaneous) | I, II, III, avF, V5-V6 | Y (IAVB) | Y | N | N | V620I | SCN5A | 53 | 280 | 84 | 432 | 406 | ICD |
| 22 | 65 | M | N | N | Y | N | N | BrS (Flecainide+) | N | Y (IAVB) | Y | Y (spontaneous) | N | F386C | CACNA1C | 55 | 200 | 116 | 484 | 464 | ICD |
| 23 | 28 | M | N | N | N | Y | N | BrS (Spontaneous/Procainamide+) | N | Y (IAVB) | N | N | N | D1080N | SCN5A | 71 | 240 | 158 | 376 | 409 | ICD |
| 24 | 43 | M | N | N | Y | N | Y | BrS (Procainamide+) | N | N | N | N | Y | R1869C | KCNJ8 | 64 | 192 | 84 | 372 | 385 | |
| 25 | 47 | F | N | N | N | N | Y | BrS/ERS3 (Ajmaline+) | II, III, avF, V4-V6 | N | Y | N | N | R1869C | CACNB2B | 56 | 18 0 | 88 | 42 4 | 40 8 | |
| Total | 42.5 ± 13.6 | F5/M20(80%) | 5 (20%) | 10 (40%) | 7 (28%) | 5 (20%) | 12 (48%) | 25 (100%) | 8 (32%) | 12 (48%) | 5 (20%) | 12 (48%) | 2 (8%) | 25 | 72.6 ± 16.3 | 193.4 ± 31.8 | 105.7 ± 18.9 | 384.4 ± 38.67 | 416.1 ± 37.4 | ICD-11 (44.0%) | |
| SCN 10A† | 41.2 ± 13.7 | F3/M16(84%) | 5 (26%) | 9 (47%) | 4 (21%) | 4 (21%) | 10 (53%) | 19 (100%) | 6 (32%) | 8 (42%) | 2 (10%) | 11 (58%) | 1 (5%) | 19 | 75.4 ± 16.8 | 18 6.0 ± 26. 9 | 10 5.9 ± 15.6 | 37 7.8 ± 32.9 | 416.9 ± 40.9 | ICD-7 (36.4%) | |
| SCN 10A and other gene† | 47.8 ± 12.8 | F2/M4(67%) | 0 (0 %) | 1 (17%) | 3 (50%) | 1 (17%) | 2 (33%) | 6 (100%) | 2 (33%) | 4 (67%) | 3 (50%) | 1 (17%) | 1 (17%) | 6 | 63. 8 ± 12.0 | 21 7.0 ± 37.0 | 10 5.0 ± 28. 7 | 40 5.3 ± 51.0 | 413.3 ± 26.4 | ICD-4 (66.7%) | |
AF: atrial fibrillation; AVB: atrioventricular block; BrS: Brugada syndrome; CCD: cardiac conduction disease; Dx: diagnosis; ER: early repolarization; ERS3: Type 3 early repolarization syndrome; F: female; HR: heart rate; ICD: implantable cardioverter-defibrillator; M: male; RBBB: right bundle branch block; SCD: sudden cardiac death; VT/VF: ventricular tachycardia/ventricular fibrillation.
FH: Family history of cardiac events/unexplained sudden death.
Other symptoms, including palpitation, dizziness, coma, et al.
Table 3.
Summary of SCN10A Rare Variants Associated with Brugada Syndrome.
