

Aggregation of intracellular amyloid-β (Aβ) is an early event in Alzheimer's disease. Here Aβ42 fused to GFP was expressed in each mutant of a Saccharomyces cerevisiae genome-wide deletion library to identify cellular processes affecting intracellular Aβ42 aggregation by assessing fluorescence of the Aβ42-GFP fusion.
Abstract
Amyloid-β (Aβ)–containing plaques are a major neuropathological feature of Alzheimer's disease (AD). The two major isoforms of Aβ peptide associated with AD are Aβ40 and Aβ42, of which the latter is highly prone to aggregation. Increased presence and aggregation of intracellular Aβ42 peptides is an early event in AD progression. Improved understanding of cellular processes affecting Aβ42 aggregation may have implications for development of therapeutic strategies. Aβ42 fused to green fluorescent protein (Aβ42-GFP) was expressed in ∼4600 mutants of a Saccharomyces cerevisiae genome-wide deletion library to identify proteins and cellular processes affecting intracellular Aβ42 aggregation by assessing the fluorescence of Aβ42-GFP. This screening identified 110 mutants exhibiting intense Aβ42-GFP–associated fluorescence. Four major cellular processes were overrepresented in the data set, including phospholipid homeostasis. Disruption of phosphatidylcholine, phosphatidylserine, and/or phosphatidylethanolamine metabolism had a major effect on intracellular Aβ42 aggregation and localization. Confocal microscopy indicated that Aβ42-GFP localization in the phospholipid mutants was juxtaposed to the nucleus, most likely associated with the endoplasmic reticulum (ER)/ER membrane. These data provide a genome-wide indication of cellular processes that affect intracellular Aβ42-GFP aggregation and may have important implications for understanding cellular mechanisms affecting intracellular Aβ42 aggregation and AD disease progression.
INTRODUCTION
Amyloid-β (Aβ) plaques are a neuropathological feature of Alzheimer's disease (AD). These extracellular plaques are primarily composed of Aβ peptide aggregates generated via amyloidogenic processing of the amyloid precursor protein (APP). According to the amyloid cascade hypothesis, the Aβ peptide may play a role in AD pathology through oligomerization of the peptide. The oligomers may be directly neurotoxic or may mediate toxicity by induction of stress and hyperphosphorylation of protein tau, leading to tau aggregation into neurofibrillary tangles, cell loss, vascular damage, and dementia (Glenner and Wong, 1984; Masters et al., 1985; Selkoe, 1991; Hardy and Higgins, 1992). Protein tau is a microtubule-associated protein that influences assembly and stabilization of microtubules. The tau protein is the main component of neurofibrillary threads and tangles (NFTs), and there is evidence supporting a key role of tau in the pathophysiology of AD. It remains to be elucidated whether tau is a bystander of amyloid toxicity or a primary mediator neurodegeneration in AD. Two major Aβ-peptide isoforms, Aβ40 and Aβ42, are generated by the amyloidogenic processing of APP, of which the latter is more hydrophobic, highly prone to aggregation and fibril formation, and more neurotoxic (Jarrett et al., 1993). Aβ42 is the predominant form of Aβ found in neurons (Gouras et al., 2000, 2005) and in the extracellular plaques of AD brains (Younkin, 1998).
