

Abstract
Cancer genome sequencing studies have identified numerous driver genes but the relative timing of mutations in carcinogenesis remains unclear. The gradual progression from pre-malignant Barrett’s esophagus to esophageal adenocarcinoma (EAC) provides an ideal model to study the ordering of somatic mutations. We identified recurrently-mutated genes and assessed clonal structure using whole-genome sequencing and amplicon-resequencing of 112 EACs. We next screened a cohort of 109 biopsies from two key transition points in the development of malignancy; benign metaplastic never-dysplastic Barrett’s esophagus (NDBE, n=66), and high-grade dysplasia (HGD, n=43). Unexpectedly, the majority of recurrently mutated genes in EAC were also mutated in NDBE. Only TP53 and SMAD4 were stage-specific, confined to HGD and EAC, respectively. Finally, we applied this knowledge to identify high-risk Barrett’s esophagus in a novel non-endoscopic test. In conclusion, mutations in EAC driver genes generally occur exceptionally early in disease development with profound implications for diagnostic and therapeutic strategies.
Keywords: ICGC, esophageal cancer, Barrett’s esophagus, whole genome sequencing
INTRODUCTION
Most epithelial cancers develop gradually from pre-invasive lesions, in some instances after an initial metaplastic conversion. Research to characterize the genomic landscape of cancer has focused on established invasive disease with the goal of developing biomarkers for personalised therapy1. However, it is becoming increasingly clear that extensive genomic heterogeneity is present in the majority of advanced cancers2. The most appropriate therapeutic targets are therefore those mutations that occur early in the development of disease and are thus clonal in the resulting malignancy. The identification of causative mutations occurring early in pathogenesis is also pivotal to developing clinically useful biomarkers. In this context mutations occurring at disease-stage boundaries, for example, the transition from non-dysplastic epithelium to dysplasia, and then to cancer would be most informative. The evidence to date on the genetic evolution of cancer from pre-malignant lesions suggests that the accumulation of mutations is step-wise3-5. In the most well-studied example, the adenoma-dysplasia-colorectal adenocarcinoma progression sequence, it has been possible to assign timings for a limited number of candidate genes by comparative lesion sequencing3. More recent studies have sought to utilize statistical algorithms to infer the life history4,5 of a tumor from single samples.
Esophageal adenocarcinoma (EAC) arises from metaplastic Barrett’s esophagus in the context of chronic inflammation secondary to exposure to acid and bile6,7. Barrett’s esophagus lends itself well to studies of genetic evolution due to the repeated sampling of the mucosa during clinical surveillance prior to therapeutic intervention8. Previous studies of EAC and Barrett’s esophagus have generally used candidate gene approaches with the goal of identifying clinical biomarkers to complement histological examination, which is an approach fraught with difficulties8,9. Data from high-density single nucleotide polymorphism (SNP) arrays and exome-sequencing studies are now accumulating with a plethora of mutations identified in many different genes10,11. However, little work has yet focused on the precise ordering of these alterations in large cohorts of patients with pre-malignant disease and associated clinical follow-up data.
Recently Agrawal et al. performed exome sequencing on 11 EAC samples and two samples of Barrett’s esophagus adjacent to the cancer. Intriguingly, the majority of mutations were found to be present even in apparently normal Barrett’s esophagus 12 similar to the observation in colorectal adenocarcinoma. This raises the possibility that prior to the progression to malignancy mutations that predict the risk of progression may be detectable within cytologically benign tissue. However it is unclear to what extent the same mutations may be present in Barrett’s esophagus tissue from patients that have not progressed to cancer. This question is important as the majority of patients with Barrett’s esophagus will not progress to cancer, and somatic alterations occurring early, prior to dysplasia, are unlikely to provide clinically discriminatory biomarkers. Biomarker research in this area is critical since the current endoscopic surveillance strategies are increasingly recognized to be ineffective13 and therefore novel approaches are required14,15.
The aims of this study were: 1) identify a list of candidate, recurrently-mutated genes in EAC; 2) to accurately resolve the stage of disease at which mutation occurs therefore providing insight as to the role of these recurrent mutations in cancer progression, and 3) test their utility in clinical applications, i.e. using the non-invasive, non-endoscopic, cell sampling device, the Cytosponge™.
RESULTS
HIGH MUTATION BURDEN AND UNUSUAL MUTATIONAL SIGNATURE IN EAC
The discovery cohort (22 EACs subject to WGS, Figure 1) reflected the known clinico-demographic features of the disease: male predominance (M:F, 4.5:1), a mean age of 68 years (range 53 to 82), and a majority with advanced disease (81.8% (18/22) > stage I). Of the 22 cases, 17 (77.3%) had evidence of Barrett’s esophagus in the resection specimen (Table 1 and Supplementary Table 1). Patient samples were sequenced to a mean coverage of 63- and 67-fold in tumor and normal samples respectively (Supplementary Table 2, normal squamous or blood was used as outlined in Supplementary Table 1).
Figure 1. Flow chart illustrating the study outline.

The number of samples used at each stage is given. The methodology used for each study phase is shown on the left hand side. EAC, Esophageal adenocarcinoma, BE, Barrett’s esophagus, HGD, high grade dysplasia.
Table 1. Demographics of the patient cohorts.
| EAC cohorts | Barrett’s esophagus cohorts | TP53 analysis on Cytosponge™ | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Discovery | Validation | Never-dysplastic Barrett’s esophagus |
Barrett’s esophagus with HGD |
No Barrett’s esophagus Controls |
Never-dysplastic Barrett’s esophagus |
Barrett’s esophagus with HGD |
||
| Number | 22 | 90 | 40 | 39 | 23 | 44 | 22 | |
|
| ||||||||
| Age (years) | 68 (53-82) | 66 (32-83) | 63 (32-81) | 71 (50-87) | 53 (28-74) | 61 (41-85) | 66 (41-82) | |
|
| ||||||||
| Sex (M:F) | 5:1 | 5 : 1 | 2 : 1 | 12:1 | 1 : 2 | 4 : 1 | 10 : 1 | |
|
| ||||||||
| Stage (%) | I | 4 (18.2) | 14 (15.6) | |||||
| II | 6 (27.3) | 14 (15.6) | ||||||
| III | 11 (50.0) | 49 (54.4) | ||||||
| IV | 1 (4.5) | 4 (4.4) | ||||||
| n/a | 0 (0.0) | 9(10.0) | ||||||
|
| ||||||||
| Barrett’s esophagus length (cm) | 4.8 (1-9) | 8.6 (2-16) | 5.8 (1-12) | 8.5 (4-16) | ||||
|
| ||||||||
| Follow up from EAC diagnosis (months) | 28.5 (5-63) | 18 (1-134) | ||||||
|
| ||||||||
| Total Barrett’s esophagus surveillance (months) | 58 (4-132) | 1 (0-45) | 56 (0-175) | 24 (0-180) | ||||
Data shown reflect mean (range) for age and Barrett’s esophagus length, number (percentage) for stage and median (range) for follow up from EAC diagnosis and total Barrett’s esophagus surveillance. Sex ratio rounded to the nearest whole number.