| Rare Variant | Reported ID | Exon | Type | Change in nucleotide | Change in amino acid | Codons | Location | Global MAF (1000genome) | Global MAF (ESP) | Number of Unrelated individuals | Number of Family members | SIFT-Score | SIFT-Prediction | Polyphen-Score | Polyphen-Prediction | Conservation (phast Cons)* | Conservation (GERP)† |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R14L | rs141207048 | 1 | missense | p.Arg14 Leu | c.41G>T | CGT/CTT | N-terminal | 0.002 | 0.002153 | 5 | 2 | 0 | deleterious | 0.998 | probably damaging | 0.987 | 2.32 |
| T137M | rs148663098 | 3 | missense | p.Thr137 Met | c.410C>T | ACG/ATG | DI-SI | 0 | 0.000077 | 1 | 0 | 1 | tolerated | 0.022 | benign | 0.077 | 3.26 |
| I206M | rs74717885 | 5 | missense | p.Ile206 Met | c.618A>G | ATA/ATG | DI-SIII | 0.048 | 0.01161 | 4 | 0 | 0.01 | deleterious | 0.082 | benign | 0.001 | -0.84 |
| S242T | rs140288103 | 6 | missense | p.Ser242 Thr | c.724T>A | TCA/ACA | DI-S4/S5 | 0 | 0.000231 | 1 | 0 | 0 | deleterious | 0.999 | probably damaging | 0.997 | 4.69 |
| F386C | rs78555408 | 9 | missense | p.Phe386 Cys | c.1157T>G | TTC/TGC | DI-S6 | 0.002 | N/A | 1 | 1 | 0 | deleterious | 1 | probably damaging | 1 | 5.21 |
| L388P | rs199734710 | 9 | missense | p.Leu388 Pro | c.1163T>C | CTG/CCG | DI-S6 | 0.001 | 0.000231 | 1 | 0 | 0 | deleterious | 1 | probably damaging | 1 | 5.21 |
| V620I | rs151303346 | 12 | missense | p.Val620 Ile | c.1858G>A | GTC/ATC | DI/DII | 0 | 0.000384 | 1 | 0 | 0.03 | deleterious | 0.999 | probably damaging | 0.818 | 4.6 |
| F938YFSX12 | N/A | 16 | Frame shift | p.Phe938 Tyr FsX12 | c.2813-2814 delinA | ‡ | DII/DIII | N/A | N/A | 1 | 1 | N/A | N/A | N/A | N/A | N/A | N/A |
| D1080N | TMPESP3 38765035 | 18 | missense | p.Asp1080 Asn | c.3238G>A | GAC/AAC | DII/DIII | 0 | 0.000077 | 1 | 0 | 0.17 | tolerated | 0.967 | probably damaging | 0.75 | 4.82 |
| I1225T | rs139638446 | 20 | missense | p.Ile1225 Thr | c.3674 T>C | ATT/ACT | DIII-S4 | 0 | 0.000461 | 2 | 0 | 0 | deleterious | 1 | probably damaging | 0.992 | 4.45 |
| R1268Q | rs138832868 | 21 | missense/near-splice | p.Arg1268 Gln | c.3803G>A | CGG/CAG | DIII-S4/S5 | 0.001 | 0.002076 | 1 | 1 | 0 | deleterious | 0.741 | probably damaging | 1 | 4.14 |
| S1337T | rs11711062 | 22 | missense | p.Ser1337 Thr | c.4009T>A | TCC/ACC | DIII-S5/S6 | 0.001 | 0.004536 | 1 | 0 | 0.61 | tolerated | 0.059 | benign | 0 | -9.34 |
| G1406D | N/A | 24 | missense | p.Gly1406 Asp | c.4217G>A | GGC/GAC | DIII-S6 | N/A | N/A | 1 | 0 | 0 | deleterious | 0.999 | probably damaging | 1 | 5.11 |
| G1662S | rs151090729 | 27 | missense | p.Gly1662 Ser | c.4984G>A | GGC/AGC | DIV-S5/S6 | 0.001 | 0.001307 | 3 | 2 | 0 | deleterious | 1 | probably damaging | 0.969 | 5.38 |
| V1697I | rs77804526 | 27 | missense | p.Val1697 Ile | c.5089G>A | GTA/ATA | DIV-S5/S6 | 0.004 | 0.010072 | 4 | 0 | 0.3 7 | tolerated | 0.098 | benign | 0.138 | 0.24 |
| N1715T | N/A | 27 | missense | p.Asn1715 Thr | c.5144A>C | AAC/ACC | DIV-S6 | N/A | N/A | 1 | 0 | 0 | deleterious | 1 | probably damaging | 1 | 4.19 |
| R1869C | rs141648641 | 27 | missense | p.Arg1869 Cys | c.5605C>T | CGC/TGC | C-terminal | 0 | 0.000923 | 2 | 0 | 0 | deleterious | 1 | probably damaging | 0.945 | 5.09 |
N/A: not available; 1000 genome: the 1000 Human Genome Project Database; ESP: Exome Sequencing Project; MAF: the Minor-Allele Frequency.
Conservation (phastCons): a number between 0 and 1 that describes the degree of sequence conservation among 17 vertebrate species; these numbers are downloaded from the University of California Santa Cruz Genome site (http://genome.ucsc.edu/).
Conservation (GERP): The Genomic Evolutionary Rate Profiling (GERP) score was obtained from the GERP website in September of 2011. It ranges from -12.3 to 6.17, with 6.17 being the most conserved.
T2813 & C2814 are replaced by an A, causing a frameshift resulting in a stop codon 12 AA later.