It is not clear whether extracellular aggregates (e.g., plaques) of Aβ lead to a protective, inert, or pathogenic mechanism. However, soluble oligomeric forms of Aβ, rather than monomeric or fibrillar forms, are the most neurotoxic species (Gouras et al., 2005; Lesne and Kotilinek, 2005; Lesne et al., 2006), and an increase of soluble oligomeric forms of Aβ42 may be an early event in AD progression (Gouras et al., 2005). The role of intracellular Aβ has received increased attention (Haass and Selkoe, 2007; LaFerla et al., 2007). Aβ42 was found in multivesicular bodies (MVBs) of neuronal cells, where it was implicated in synaptic pathology (Takahashi et al., 2002). The peptide localizes to the outer membranes of MVBs (Takahashi et al., 2002; Langui et al., 2004) and is most often located in the perinuclear region (Langui et al., 2004). Aβ accumulation also directly inhibits the proteasome (Oh et al., 2005; Almeida et al., 2006), indicating that soluble Aβ may be responsible for induction of toxicity, which may increase with impaired proteasomal function. Gradual accumulation of Aβ in mitochondria (Manczak et al., 2006) has also been associated with diminished activity of electron transport chain complexes III and IV and reduced rates of oxygen utilization (Caspersen et al., 2005). Intracellular accumulation of Aβ precedes extracellular plaque formation, and these findings support the view that it may be an early event in the progression of AD (Gouras et al., 2005). The mechanism(s) contributing to the intracellular aggregation and localization of the Aβ42 peptide in patients remains unclear. Because preventing Aβ aggregation and/or low-order oligomerization has been proposed as a potential therapeutic method (Gouras et al., 2005), improved understanding of cellular processes involved in Aβ42 aggregation may help understand AD disease progression and lead to development of therapeutic strategies.
The budding yeast Saccharomyces cerevisiae is an important model organism for understanding many aspects of eukaryotic molecular biology. S. cerevisiae has been exploited to study proteins implicated in neurodegenerative disorders including Huntington's disease (Willingham et al., 2003; Giorgini et al., 2005) and Parkinson's disease (Zhang et al., 1994; Outeiro and Lindquist, 2003; Flower et al., 2005). Yeast model systems have been exploited to study toxicity, aggregation, and localization of Aβ or as facile systems for identification of compounds influencing Aβ oligomerization (Zhang et al., 1994, 1997; Komano et al., 1998; Caine et al., 2007; Macreadie et al., 2008; Winderickx et al., 2008; Treusch et al., 2011). As indicated, protein tau has also been strongly correlated with several neuropathies, including AD. Studies of wild-type human tau in yeast have shown that the model system recapitulates several key pathological features of tau, including tau hyperphosphorylation, attainment of pathological confirmations of tau, and tau aggregation (Vandebroek et al., 2005, 2006). Disruption of yeast Pho85p or Mds1p, which are orthologues of human glycogen synthase kinase-3B and cdk5, influences formation of pathological phosphoepitopes of tau in yeast and their binding affinity for microtubules (Vandebroek et al., 2006). Furthermore, deletion of PHO85 enhances phosphorylation of the S409 residue in wild-type tau but also in tau variants associated with frontotemporal dementia with parkinsonism (Vanhelmont et al., 2010).
Green fluorescent protein (GFP)-fusion protein folding has been exploited to study the kinetics of Aβ aggregation in Escherichia coli. An aggregation reporter assay based on fluorescence of a fusion between Aβ42 and GFP has been developed (Waldo et al., 1999). Aggregation of the GFP-fusion protein before the folding of GFP quenches its fluorescence. Expression of wild-type Aβ42 fused to GFP led to formation of insoluble aggregates in which GFP was inactive (Wurth et al., 2002). Replacement of Aβ42 in the fusion protein with the less-aggregation-prone peptide Aβ40 led to increased GFP-associated fluorescence. This approach was exploited to identify variants of Aβ42 that affect aggregation of Aβ (Wurth et al., 2002; Kim and Hecht, 2005). Fluorescence intensity of Aβ-GFP fusions was inversely correlated with the aggregation propensity of the Aβ moiety, demonstrating the efficacy of using Aβ-GFP fusion-based approaches to identify factors affecting Aβ aggregation. Yeast cells expressing Aβ exhibited lower growth yield and a heat shock response, indicating that Aβ fusions cause stress in cells (Caine et al., 2007). Yeast can therefore serve as a model system to screen for modifiers of intracellular Aβ aggregation, which has relevance in understanding the role of Aβ in the death of neuronal cells. Here we exploit an aggregation reporter assay by expressing Aβ42 fused to GFP (Aβ42-GFP) in each mutant of the S. cerevisiae genome-wide deletion library of nonessential genes (Winzeler et al., 1999) to identify the cellular processes and metabolites that affect intracellular Aβ42 aggregation.