We identified a median of 16,994 somatic SNVs (range: 4,518-56,528) and 994 small indels per sample (range 262-3,573). From this final dataset a total of 1,086 coding region mutations were subject to verification as part of a larger pipeline bench marking study (see online Methods). We used ultra-deep targeted re-sequencing, achieving a median coverage of >13,000 fold, and confirmed 1,081 (99.5%) as somatic. Using Sanger sequencing, 23/25 (92%) indels were verified as real and somatic. As observed by Dulak et al in the intervening time since our study commenced11, the most frequent mutation type across the discovery cohort was T:A>G:C transversions with a striking enrichment at CTT trinucleotides (Supplementary Figure 1). This enrichment for T:A>G:C transversions differentiates EAC from other cancers that have been studied by WGS, including breast, colorectal and hepatocellular16-18.
TARGETED AMPLICON RESEQUENCING IN A VALIDATION COHORT OF EACs
To highlight genes most likely to be relevant in the development of EAC in Barrett’s esophagus, we sought to determine the degree to which mutated genes identified in our discovery cohort (n=22 cases) were representative of the spectrum of mutations in an expanded cohort. Hence, a final list of 26 genes that were either mutated significantly above the background rate or in pathways of interest were selected (Supplementary Note) and tested in a larger cohort (90 additional EACs, Table 1 and Supplementary Table 3), using targeted amplicon-resequencing. The findings confirmed and extended those of our discovery cohort and previous work from others11,12,19, including the identification of recurrent mutations in the SWI/SNF complex, such as ARID1A (Supplementary Figure 2). Analysis of ARID1A protein expression loss by immunohistochemistry in a cohort of 298 additional EACs identified absent or decreased expression in 41% (122/298). This suggests alternative mechanisms of down regulation may be present though we did not identify any large-scale structural variants within the WGS data from our discovery cohort (data not shown).
We next combined the data from both the discovery and validation cohorts and identified 15 genes that were mutated in four or more samples (Figure 2). These included those previously identified as EAC candidate genes, and several novel candidates: MYO18B, SEMA5A and ABCB1. Comparison with the recent EAC exome sequencing from Dulak et al confirmed that these genes were recurrently mutated in an external data set (Supplementary table 4). TP53 was mutated in the majority of cases; however 31% of cases are wild type for TP53. Although we do not have enough power to detect mutually-exclusive mutations in our cohort, we can detect significantly co-occurring mutations. SEMA5A and ABCB1 mutations occurred more commonly in the same tumor than would be expected by chance (Benjamini-Hochberg-adjusted p-value = 0.0021) although the reason for this association remains unclear.
Figure 2. Mutation in esophageal adenocarcinoma.

The bar graph on the top indicates the percentage of samples with aberrations for a given gene. The number in bold denotes the total number of mutations for each gene. Genes with four or more mutations in our EAC discovery and validation cohort (combined total of 112 patients) were included. The proportion of missense, nonsense/splice and indel mutations are shown. The matrix below shows the number of samples with mutations in both genes for each possible pairing of genes. The red highlighted box indicates significantly co-occurring mutations (Significance was assessed empirically from 100,000 permutations. False discovery rate was controlled using the Benjamini-Hochberg procedure.).
SIMILAR MUTATION FREQUENCY ACROSS DISEASE STAGES
The stage specificity of mutations can be derived from patients at discrete stages of Barrett’s esophagus carcinogenesis. Mutations occurring at disease-stage boundaries would be candidate biomarkers of malignant progression. In addition, mutations occurring early in the development of disease should represent ideal targets for novel therapeutic interventions due to their presence in the majority of cells in more advanced lesions due to clonal expansion early in the natural history. We therefore sought to identify the mutation status of the 26 genes in our panel in Barrett’s esophagus samples obtained from a prospective cohort of patients undergoing endoscopic surveillance. This included 109 Barrett’s esophagus biopsies from 79 patients (Figure 1). We selected 66 never-dysplastic Barrett’s esophagus samples from 40 Barrett’s esophagus patients for whom there was no evidence for progression to dysplasia or malignancy (median follow-up time 58 months, range 4-132), and 43 Barrett’s esophagus biopsy samples (from 39 patients) of histopathologically confirmed high grade dysplasia (HGD), the stage just prior to the development of invasive EAC (Table 1). We did not include low-grade dysplasia due to the poor agreement on the histopathological grading of this lesion20.
The findings were striking and unexpected. For the never-dysplastic Barrett’s esophagus cohort, 21/40 (53%) patients were found to have mutations within their Barrett’s esophagus segment (Figure 3a), with several biopsies containing multiple mutations (Supplementary Table 5). In total, we identified 29 SNVs and 7 indels within this cohort. Importantly, the mutations identified in never-dysplastic Barrett’s esophagus occurred in several genes previously identified as drivers in EAC11,19 and other cancers21,22, including SMARCA4, ARID1A, and CNTNAP5 (Figure 3b). Of interest, seven of these 29 SNVs were mutations at T:A base pairs. Of these, 5/7 (71%) occurred at TT dinucleotide sequences, the mutational context identified as highly enriched in the EAC WGS data. Thus, this mutational process may well be active at the earliest stages of disease. Of the 43 HGD biopsy samples, 39 (91%) were found to have mutations in at least one of the genes in our panel with a total of 67 SNVs and 7 indels. Hence, rather than the frequency of mutation in a given gene increasing across disease stages, we observed that for the vast majority of genes the mutational frequency was not significantly different between never-dysplastic Barrett’s esophagus, HGD and EAC (Fisher’s exact test with Benjamini-Hochberg correction for multiple testing, Figure 3b and Supplementary Table 6). For two genes, MYO18B and ARID1A we performed amplicon sequencing in an additional 25 NDBE samples and 11 HGD increasing the cohort to a total of 91 NDBE and 54 HGD, but did not identify any significant difference in frequency of mutation between disease stages (Supplementary table 7). Only TP53 (p<0.0001) and SMAD4 p=0.0061) (Figure 3b and c) exhibited mutational frequencies that would distinguish between disease stages and thus identify progression towards malignancy. TP53 was found to be recurrently mutated in both HGD (72%) and EAC (69%) samples, but only in a single case (2.5%) of never-dysplastic Barrett’s esophagus. SMAD4 was mutated at a lower frequency (13%) and intriguingly was only found in EAC, the invasive stage of disease.