Among the 25 SCN10A mutation or rare variant carriers, 6 carried a secondary mutation in 1 of the 12 known BrS-susceptibility genes (24.0%, Table 2). F938YFSX12, G1406D and N1715T are novel variants in SCN10A, not previously reported (Table 3). A majority of missense mutations (13/16) were in highly conserved residues and showed minor allele frequencies (MAF) of 0 to 0.002 in control databases. None were found in more than 400 reference alleles in our healthy controls. All but 1 (T137M) of these 13 mutations were predicted to be damaging by in silico prediction tools (Table 3). The MAF of S1337T was 0.0047, that of V1697I was 0.0044, and that of I206M was 0.0046 in our controls. All 4 cases carrying these 3 rare polymorphisms were middle-aged males (31-58 y/o) and 3 were symptomatic.
Overlapping phenotypes of probands with SCN10A variants
With a positive proband yield of 16.7%, the prevalence of SCN10A in BrS probands is approaching our historical yield for SCN5A mutations, which is 20.1% (Fig. 2D). In 25 SCN10A+ BrS cases, 23 (92.0%) displayed overlapping phenotypes (Table 2). In cases of BrS with overlapping phenotypes (such as CCD and early repolarization/ER patterns in leads other than V1-V3), SCN10A+ positive proband yield was greater (Table 1). BrS patients with SCN10A mutations were more symptomatic (syncope, SCD, chest pain) and displayed longer PR and QRS intervals (193.4±31.8 ms and 105.7±18.9 ms) than SCN10A- BrS probands (171.5±38.4 ms and 97.3±17.3 ms, p<0.05 respectively). No difference in HR, QT, or Bazett corrected QT interval (QTc) was observed. The yield of SCN10A+ BrS probands was greater in male (19.8%) than in female (10.2%, Fig. 2F) subjects in general. This difference was not observed in the subgroup of BrS with CCD but was more obvious in BrS cases without CCD. Figure 2E shows yield as a function of age. The yield of probands with spontaneous Type 1 Brugada ECG pattern was 15.8%, which was similar to that in BrS cases unmasked with a sodium channel blocker (14.5%). Interestingly, BrS probands diagnosed during fever showed a much higher yield (5 out of 17; 29.4%) for SCN10A variants; all were male.
The average PR interval (PRI) for BrS probands with CCD was 218.3±34.59 ms (maxium PRI, 328 ms). The yield of SCN10A+ in this cohort was significantly higher (33.3%) than those without CCD (11.4%; P<0.01). Compared with SCN10A- subjects, SCN10A+ CCD and BrS cases had a higher incidence of VT/VF, SCD, and chest pain (Table 1).
Also, 24 BrS cases displayed an ER pattern in leads other than V1-V3. Seven of these probands and 2 family members were positive for SCN10A mutations, including 5 probands with global J-point/wave elevation (ERS3, 71.4%), indicating a higher correlation of SCN10A with BrS and ERS compared with BrS phenotype alone. In the case pictured in Fig. 1D, the proband presented with global J-point elevation (ERS/BrS), bradycardia, and a family history of SCD. He and his affected family members carried the same SCN10A-G1662S mutation. (Details in the Supplemental Materials).
Among 33 BrS patients presenting with VT/VF, SCN10A mutations were identified in 12. BrS appeared spontaneously in 5 cases (41.7%), 2 were unmasked during fever (16.7%) and the rest were unmasked using sodium channel blockers (41.7%). Including those with pediatric bradycardia, the average heart rate of 24 probands with bradycardia and BrS was 51.4±1.7 bpm. SCN10A mutations were identified in five cases. Four family members in 3 families also were positive for SCN10A mutations (G1662S for 2, R14L for 1, and F938Y FSX12 for 1), indicating clear genetic penetrance. SCN10A-S1337T and R1869C were found in 2 AF probands with BrS phenotypes. The SCN10A-N1715 mutant carrier presented with BrS and RBBB ECG pattern, an overlapping phenotype recently highlighted by Aizawa et al. (26). (Fig. 1F).
Functional expression studies
For functional characterization, SCN5A/WT, SCN10A/WT, or SCN5A/WT+SCN10A/WT were co-expressed with SCN3B/WT in HEK293 cells (Fig. 3A). Peak INaamplitude at -35mV was -462.8±83.2 pA/pF for SCN5A/WT+ SCN3B/WT. Addition of SCN10A/WT yielded a near doubling of peak INato -859.7±98.9 pA/pF (P<0.01). In contrast, co-expression of SCN10A/WT+SCN3B/WT alone generated very low amplitude current (-12.2±3.3 pA/pF, P<0.01 compared with the other 2 groups, Fig. 3B). Co-expression of the SCN10A mutants, R14L and R1268Q, with SCN5A/WT and SCN3B/WT caused a major loss of function of INa (Fig. 3C-I). SCN10A-R14L reduced peak INa density to -177.5±49.5 pA/pF (P<0.01 vs. SCN10A-WT) and caused a significant positive shift of half-activation voltage (V1/2, P<0.05). SCN10A-R1268Q reduced current density to -133.9±36.6 pA/pF (P<0.01 vs. SCN10A/WT) with no change in activation parameters. The half-inactivation voltage (V1/2) of SCN10A-R1268Q was 7.7 mV more negative than that of SCN10A-WT when co-expressed with SCN5A-WT+SCN3B-WT (P<0.05). Recovery from inactivation was similar in the two mutant groups, but both were slower than WT channels (P<0.05 respectively in both τfand τs). The gating defects caused by SCN10A-R14L and SCN10A-R1268Q served to reduce sodium channel availability. (Details in Table S2 of the Online Supplement).
Figure 3. Electrophysiology Effect of SCN10A on Cardiac Sodium Channel Current (INa) when Co-Expressed with SCN5A and SCN3B in HEK293 Cells.
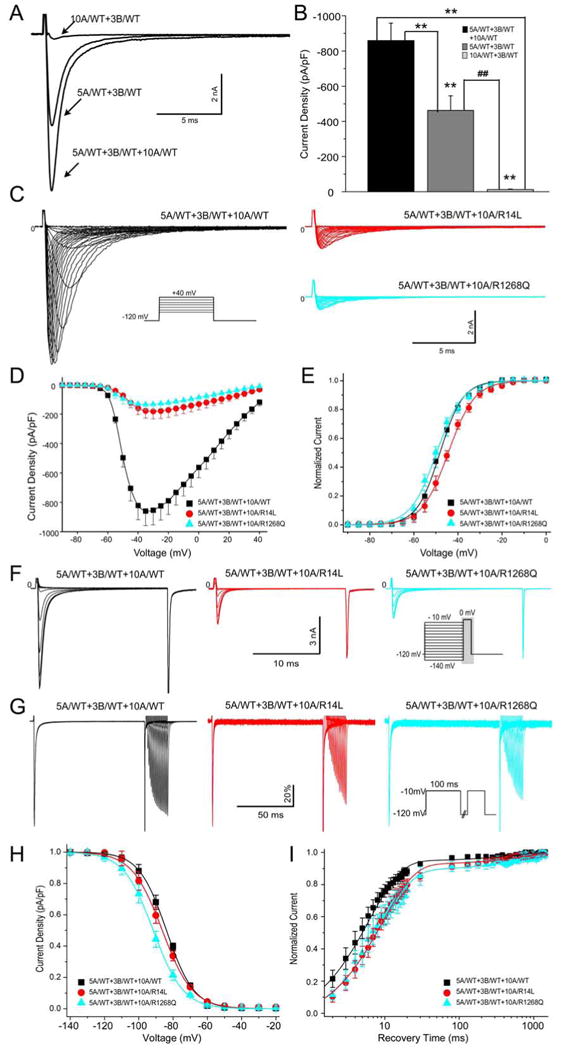
A&B: Superimposed traces and bar graph depicting peak INa recorded from co-expression of SCN10A/wild type (WT)+SCN3B/WT, SCN5A/WT+SCN3B/WT and SCN5A/WT+SCN10A/WT+SCN3B/WT. **P<0.01 vs. SCN5A/WT+SCN10A/WT+SCN3B/WT, ##P<0.01 vs. SCN5A/WT+SCN3B/WT. C-E: Representative INa traces, current-voltage relationship and voltage dependence of activation for SCN10A/WT, SCN10A/R14L and SCN10A/R1268Q when co-expressed with SCN5A/WT+SCN3B/WT. F&G: Representative steady-state inactivation and recovery traces recorded from WT and mutant channels. H&I: Boltzmann distributions of voltage-dependent channel inactivation and recovery curve with a double-exponential fit for the 3 groups. All related values and the number of cells used are presented in Table S2 in the Supplemental Materials.
Co-IP Study
We examined the capability of Nav1.5 to physically interact with Nav1.8 using Co-IP. The channels were expressed in HEK293 cells either alone or in combination and isolated by pull-down using an antibody to the FLAG on SCN10A. Figure 4A shows the protein input for each condition, demonstrating the presence of the transfected proteins under the appropriate conditions. Figure 4B demonstrates the association between Nav1.5 and Nav1.8 when co-expressed (Lane 5, bottom). This interaction was lost when the pull-down antibody was omitted (Lane 4, bottom) and did not occur due to in vitro mixing of the protein lysates (Lane 6, bottom).
Figure 4. Representative Experiments Demonstrating Physical Interaction between Nav1.8 and Nav1.5.