Figure 3. TP53 and SMAD4 mutations accurately define the boundaries in the progression towards cancer whilst other mutations appear to occur independent of disease stage.

A. Bar graph showing the number of never-dysplastic Barrett’s esophagus patients (NDBE) (n=40), Barrett’s esophagus patients with high grade dysplasia (HGD) (n=39) and EAC patients (n=112) with at least one mutation in our panel consisting of 26 genes. B. Percentage of never-dysplastic Barrett’s esophagus, Barrett’s esophagus with HGD and EAC samples with mutations in recurrently-mutated genes (mutated in ≥4 samples) identified in the EAC discovery cohort and EAC Validation cohort. TP53 and SMAD4 are the only genes for which mutations separate the boundaries between never-dysplastic and dysplastic Barrett’s esophagus (TP53) or cancer (SMAD4) (two-tailed Fisher’s exact test with Benjamini-Hochberg correction for multiple testing, * p<0.05). C. Proposed model for the boundary-defining mutations in Barrett’s esophagus carcinogenesis. The hashed box depicts multiple other mutations which may occur and provide selective advantage at any stage of disease.
CLONAL ANALYSIS OF RECURRENT MUTATIONS
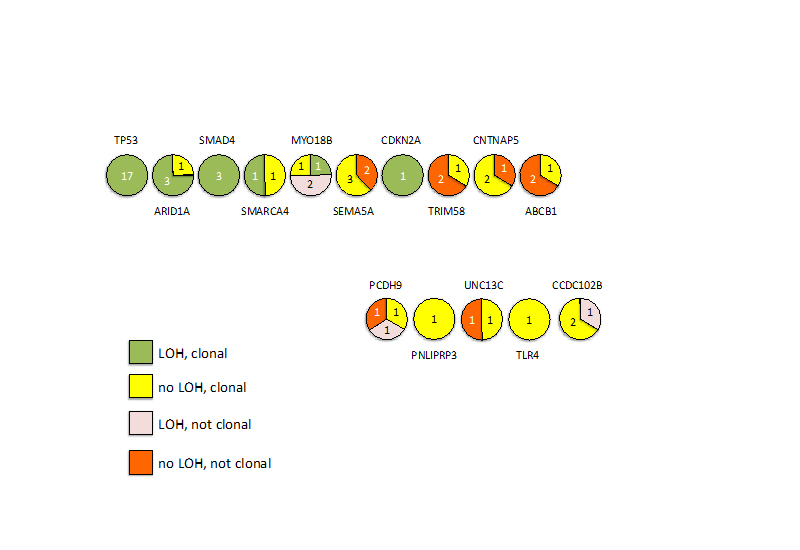
Having identified the occurrence of mutations in the earliest stages of disease development we next sought to identify whether these mutations were fully-clonal or sub-clonal in our original discovery cohort of 22 EACs. For each of the 15 genes mutated in ≥4 samples from our expanded cohort we combined our high-depth resequencing of SNVs, copy number variant data and LOH analysis to determine the fraction of tumor cells containing the mutation (Supplementary Note). If mutation occurs at the earliest stage of disease development, prior to the clonal expansion of the malignancy, we would expect that the mutation would be present in all cells of the tumor. For 7/15 genes; SMAD4, TP53, ARID1A, SMARCA4, TLR4, CDKN2A and PNLIPRP3 this was the case. Mutation in the other 8 genes (MYO18B, TRIM58, CNTNAP5, ABCB1, PCDH9, UNC13C and CCDC102B) was not always present in the major clone (Supplementary Figure 3), suggesting that mutation of these genes may be selected for at multiple stages of tumorigenesis.
APPLICATION OF MUTATIONAL KNOWLEDGE TO A DIAGNOSTIC TEST
The current clinical strategy for patients with Barrett’s esophagus involves regular endoscopic examinations to try and identify patients with dysplasia who are at high risk of progression to adenocarcinoma. This approach is highly controversial due to the inherent difficulties in accurate identification of dysplastic lesions, and recent data suggest that endoscopic surveillance of Barrett’s esophagus is not effective13,23. The difficulties involved in endoscopic surveillance for Barrett’s esophagus include sampling bias inherent in random biopsies protocols and the subjective and time-consuming histopathological diagnosis of dysplasia. We therefore developed a novel approach which has the potential to overcome these limitations of Barrett’s esophagus surveillance. The strategy comprises a non-endoscopic device called the Cytosponge™ which can be provided to patients in the primary care setting. This device collects cells from the entire esophageal mucosa, thus avoiding sampling bias and can be combined with objective biomarkers for diagnosis24,25. To date our focus has been on a biomarker for diagnosing Barrett’s esophagus, however, since most Barrett’s esophagus patients will not progress to EAC, this Barrett’s esophagus biomarker needs to be combined with a biomarker (or a panel of biomarkers) to identify the high-risk dysplastic patients. From the aforementioned sequencing data, TP53 mutations fit the criteria of a good risk stratification candidate marker, since TP53 mutations discriminate between patients with and without high grade dysplasia, the key point of therapeutic intervention. Though the device samples abnormal tissue, the majority of cells collected are from normal gastric glandular tissue at the top of the stomach as well as normal squamous areas of the esophagus, and therefore any mutant DNA would theoretically be in the minority, requiring a very sensitive assay (Supplementary Figure 4). This situation is analogous to the detection of tumor cell-free DNA in blood as a biomarker in advanced malignant disease: sensitive assays have been developed to detect extremely low levels of mutant DNA against normal background26,27. We therefore took an analogous approach to detecting mutations in Cytosponge™ material.
To determine whether mutations within Barrett’s esophagus lesions could be detected in material collected from the Cytosponge™, we first tested mutations previously identified in endoscopic Barrett’s esophagus biopsies. Four patients with HGD dysplasia had TP53 mutations and had also swallowed the Cytosponge™ (twice in patient 4). For all four patients, the specific TP53 mutations were detected at an allele fraction (proportion of variant reads) of between 0.04 and 0.24 (Table 2).
Table 2. Allele fractions for known TP53 mutations, previously identified by sequencing TP53 on diagnostic biopsies.