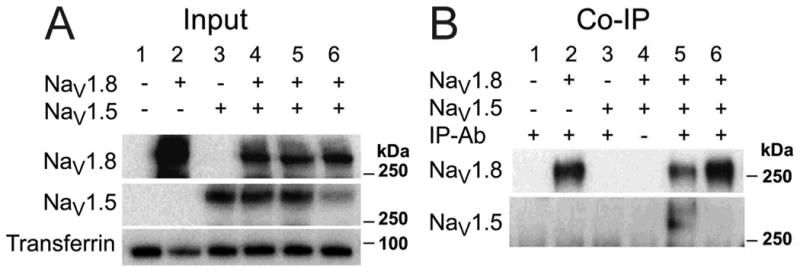
Panel A shows protein input and panel B shows protein isolated by the antibody pull-down co-immunoprecipitation (Co-IP). Lanes 1-6 correspond to the following experimental conditions: (1) non-transfected, (2) Nav1.8 expressed alone, (3) Nav1.5 expressed alone, (4 and 5) Nav1.8 and Nav1.5 co-expressed, (6) mixed lysates from lanes 3 and 4. Panel B shows the pull-down of Nav1.5 is specific to Nav1.8 cellular co-expression.
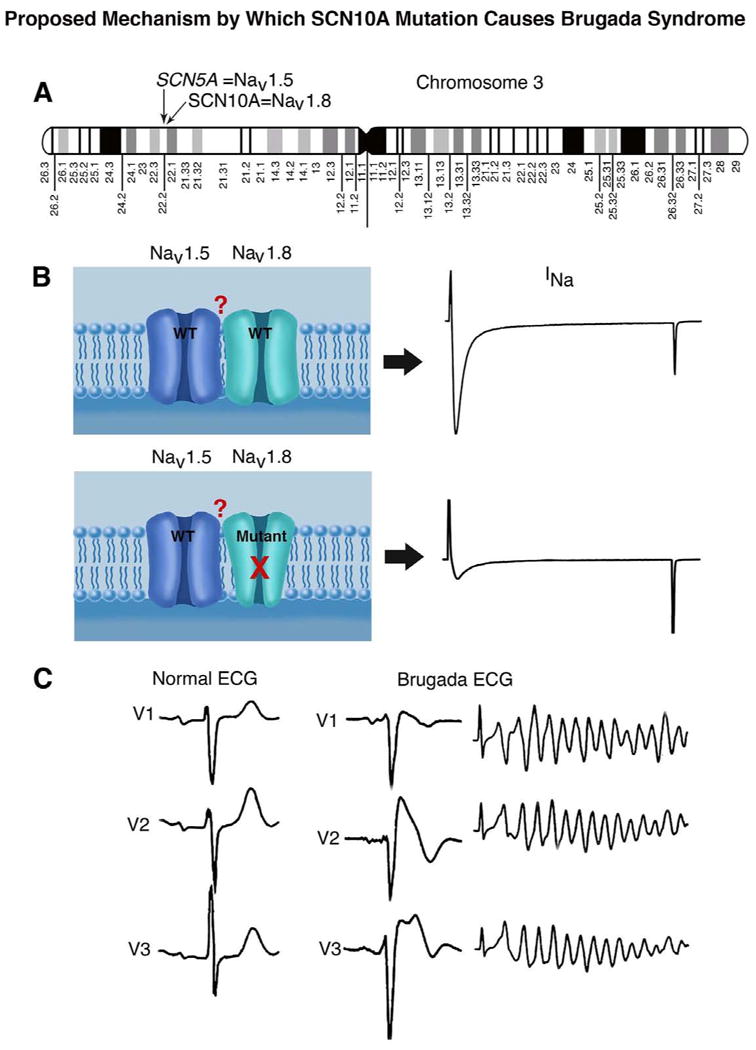
Central Illustration: SCN5A and SCN10A, genes encoding cardiac and neuronal sodium channels, are found in close proximity on chromosome 3 (A). Our study suggests that mutations in SCN10A can lead to a loss of function in sodium channel current (INa) and thus contribute to the manifestation of Brugada syndrome (BrS), a sudden cardiac death syndrome. The data suggest physical association of the two channel proteins (NaV1.5 and NaV1.8) in the plasma membrane (B). Our study identifies SCN10A as a major susceptibility gene for BrS, thus greatly enhancing our capability to genotype and risk stratify probands and family members (C).
Discussion
SCN10A in the heart and its role in arrhythmogenesis
SCN5A and SCN10A located in close proximity to each other in chromosome 3p22. In 1997, SCN10A protein (also referred to as PN3, SNS, and hereafter, Nav1.8) was initially shown to be specifically expressed in rat and human dorsal root ganglia (27). Real-time polymerase chain reaction and immunostaining methodologies have detected a low level of expression of the SCN10A gene product in mouse and human heart tissues with somewhat higher levels in the Purkinje system (12,15,18). Nav1.8 immunoreactivity was detected in intra-cardiac neurons and ganglia in human myocardium (28). With in situ hybridization method, SCN10A displayed a similar distribution pattern Scn5a in mouse hearts (10). These findings notwithstanding, some researchers deny the existence of Nav1.8 in cardiac myocytes. For example, Veldkamp and colleagues reported that SCN10A expression modulates cardiac electrical activity primarily by regulating the firing patterns of intracardiac neurons (11). Conflicting data also resulted from other in vivo and in vitro experimental studies in the animal models (12,15).
The localization, expression level, and function role of Nav1.8 in the heart remain highly controversial. Nonetheless, our results support the conclusion that SCN10A variants play a key role in developing arrhythmogenic J-wave syndromes, including both BrS and ERS, likely through a direct effect on Nav1.5-mediated cardiac INa(Central Illustration). A key role for Nav1.8 in human cardiac electrophysiology is supported by GWAS, showing that SCN10A plays an important role in cardiac conduction disease, by influencing PRI and QRS duration, as well as heart rate and arrhythmic risk. Several independent loci within SCN10A have been identified, including rs6795970 (13-18), rs6798015 (16,19), rs6800541 (16,20), rs7430477 (16), and rs12632942 (15). A recent genome-wide association study of 312 individuals with BrS and 1,115 controls reported a significant association signal at a SCN10A locus rs10428132, providing additional support for a role for SCN10A variants, in this case 3[prime]-UTR or intronic, in the development of BrS (21).
Clinical and genetic findings related to SCN10A
We identified 17 putative pathogenic SCN10A variants in 25 of the 150 BrS probands screened. A positive proband yield of 16.7% is approaching our historical yield of 20.1% for SCN5A and a yield of 11% to 28% (21% average) reported in the international compendium of SCN5A mutations (5). In our study, as in the international compendium study, there was a male predominance of the BrS phenotype (67% vs.78%). The latter has a similar yield between males and females (20% vs. 22%, respectively).This was not the case in our screen for SCN10A mutations, where the yield was greater in the case of males (20% vs. 10%).
In our study, 66.7% of SCN10A mutations were localized to transmembrane and pore-forming domains; this is in comparison to the nearly 75% reported in the SCN5A compendium. Of all BrS-related SCN10A variants, one was a frameshift and the rest were missense mutations (94.1%), whereas in the compendium of SCN5A mutations, two-thirds were reported to be missense mutations.
In 25 of the cases reported, 6 also were found to carry a second potentially pathogenic BrS mutation (Table 2). As such, the number of SCN10A variants that we count as potentially responsible for the clinical phenotype could be an overestimate. This notwithstanding, the 3 mutations in calcium channel genes were found in patients displaying a prolonged PRI (>180 ms) and normal QTc interval, pointing to a clear predominance of the SCN10A mutation leading to a loss of function of INa. The KCNJ8 mutation likewise was accompanied by a prolonged PRI. The two SCN5A mutations were both accompanied by very prolonged PRI (240-280 ms) suggesting that both the SCN5A and SCN10A variants contributed to the clinical phenotype. Interestingly, the yield of BrS probands unmasked by fever is much higher in the case of SCN10A vs. SCN5A mutation (29.4% vs. 17.2%, unpublished data from Dan Hu et al.). There was a higher association with SCD and syncope in the case of SCN10A vs. SCN5A mutations. Also interesting is the larger number of complaints of chest pain in the SCN10A+ group than the SCN10A- cases, which is not observed when SCN5A+ and SCN5A- cases are compared.
Greater than 90% of the SCN10A+ BrS subjects presented with mixed phenotypes, the most common of which was CCD. It is not surprising that, as with SCN5A mutations (39% when PR>200 ms vs. 8% when PR<200 ms (6)), the yield of SCN10A mutants was much higher in BrS probands with prolonged PRI (31% in PR>200 ms vs.11% in PR<200 ms).
Our observations of a high prevalence of SCN10A variants associated with BrS and ERS, most of which: 1) are in amino acid residues that are highly conserved in mammalian species; 2) exhibit a very low MAF in controls; 3) are predicted by in silico models to be pathogenic; 4) show good genotype-phenotype correlation in cases in which family pedigrees are available; and 5) show a major loss of function in INa in the 2 cases in which the variants were functionally co-expressed with SCN5A, suggesting that SCN10A is an important susceptibility gene for BrS and as well as for other cardiac syndromes including CCD, ERS, AF, VT/VF, RBBB, and bradycardia. SCN10A is known to be involved in nociception (29). Our referring physicians did not report altered nociception other than an increased incidence of chest pain.