For these four patients the mutation can also be detected in material collected using the Cytosponge™. Patient 4 swallowed the Cytosponge™ on two different occasions, 8 months apart, and the data for both Cytosponge™ samples is shown. N/A = Not applicable as no sample was taken, AF= allele fraction.
| AF on Biopsy | AF on Cytosponge™ | ||||
|---|---|---|---|---|---|
|
| |||||
| Patient | Mutation | #1 | #2 | #1 | #2 |
| HGD_01 | Chr17: 7574003 G>A | 0.35 | N/A | 0.04 | N/A |
| HGD_40 | Chr17: 7577538 C>T | 0.23 | 0.52 | 0.10 | N/A |
| HGD_03 | Chr17: 7578406 C>T | 0.51 | 0.72 | 0.06 | N/A |
| HGD_04 | Chr17: 7577551 C>T | 0.19 | N/A | 0.14 | 0.24 |
We then tested whether we could detect unknown TP53 mutations within material collected using the Cytosponge™ as this would be required for a clinical test. We amplified the majority of the coding region of TP53 (1019/1182 bp (86%)) by multiplexed PCR and sequenced the amplified DNA by massively-parallel sequencing. TP53 mutations were called de novo using TAm-Seq26 on samples from control patients (no Barrett’s esophagus), Barrett’s esophagus patients with no dysplasia as well as Barrett’s esophagus patients with high grade dysplasia. As we expected, no TP53 mutations were identified in samples from control patients or Barrett’s esophagus patients with no dysplasia (Figure 4a), demonstrating 100% specificity in differentiating between patients with HGD and no dysplasia. In contrast, TP53 mutations were identified in 19/22 (86%) HGD patients (details of individual mutations can be found in Supplementary table 8). The allele fractions of the TP53 mutations varied widely (between 0.006 to 0.357) but anything in this range can be called successfully and mutations were mostly clustered in the DNA binding domain, as expected (Figure 4b).
Figure 4. TP53 mutations can be used to diagnose Barrett’s esophagus with prevalent high-grade dysplasia on the Cytosponge™.

A. The allele fraction of TP53 mutations identified in Cytosponge™ samples is shown for the three patients groups: no Barrett’s esophagus (n=23), Barrett’s esophagus with no dysplasia (n=44) and Barrett’s esophagus with high grade dysplasia (HGD) (n=22). B. The positions of the TP53 mutations identified on the Cytosponge™ samples are shown above the gene diagram compared with those found in the EAC and Barrett’s esophagus HGD biopsy cohorts. The dotted line on the gene outline denotes the two small areas not covered by the multiplex PCR assay (amino acids 1-27 and 361-393). TA, transcription activation domain; OD, oligomerization domain.
DISCUSSION
Barrett’s esophagus is the only known precursor lesion of EAC, co-occurring in >80% of cases presenting de novo28, however the majority of Barrett’s esophagus patients will never progress to invasive disease29. There is therefore a need for sensitive and specific biomarkers that can identify those patients at risk of progression. As long ago as the Nowell hypothesis, a stepwise selection of genomic mutations has been assumed necessary for cancer development30. Somatic genomic variants should therefore be highly sensitive and specific markers of disease stage. By screening for our panel of recurrently-mutated genes in a cohort of patients with Barrett’s esophagus who had never developed dysplasia, and a cohort of those with HGD, we hoped to identify a step-wise accumulation of mutations across these disease stages. Surprisingly we identified numerous mutations occurring in never-dysplastic Barrett’s esophagus at detectable allele fractions (>10%). Intriguingly the most prevalent gene mutations in EAC were also present at a similar frequency in Barrett’s esophagus and HGD samples, including mutations within cancer-associated genes, for example ARID1A and SMARCA4, members of the SWI/SNF complex. These data demonstrate the complex mutational landscape that may be present even within tissue with a very low risk of malignant progression which has an entirely benign histopathological appearance. The exact role of these mutations at such an early stage of disease development remains unclear. However, it is known that clonal expansions occur frequently in Barrett’s esophagus and it is possible that these mutations provide an increase in fitness of a clone without leading to disruption of the epithelial architecture or providing the necessary cellular characteristics for invasion. A similar observation has been reported in endometrial cancer. In the normal population ~35% of women harbour PTEN mutant glands in their endometrial tissue yet the lifetime risk of endometrial cancer is ~2.5%31.
Our result has substantial implications for the specificity of tests aiming to use highly sensitive detection of mutations for the early diagnosis of malignancy32. Biomarkers predicting individuals at risk for cancer need to have substantial predictive power to distinguish between those who will and will not develop cancer. In our study almost all recurrently mutated genes in EAC, including ABCB1, CNTNAP5, MYO18B amongst others, are ruled out for use as surveillance tools for progression risk. Only mutation in TP53 and SMAD4 accurately defined the boundaries of disease states. The fact that mutation of SMAD4 was only found in EAC provides a clear genetic distinction between EAC and HGD. However, the low frequency of SMAD4 mutation (13%) makes it a sub-optimal candidate for biomarker development. Furthermore, HGD, rather than EAC, is now the ideal point of clinical intervention due to the advent of endoscopic therapy. We therefore focused on TP53 for the proof-of-principle Cytosponge™ study. Sequencing technologies are now being introduced to routine clinical use, and genes of interest can be sequenced rapidly and with exquisite sensitivity, providing a quantitative read-out26.
We detected mutations in 86% of HGD Cytosponge™ samples using a simple, clinically applicable test. To improve the sensitivity of any early detection programme, it will also be key to identify the genetic or epigenetic changes that drive HGD and EAC in the minority of patients without a detectable TP53 mutation. In addition, since genetic diversity has been shown to predict progression to Barrett’s esophagus it may be possible to perform somatic mutation testing looking at both presence and relative proportions of mutations in a panel of genes, to identify patients with high-risk disease33.
In conclusion, never-dysplastic Barrett’s esophagus harbours frequent mutations affecting recurrently-mutated genes in EAC. Given the low rate of progression to malignant disease in never-dysplastic Barrett’s esophagus, the role of these mutations on the road to malignancy is unclear. It is generally accepted that the mutations observed in a tumor are accrued in a linear progression with each step bringing the clone closer to the invasive endpoint. Our observation of mutations in almost all of the recurrently-mutated genes in the tissue of patients who have not gone on to develop malignancy argues against a major role of these mutations in the progression towards cancer. Though their recurrent nature suggests a role in clonal expansion at the pre-malignant stage they do not seem to provide any long term increase in the likelihood of malignant progression. It is likely that additional sequencing cohorts with greater sample numbers and differing demographics will identify further recurrently-mutated genes in EAC, and these too will need careful analysis to determine the disease stage at which they occur.