Mechanisms underlying SCN10A modulation of electrical function of the heart
α-subunit interactions have previously been shown to aggravate as well as ameliorate disease phenotypes. The combination of SCN2A and KCNQ2 mutations cause severe seizure manifestations (30), an Scn8a mutation has been shown to compensate for haploinsufficiency of Scn1a (31), and SCN9A mutations are known to modify the severity of SCN1A-related Dravet's syndrome (32). A recent BrS study reported a dominant-negative effect of SCN5A mutant channels interacting with SCN5A-WT channels (33). Given their proximity to one another, SCN5A and SCN10A may be subject to common regulatory mechanisms, such as transcriptional control by TBX3 and TBX5 (10).
We hypothesize that SCN10A modulates the activity of the canonical cardiac sodium channel encoded by SCN5A in the heart. Our co-expression studies provide evidence in support of this hypothesis showing that Nav1.5 and Nav1.8 co-associate when expressed together. The observed functional interaction between Nav1.5 and Nav1.8 may suggest either a direct physical interaction between the two channels or an indirect interaction within a larger protein complex. SCN10A-WT causes a gain of function in Nav1.5 current, whereas SCN10A-mutants (R14L and R1268Q) cause loss-of-function of Nav1.5 current, which is expected to reduce excitability and lead to development of the arrhythmogenic substrate responsible for BrS and ERS, as well as CCD, VT/VF, AF, RBBB, and bradycardia.
Limitations and future directions
Of the 16 missense mutations uncovered in this study, only 2 were functionally characterized. Despite these limitations, it is important to note that 13 of these variants were totally absent from our own ethnically matched controls and are either absent or negligibly present in all available public databases. Moreover, these mutations are located in highly conserved residues and are predicted to be pathogenic by in silico prediction tools.
Our co-immunoprecipitation data pointing to co-association of Nav1.5 and Nav1.8 proteins were performed following co-expression of SCN5A and SCN10A in HEK cells. Ideally, these studies should be performed in native human ventricular myocytes. This, however, must await the availability of more reliable Nav1.8-specific antibodies. Additional studies are needed to expand the size of the cohort and to conduct functional expression of WT and mutant SCN10A in native myocytes or alternatively in induced pluripotent stem cell-derived cardiomyoctyes.
Conclusions
The findings of this study extend our knowledge of the role of Nav1.8 in the heart and provide an explanation for why SCN10A variants cause conduction and rhythm disturbances, some previously identified by GWAS. Our data identify SCN10A as a new BrS susceptibility gene and as a potential target for genetic screening and antiarrhythmic intervention. We demonstrate co-localization and co-association of Nav1.8 and Nav1.5 in the plasma membrane and a gain of function of SCN10A-WT and loss-of-function of SCN10A-mutants on Nav1.5 INa. BrS males between 11-50 years old, presenting with a prolonged PRI and QRS prolongation, VT/VF, ERS, and/or symptoms (syncope, SCD, chest pain), have the highest probability of carrying an SCN10A variant. The spectrum of SCN10A arrhythmic phenotypes, including BrS, ERS, CCD, VT/VF, AF, RBBB, and bradycardia, is similar to that of SCN5A variants. With a yield of 16.7% for SCN10A, a genotype can now be identified by us in more than 50% of BrS probands.
Perspective
Competency in Medical Knowledge
Brugada (BrS) and early repolarization (ERS) syndromes are responsible for ventricular fibrillation (VF) and sudden cardiac death (SCD) of young adults. Fewer than 35% of BrS probands have genetically identified pathogenic variants. The identification of SCN10A as a major susceptibility gene for BrS and ERS greatly enhances the capability to risk-stratify probands and family members by genotyping.
Translational Outlook 1
The ability of mutant neuronal sodium channels to cause a loss of cardiac sodium channel activity provides insights into mechanisms by which SCN10A variants may contribute to overlap syndromes including BrS, ERS, cardiac conduction disease, and various bradycardia phenotypes
Translational Outlook 2
These findings help to delineate the role of neuronal sodium channels in the electrical function of the heart.
Supplementary Material
Table S1. Sequences of Primers in SCN10A.
Table S2. Effects of SCN10A, SCN5A and SCN3B Co-Expression on Equilibrium Gating
Acknowledgments
The authors are grateful to Drs. Philip J. Iuliano and Ramon Saldana for clinical assistance, Judy Hefferon and Robert J. Goodrow, Jr. for technical assistance and Susan Bartkowiak for maintaining the MMRL genetic database.
Funding: This study was supported by grants from NIH/NHLBI HL47678 (CA); National Research Service Award fellowship #F32-HL107029 (MJB); CONACYT #FM201866 (HBM and DH); New York Stem Cell Foundation #C026424 (CA) and the Masons of New York, Florida, Massachusetts, Connecticut, Maryland, Wisconsin and Rhode Island.