From a clinical perspective, because the vast majority of recurrently-mutated genes in EAC do not differentiate between the pre-malignant and malignant stages of disease, they therefore cannot be applied in a simple binary test, i.e. mutant or non-mutant, as biomarkers of malignant progression. The Cytosponge™ provides a representative sample of the entire esophageal mucosa and coupled with high-throughput sequencing is capable of sensitive and objective detection of HGD. This approach could be readily adapted as our understanding of the genetic basis for this disease evolves. Furthermore, our systematic molecular approach to identify key mutations involved in the steps distinguishing pre-inasive from invasive disease has applicability to other epithelial cancers amenable to early detection.
ONLINE METHODS
Sample Collection, Pathology Review and Extraction
The study was approved by the Institutional Ethics Committees (REC Ns 07/H0305/52 and 10/H0305/1) and all patients gave individual informed consent. For the discovery cohort, esophageal adenocarcinoma (EAC) patients were recruited prospectively and samples were obtained either from surgical resection or endoscopic ultrasound (EUS). This was an exploratory pilot with no pre-specified effect size. Blood or normal squamous esophageal samples, distant at least 5cm from the tumor, were used as germline reference. All tissue samples were snap-frozen in liquid nitrogen immediately after collection and stored at −80°C. Prior to DNA extraction, one section was cut from each oesophageal tissue sample and H&E staining was performed. Cancer samples were deemed suitable for DNA extraction only after consensus review by two expert pathologists had confirmed tumor cellularity ≥70%. Where blood was not available the same review process was applied to the normal esophageal samples to ensure that only squamous epithelium was present. For the Discovery cohort 127 cases were screened from two centers (Cambridge and Southampton). 63 cases had 70% cellularity required to meet ICGC criteria and of these 22 tumor:normal pairs had sufficient quality and quantity of DNA extracted (total yield ≥ 5μg), and were submitted for whole genome sequencing. From the remaining 105 cases available, 90 had >50% cellularity and all of these had sufficient DNA for the amplicon sequencing. For all cases in the discovery and validation cohort there was a 260/280 ratio 1.8-2.1. For the pre-invasive disease cohort we screened our entire 10 year prospective Barrett’s cohort of >500 patients and selected cases in which there was frozen material available and for which review of the frozen section revealed a homogeneous grade of dysplasia following expert histopathological review. The Cytosponge™ samples were all those available as part of an interim analysis from an ongoing prospective case-control study (BEST2).
DNA was extracted from frozen esophageal tissue using the DNeasy kit (Qiagen) and from blood samples using the Nucleon™ Genomic Extraction kit (Gen-Probe) according to the manufacturer’s instructions. For validation DNA was extracted using the AllPrepDNA/RNA Mini Kit (Qiagen) according to the manufacturer’s instructions.
Whole Genome Sequencing
A single library was created for each sample, and 100bp paired-end sequencing was performed under contract by Illumina to a typical depth of at least 50x, with 94% of the known genome being sequenced to at least 8x coverage while achieving a PHRED quality of at least 30 for at least 80% of mapping bases. Typically, 5 lanes of a HiSeq-2000 (Illumina) flow cell achieved this, but samples were not multiplexed, so some exceeded these minimum standards by a large margin. Filtered read sequences were mapped to the human reference genome (GRCh37) using Burrows-Wheeler Alignment (BWA)34, and duplicates marked using Picard (http://picard.sourceforge.net). As part of an extensive quality assurance process, QC metrics and alignment statistics were computed on a per lane basis. Aggregated QC for each discovery cohort sample is given in Supplementary Table 9. Details of any tiles within flow cells that were removed post-QC are shown in Supplementary Table 10.
The FastQCpackage (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to assess the quality score distribution of the sequencing reads, and enabled the identification of three lanes of sequencing that required trimming due to a drop in quality in the later cycles of sequencing (see details in Supplementary Table 11).
WGS Mutation Calling
Somatic single nucleotide variants (SNVs) were predicted using SomaticSniper V1.0.235 run with the following command: somaticsniper -q 1 -Q 15 -F vcf -J -r 0.001000 -T 0.850000 -N 2 -s 0.01 -f The output from SomaticSniper was then filtered using the following criteria derived from comparison of heuristic filters applied to SomaticSniper and VarScan 236 and implemented using scripts provided in Koboldt et al36 and custom scripts (homopolymer filter). The filtering criteria were; 1) germline and tumor sample coverage ≥10, 2) average variant position in read between positions 10 and 90, 3) percentage of supporting reads from each strand ≥1% and ≤99%, 4) total number of supporting reads ≥4, 5) average distance of variant base from effective 3′ end of supporting reads ≥20 bp, 6) average mapping quality difference between reference and variant supporting reads <30, 7) average difference in length of trimmed sequences between reference and variant reads <25bp, 8) mismatch quality sum difference <100 between reference and variant reads, 9) adjacent homopolymer <5bp away, and 10) nearest indel ≥ 40bp away. In addition, all variants were compared to dbSNP129 and removed if overlapping with predicted germline SNPs. A median of 99.7% of the mappable genome was covered to at least 10-fold coverage in the tumor and matched germline sample and so was defined as callable (Supplementary Table 12).
Candidate somatic indels were taken as the consensus between SAMtools37 and Pindel38, filtered to exclude those indels present in the matched normal genome of any of the 22 samples (including non-consensus indel calls). Indels falling within coding regions and splice sites were manually inspected to generate a final list of calls. Variants were annotated with sequence ontology terms to describe consequence and position relative to Ensembl gene annotations. SNVs and indels were also annotated with matching or nearest features in UniSNP.
Verification of indel variants by PCR
A total of 25 coding indels, confirmed by manual review, were randomly selected for verification. Primers (sequences available on request) were designed to amplify the predicted variant location. PCR was performed on both the tumor and normal DNA and the resulting products were Sanger sequenced. All traces were visualized using Chromas lite 2.01 and were manually reviewed for presence of the variant. An indel was considered somatic if it was present only in the tumor trace.
Verification of single nucleotide variants by targeted re-sequencing
As part of a larger benchmarking exercise of our SNV calling pipeline we selected 2007 SNVs to be verified. These SNVs included those that had failed filters and those that had been predicted using the Illumina pipeline, ELAND alignment plus STRELKA. The complete analysis of these data is ongoing with the overall aim of optimizing the sensitivity of our SNV calling pipeline. Following a preliminary analysis and comparison to the ICGC benchmarking exercise we chose to increase the stringency of our filters for this pilot dataset (detailed above). The verification data in this manuscript is for only those SNVs passing these additional filters. Putative non-synonymous SNVs (1330 in total) underwent ultra-high-depth targeted sequencing. For eight samples all non-synonymous variants were sent for verification. In the remaining 14 cases, the selected SNVs were restricted to non-synonymous variants in genes mutated in more than one sample. Amplicons were generated, indexed and pooled, and libraries constructed as per Shah et al39. Samples were pooled separately and a single lane of HiSeq-2000 data was generated for each, leading to a typical depth of coverage of 13,855 (IQR:3,408 to 39,059 for the amplicons). For 1086 of these ≥50-fold coverage was generated for both tumor and normal. An SNV was confirmed as somatic if the variant allele frequency was ≤ 1% in the matched normal and ≥ 2% in the tumor, and 1081 SNVs met these criteria giving a verification rate of 1081/1086 (99.5%).
Mutation validation in independent samples
Mutation validation was performed in a cohort of 90 additional EACs and 109 Barrett’s esophagus biopsies, including 43 Barrett’s esophagus biopsies with histopathologically confirmed HGD and 66 with no dysplasia. The Access Array microfluidics PCR platform (Fluidigm) together with high-throughput sequencing (Illumina) was used for the targeted re-sequencing.
Amplicons with a median size of 180bp (range 100-200bp) were designed using Fluidigm in-house software (primers available on request)26. After two iterations of primer design, one gene remained uncovered by suitable amplicons (DIRC3) and this was removed from further analysis. Hence, in total 26 genes were selected (Supplementary Table 13 and 14). All primers were synthesised with universal sequences (termed CS1 and CS2) appended at the 5′-end.
Target amplification and sample barcoding was performed using the manufacturer’s standard multiplex protocol (Fluidigm, Access Array User Guide). Primers were combined into multiplex pools ranging from 1 to 12 primer pairs. The Access Array system was used to combine PCR reagents (FastStart High Fidelity PCR System, Roche) with 47 DNA samples (50ng) plus a single negative control and 48 sets of multiplexed primers into 2,304 unique 35nL PCR reactions. Thermal cycling was then applied to amplify all selected targets by PCR. Post-PCR, a harvesting reagent was used to collect the amplified products of the 48-multiplex reactions, per sample, through the sample inlets, for subsequent sequencing. Illumina sequencing adaptors and a 10bp sample specific barcode were attached through an additional 15 cycles of PCR. After the PCR products were barcoded, the PCR products from a small number of samples, as well as the water controls, were analyzed using the Agilent 2100 BioAnalyzer to ensure the expected amplicon size was obtained and that there was no contamination across the PCR reactions. They were then pooled together and purified using AMPure XP beads using a bead to amplicon ratio of 1.8:1.0. The library was quantified using the Agilent BioAnalyzer and subjected to Illumina cluster generation. One-hundred to 150bp paired-end sequencing was performed on a HiSeq 2000 or MiSeq with a 10-base indexing (barcode) read, using custom sequencing primers targeted to the CS1 and CS2 tags for both read1, read 2 (index read) and read 3, according to manufacturer’s recommendations.
Methods used for analysis of targeted sequencing data generated using TAm-Seq have been reported previously26. Reads were de-multiplexed using a known list of barcodes allowing zero mismatches. Each set of reads was aligned independently to the hg19 reference genome using BWA in the paired-end mode34. Using expected genomic positions, each set of aligned reads was separated further into its constituent amplicons. A pileup was generated for each amplicon using SAMtools v1.1737. Using a base quality and a mapping quality cut-off of 30, observed frequencies of non-reference alleles for every sequenced locus across all amplicons and barcodes were calculated. For each locus and base, the distribution of non-reference background allele frequencies/reads was modeled and the probability of obtaining the observed frequency/number of reads (or greater) was calculated. Putative substitutions were identified based on a probability cut-off (confidence margin) of 0.9995. Known SNPs obtained from the 1000 Genomes project, dbSNP version 135 and regions covering amplification primers were discarded. Any substitutions observed at >5% allele frequency in more than half of the sequenced samples were discarded. Small insertions and deletions of sequence were predicted using GATK (this tool was preferred to samtools/pindel for the higher depth of sequencing). All remaining putative mutations were annotated with sequence ontology terms to describe consequence and position relative to Ensembl gene annotations. In the final list, all nonsense or missense exonic mutations and splicing mutations with an allele frequency of 10% or greater at loci covered at least 100-fold were retained. Three genes were removed at this stage due to poor sequence coverage in all samples, TLR1, TLR7 and TLR9, leaving a total of 23 genes for further analysis (Supplementary Table 14).
In order to verify the called mutations, all nonsynonymous mutations identified from the Fluidigm Access Array sequencing were re-amplified using the CS1-/CS2-tagged primer pair targeting the region and DNA from the original sample. Where available, DNA from a matched normal sample (blood, duodenum or normal squamous epithelium) was also amplified using the identical, tagged primer pair. Amplification was performed in 5 μl reactions (0.1 Phusion® High-Fidelity DNA Polymerase (New England BioLabs), 1x Phusion Buffer, 4.5 mM MgCl2, 5% DMSO, 0.2 mM dNTPs, 1 μM forward and reverse primer, 25 ng genomic DNA. The PCR cycling conditions were as follows; 50°C for 2 minutes, 70°C for 20 minutes, 95°C for 10 minutes followed by 10 cycles of 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 1 minute, followed by 2 cycles of 95°C for 15 seconds, 80°C for 30 seconds, 60°C for 30 seconds and 72°C for 1 minute, followed by 8 cycles of 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 1 minute followed by 2 cycles of 95°C for 15 seconds, 80°C for 30 seconds, 60°C for 30 seconds and 72°C for 1 minute, and 8 cycles of 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 1 minute followed by 5 cycles of 95°C for 15 seconds, 80°C for 30 seconds, 60°C for 30 seconds and 72°C 1 minute. Following amplification, 2μl of each PCR reaction were collected and pooled in batches of 12 reactions such that only unique amplicons were contained within each pool. Thereafter, 5μl of the pooled reaction mix was added to 2μl of ExoSAP-IT® (Affymetrix). The samples were incubated at 37°C for 15 minutes followed by 80°C for 15 minute. The resulting product was diluted 1:100 in sterile water and Illumina sequencing adaptors and a 10bp barcode was attached to each pool using an additional 15 cycles of PCR (0.1 unit Phusion® High-Fidelity DNA Polymerase (New England BioLabs), 1x Phusion Buffer, 4.5mM MgCl2, 5% DMSO, 0.2mMdNTPs, 1 μM forward and reverse barcoding primers, 1μl ExoSAP-IT®-treated PCR product (1:100 dilution). Cycling conditions were as follows: heat activation at 95°C for 2 minutes, followed by 15 cycles of 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 1 minute, followed by a final elongation step of 72°C for 3 minutes.