Abbreviations and Acronyms
- BrS
Brugada syndrome
- CCD
cardiac conduction defect
- Co-IP
co-immunoprecipitation
- ERS
early repolarization syndrome
- GWAS
genome-wide association studies
- INa
sodium channel current
- MAF
minor allele frequency
- PRI
PR interval
- SCD
sudden cardiac death
- WT
wild type
Footnotes
Disclosures: Dr. Antzelevitch is a paid consultant to Gilead Sciences, Inc.; Drs. Belardinelli, Kahlig, and Rajamani, are employees of Gilead. All other authors have reported no financial relationships to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Yan GX. J wave syndromes. HeartRhythm. 2010;7:549–58. doi: 10.1016/j.hrthm.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J. 2012;76:1054–65. doi: 10.1253/circj.cj-12-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu D, Barajas-Martinez H, Terzic A, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171:431–42. doi: 10.1016/j.ijcard.2013.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. HeartRhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crotti L, Marcou CA, Tester DJ, et al. Spectrum and prevalence of mutations Involving BrS1-through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J Am Coll Cardiol. 2012;60:1410–8. doi: 10.1016/j.jacc.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) HeartRhythm. 2011;8:1308–39. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Sangameswaran L, Delgado SG, Fish LM, et al. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–6. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 9.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–62. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 10.van den Boogaard M, Wong LY, Tessadori F, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–30. doi: 10.1172/JCI62613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verkerk AO, Remme CA, Schumacher CA, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111:333–43. doi: 10.1161/CIRCRESAHA.112.274035. [DOI] [PubMed] [Google Scholar]
- 12.Yang T, Atack TC, Stroud DM, et al. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–32. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeff JM, Ritchie MD, Denny JC, et al. Generalization of variants identified by genome-wide association studies for electrocardiographic traits in African Americans. Ann Hum Genet. 2013 doi: 10.1111/ahg.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie MD, Denny JC, Zuvich RL, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–85. doi: 10.1161/CIRCULATIONAHA.112.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sotoodehnia N, Isaacs A, de Bakker PI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–76. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denny JC, Ritchie MD, Crawford DC, et al. Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation. 2010;122:2016–21. doi: 10.1161/CIRCULATIONAHA.110.948828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–22. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 18.Chambers JC, Zhao J, Terracciano CM, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–52. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 19.Smith JG, Magnani JW, Palmer C, et al. Genome-wide association studies of the PR interval in African Americans. PLoS Genet. 2011;7:e1001304. doi: 10.1371/journal.pgen.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeufer A, van NC, Marciante KD, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–9. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bezzina CR, Barc J, Mizusawa Y, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–9. doi: 10.1038/ng.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu D, Barajas-Martinez H, Kahlig K, et al. Genetic variants in SCN10A associated with Brugada sydrome, right bundle branch block and atrioventricular block. HeartRhythm. 2012;9:S395. Abstract. [Google Scholar]
- 23.Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference. HeartRhythm. 2005;2:429–40. doi: 10.1016/j.hrthm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Antzelevitch C, Yan GX. J-wave syndromes. From cell to bedside. J Electrocardiol. 2011;44:656–61. doi: 10.1016/j.jelectrocard.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–8. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aizawa Y, Takatsuki S, Sano M, et al. Brugada syndrome behind complete right bundle-branch block. Circulation. 2013;128:1048–54. doi: 10.1161/CIRCULATIONAHA.113.003472. [DOI] [PubMed] [Google Scholar]
- 27.Tzoumaka E, Novakovic SD, Haraguchi M, et al. PN3 sodium channel distribution in the dorsal root ganglia of normal and neuropathic rats. Proc West Pharmacol Soc. 1997;40:69–72. [PubMed] [Google Scholar]
- 28.Facer P, Punjabi PP, Abrari A, et al. Localisation of SCN10A gene product Na(v)1.8 and novel pain-related ion channels in human heart. Int Heart J. 2011;52:146–52. doi: 10.1536/ihj.52.146. [DOI] [PubMed] [Google Scholar]
- 29.Leo S, D'Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav Brain Res. 2010;208:149–57. doi: 10.1016/j.bbr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 30.Kearney JA, Yang Y, Beyer B, et al. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006;15:1043–8. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- 31.Martin MS, Tang B, Papale LA, et al. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–9. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- 32.Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clatot J, Ziyadeh-Isleem A, Maugenre S, et al. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 alpha-subunits. Cardiovasc Res. 2012;96:53–63. doi: 10.1093/cvr/cvs211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of Primers in SCN10A.
Table S2. Effects of SCN10A, SCN5A and SCN3B Co-Expression on Equilibrium Gating