As previously, PCR products following barcoding were first analyzed using an Agilent 2100 BioAnalyzer to ensure the expected amplicon size was obtained. They were then pooled together and purified using AMPure XP beads using a bead to amplicon ratio of 1.8 to 1.0. The library was quantified using the KAPA-Library Quantification Kit (KAPA Biosystems) on a Lightcycler® 480 (Roche), diluted to 2nM and subjected to Illumina cluster generation and sequencing on the Illumina MiSeq (150bp paired-end). Reads were de-multiplexed using a known list of barcodes allowing zero mismatches. Each set of reads was aligned independently to the hg19 reference genome using BWA in the paired-end mode34. Samtoolsmpileupv1.1737 was used to generate counts for each nucleotide at the position of the putative somatic mutation. Samples with a mutant allele frequency ≥3% and a depth of coverage ≥ 50 were considered as verified mutations. In addition, mutant allele frequency in the matched normal was required to be <1%. We additionally removed all mutations from those samples without a matched normal that were confirmed as germline in the cohort of samples with sequenced matched normal.
Processing of the capsule sponge specimens
Cytosponge™ capsules were swallowed by patients and then placed directly into preservative solution at 4°C until processed further. The samples were vortexed extensively and shaken vigorously to remove any cells from the sponge material. The preservative liquid containing the cells was centrifuged at 1000 RPM for 5 minutes to pellet the cells. The resulting pellet was re-suspended in 500 μL of plasma and thrombin (Diagnostic reagents, Oxford, UK) was then added in 10 μL increments until a clot formed. The clot was then placed in formalin for 24h prior to processing into a paraffin block. Eight times ten micrometer sections were cut and placed in a tube for DNA extraction.
DNA extraction from the Cytosponge samples
Genomic DNA was extracted from 8 × 10μm sections of the processed Cytosponge™ FFPE clot using Deparaffinization Buffer (Qiagen) and the QIAamp FFPE DNA Tissue Kit (Qiagen). The protocol was followed as described by the manufacturer with the exception that samples were incubated at 56°C for 24 hours instead of the described 1 hour, and 10 μl of extra Proteinase K was added to the samples roughly half way through the 24 hour incubation. After extraction, DNA was quantified using the Qubit™ dsDNA HS Assay Kits (Invitrogen)
Sequencing for TP53 mutations
A multiplex TP53 PCR assay was used to sequence the coding region of the TP53 gene. The multiplex consisted of 14 primer pairs26 and these 14 primer pairs were divided into two different pools. The sequences of each of the primers, the genomic region that they amplify (co-ordinates are correct for the hg19 version of the human genome) as well as which pool they were part of are described in Supplementary Tables 15 and 16.
All p53 multiplex PCRs were performed in duplicate using Q5 Hot start High-Fidelity 2X Master Mix (New England Biolabs). The coding region of the TP53 gene was first amplified using a PCR mix consisting of: 1 × Q5 master mix, 5% DMSO, final concentration of 50 nM of each primer pair and up to 70 ng of FFPE DNA extracted from Cytosponge samples. The cycling conditions for the PCR were: Initial denaturation at 95°C for 30 seconds followed by 30 cycles of 95°C for 10 seconds, 60°C for 10 seconds and 72°C for 15 seconds. A final extension at 72°C for 2 minutes was also included to ensure elongation of all PCR products.
After the first round of PCR, 2.5 ul of Pool 1 and 2.5ul of Pool 2 were pooled together. Two microlitres of IllustraExostar, 1-step (GE Healthcare UK Ltd) was added to the 5 ul of pooled PCR products and the Exostar reaction was performed (15 minutes at 37°C followed by 15 minutes at 80°C) to degrade the primers from the first reaction. One microlitre of the pooled, Exostar-treated products was then added to the barcode PCR in order to add a unique barcode as well as add the sequencing primers onto the PCR products. The barcodes used for this second PCR were taken from Forshew et al26 and the core sequence of the barcode primers can be found in Table 17. The Fluidigm barcode primers were used as they contain a sequence that binds to the CS1 and CS2 sequences present in the first p53 primers as well as the Illumina adapters. The barcode PCR mix consisted of 1 × Q5 master mix, 5% DMSO, final concentration of 400 nM of each barcode primer pair and 1 ul of undiluted, Exostar-treated DNA. The cycling conditions for the PCR were: Initial denaturation at 98°C for 30 seconds followed by 14 cycles of 98°C for 10 seconds, 60°C for 10 seconds and 72°C for 30 seconds. A final extension at 72°C for 2 minutes was also included to ensure elongation of all PCR products.
TAm-seq SNV and indel calling for detecting TP53 mutations on Cytosponge™ samples
Indels were called by selecting outliers from locus-specific distributions of background mutation rates. Candidate insertions and deletions in each sample were compared with insertion and deletion rates at the same locus in samples from every other patient, and scored by means of z-scores. Indels with a z-score greater than or equal to 10, at least 200x coverage and at least 5 supporting reads were retained.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The whole genome sequencing of esophageal adenocarcinoma is part of the International Cancer Genome Consortium through the Esophageal Cancer Clinical And Molecular Stratification (OCCAMS) consortium and is funded by Cancer Research UK (CRUK). We thank the ICGC members for their input on verification standards as part of the bench-marking exercise. Cytosponge™ samples were collected as part of the CRUK-funded BEST2 trial. We thank Prof M. Griffin, Dr L. Lovat and Prof K. Ragunath for their contribution to Cytosponge™ collection. The MRC developed the Cytosponge™ and also funded the laboratory work through a program grant to RCF. JMJW was funded by a Wellcome Trust Translational Medicine and Therapeutics grant. RCF and CC are supported with additional clinical research infrastructure funding from the NHS National Institute for Health Research, Experimental Cancer Medicine Centre Network and the National Institute for Health Research Cambridge Biomedical Research Centre. Bioinformatics work was also supported by a CRUK program grant to ST.
We thank the Genomics Core at the CRUK Cambridge Institute for their help with processing some of the Access Array experiments as well as for running the targeted re-sequencing experiments. We thank the IT department at the CRUK Cambridge Institute for their support. We thank Francesco Marass for assistance with data analysis. We thank the Human Research Tissue Bank, supported by the NIHR Cambridge Biomedical Research Centre, from Addenbrooke’s Hospital as well as the University Hospital of Southampton Trust and the Southampton Experimental Cancer Medicine Centre. We are grateful to all patients who provided written consent for participation in this study and the staff at Addenbrooke’s and the University of Southampton Tissue Bank.
THE OCCAMS CONSORTIUM MEMBERS
Stephen J Hayes10,16, Ang Yeng10, Anne-Marie Lydon10, Soney Dharmaprasad10, Sandra Greer11, Shaun Preston12, Sarah Oakes12, Vicki Save13, Simon Paterson-Brown13, Olga Tucker14,17, Derek Alderson14, Philippe Taniere14, Jamie Kelly15, James Byrne15, Donna Sharland15, Nina Holling15, Lisa Boulter15, Fergus Noble15, Bernard Stacey15, Charles Crichton17, Hugh Barr18, Neil Shepherd18, L. Max Almond18, Oliver Old18, Jesper Lagergren19,28,29, James Gossage19,28,29, Andrew Davies19, 28,29, Robert Mason19,28,29, Fuju Chang19,28, Janine Zylstra19,28, Grant Sanders20, Tim Wheatley20, Richard Berrisford20, Tim Bracey20, Catherine Harden20, David Bunting20, Tom Roques21, Jenny Nobes21, Suat Loo21, Mike Lewis21, Ed Cheong21, Oliver Priest21, Simon L Parsons22, Irshad Soomro22, Philip Kaye22, John Saunders22, Vincent Pang22, Neil T Welch22, James A Catton22, John P Duffy22, Krish Ragunath22, Laurence Lovat23, Rehan Haidry23, Haroon Miah23, Sarah Kerr23, Victor Eneh23, Rommel Butawan23, Laszlo Igali24, Hugo Ford25, David Gilligan25, Peter Safranek25, Andy Hindmarsh25, Vijayendran Sudjendran25, Andy Metz25, Nick Carroll25, Michael Scott26, Alison Cluroe3, Ahmad Miremadi3, Betania Mahler-Araujo3, Olga Knight1, Barbara Nutzinger1, Chris Peters19, Zarah Abdullahi1, Irene Debriram-Beecham1, Shalini Malhotra3, Jason Crawte1, Shona MacRae1, Ayesha Noorani1, Rachael Fels Elliott1, Xiaodun Li1, Lawrence Bower2, Achilleas Achilleos2, Jan Bornschein1, Sebastian Zeki1, Hamza Chettouh1, Maria Secrier2, Nadeera de Silva1, Eleanor Gregson1, Tsun-Po Yang1 and J. Robert O’Neil27.
10. Salford Royal NHS Foundation Trust, Salford, UK
11. Wigan and Leigh NHS Foundation Trust, Wigan, Manchester, UK
12. Royal Surrey County Hospital NHS Foundation Trust, Guildford, UK
13. Edinburgh Royal Infirmary, Edinburgh, UK
14. University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK
15. Southampton General Hospital, Southampton, UK
16. Faculty of Medical and Human Sciences, University of Manchester, UK
17. Department of Computer Science,University of Oxford,UK
18. Gloucester Royal Hospital, Gloucester, UK
19. St Thomas’s Hospital, London, UK
20. Plymouth Hospitals N H S Trust, Plymouth, UK
21. Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, UK
22. Nottingham University Hospitals NHS Trust, Nottingham, UK
23. University College London, London, UK
24. Norfolk and Waveney Cellular Pathology Network, Norwich, UK
25. Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK
26. Department of Pathology, Wythenshawe Hospital, Manchester, UK
27. Edinburgh University, Edinburgh, UK
28. King’s College London, London, UK
29. Karolinska Institutet, Stockholm, Swedan
Footnotes
URLS: http://dcc.icgc.org/
DATABASE ACCESSION NUMBERS: Whole genome sequencing data is available at the EGA (Accession number: EGAD00001000704), however data access approval is required via the ICGC data portal.
COMPETING FINANCIAL INTERESTS: RCF developed the Cytosponge technology with MRC-Technology which provided devices for research. The technology has recently been licensed to Covidien, RCF has no direct financial relationship with Covidien.
REFERENCES
- 1.Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17:297–303. doi: 10.1038/nm.2323. [DOI] [PubMed] [Google Scholar]
- 2.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 6.Goh XY, et al. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut. 2011;60:1317–26. doi: 10.1136/gut.2010.234179. [DOI] [PubMed] [Google Scholar]
- 7.Quante M, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21:36–51. doi: 10.1016/j.ccr.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varghese S, Lao-Sirieix P, Fitzgerald RC. Identification and clinical implementation of biomarkers for Barrett’s esophagus. Gastroenterology. 2012;142:435–441 e2. doi: 10.1053/j.gastro.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Dulak AM, et al. Gastrointestinal Adenocarcinomas of the Esophagus, Stomach, and Colon Exhibit Distinct Patterns of Genome Instability and Oncogenesis. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dulak AM, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrawal N, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012 doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corley DA, et al. Impact of Endoscopic Surveillance on Mortality From Barrett’s Esophagus-Associated Esophageal Adenocarcinomas. Gastroenterology. 2013;145:312–319 e1. doi: 10.1053/j.gastro.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaheen NJ, Hur C. Garlic, Silver Bullets, and Surveillance Upper Endoscopy for Barrett’s Esophagus. Gastroenterology. 2013;145:273–6. doi: 10.1053/j.gastro.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 15.Hayes DF, et al. Breaking a vicious cycle. Sci Transl Med. 2013;5:196cm6. doi: 10.1126/scitranslmed.3005950. [DOI] [PubMed] [Google Scholar]
- 16.Nik-Zainal S, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 18.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43:964–8. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Streppel MM, et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene. 2013 doi: 10.1038/onc.2012.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curvers WL, et al. Low-grade dysplasia in Barrett’s esophagus: overdiagnosed and underestimated. Am J Gastroenterol. 2010;105:1523–30. doi: 10.1038/ajg.2010.171. [DOI] [PubMed] [Google Scholar]
- 21.Wang K, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010;10:87–101. doi: 10.1038/nrc2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadri SR, et al. Acceptability and accuracy of a non-endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ. 2010;341:c4372. doi: 10.1136/bmj.c4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lao-Sirieix P, et al. Non-endoscopic screening biomarkers for Barrett’s oesophagus: from microarray analysis to the clinic. Gut. 2009;58:1451–9. doi: 10.1136/gut.2009.180281. [DOI] [PubMed] [Google Scholar]
- 26.Forshew T, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 27.Dawson SJ, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 28.Theisen J, et al. Preoperative chemotherapy unmasks underlying Barrett’s mucosa in patients with adenocarcinoma of the distal esophagus. Surg Endosc. 2002;16:671–3. doi: 10.1007/s00464-001-8307-3. [DOI] [PubMed] [Google Scholar]
- 29.Bhat S, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst. 2011;103:1049–57. doi: 10.1093/jnci/djr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 31.Mutter GL, et al. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001;61:4311–4. [PubMed] [Google Scholar]
- 32.Kinde I, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5:167ra4. doi: 10.1126/scitranslmed.3004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maley CC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson DE, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah SP, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.