RESULTS
The Aβ42-GFP fusion protein expressed in wild-type S. cerevisiae is not fluorescent, and the less amyloidogenic Aβ42-GFP variants exhibit increased fluorescence
In E. coli, the fluorescence intensity exhibited by Aβ-GFP fusions is inversely correlated with the aggregation propensity of the Aβ moiety (Wurth et al., 2002; Kim and Hecht, 2005). Mutation of Aβ residues 41 and 42 (I41E/A42P) generated a variant (AβEP-GFP) that was less aggregation prone and exhibited higher fluorescence than Aβ42-GFP or Aβ40-GFP (Wurth et al., 2002). Before commencing genome-wide screening in S. cerevisiae, it was important to determine whether the correlations demonstrated in E. coli (Wurth et al., 2002; Kim and Hecht, 2005) between fluorescence and aggregation propensity of Aβ-GFP fusions could be recapitulated in S. cerevisiae cells.
Wild-type Aβ42, Aβ40, or AβEP (Aβ-I41E/A42P) sequences fused to the N-terminus of GFP were expressed in the cytosol of wild-type S. cerevisiae cells. Microscopic analysis (Figure 1A) indicated that the proportion of fluorescent cells and relative fluorescence intensity per cell decreased in the order AβEP-GFP > Aβ40-GFP > Aβ42-GFP, which inversely correlates with the aggregation propensity of the Aβ moiety of the fusion protein. In comparison to GFP expression, which yielded fluorescence in ∼85% of wild-type cells, expression of AβEP-GFP, Aβ40-GFP, and Aβ42-GFP led to fluorescence in ∼60, 50, and 5% of cells, respectively (Figure 1C). Expression of AβEP-GFP led to diffuse fluorescence distributed throughout the cytosol, whereas expression of Aβ42-GFP was associated with trace levels of cytosolic fluorescence and the presence of one to three fluorescent puncta per cell. Relative to Aβ42-GFP, expression of Aβ40-GFP led to an increase in the cytosolic fluorescence intensity and one to three puncta per cell.
FIGURE 1.

Fluorescence and corresponding light microscope images of wild-type cells expressing a GFP control vector, Aβ42-GFP, Aβ40-GFP, or AβEP-GFP. (A) Wild-type cells expressing GFP, Aβ42-GFP, Aβ40-GFP, or AβEP-GFP were induced in SC-galactose medium, and fluorescence was analyzed in exponential phase (OD600 of 1.5). Bar, 5 μm. (B) Western blot analysis (using 6E10 antibody) and quantification of relative band intensity of soluble (supernatant) and insoluble (pellet) cell extracts from wild-type cells expressing Aβ42-GFP, Aβ40-GFP, and AβEP-GFP grown to exponential phase (OD600 of 1.5). Aβ-GFP bands, ∼31 kDa. (C) Proportion of fluorescent wild-type cells expressing Aβ42-GFP, Aβ40-GFP, AβEP-GFP, or GFP (correlating with A). Nine hundred cells were counted per sample, and data shown are averages of three independent experiments. *p < 0.01.
To assess whether reduced fluorescence of Aβ42-GFP relative to Aβ40-GFP and AβEP-GFP was due to decreased levels of soluble (nonaggregated) Aβ-GFP, cells expressing each of the foregoing Aβ-GFP fusion constructs were grown to exponential phase in SC-galactose (inducing) medium and lysed, and soluble and insoluble proteins were fractionated by ultracentrifugation. A single band of approximately 31-kDa molecular mass reacting with an anti-Aβ antibody was detected in the insoluble pellet fraction for all strains (Figure 1B), corresponding to the predicted size of the Aβ-GFP fusion protein. A band of identical molecular mass was also observed in the soluble supernatant fraction of cells expressing the Aβ40-GFP and AβEP-GFP fractions. In contrast, no band could be detected in the supernatant of cells expressing Aβ42-GFP. This result indicates that the very low levels of fluorescence in Aβ42-GFP cells were not due to lack of expression or complete proteolysis of the fusion protein. In addition, fusion protein levels in the soluble supernatant fraction were positively correlated with fluorescence, validating use of Aβ-GFP fluorescence as measure of the propensity of the fusion protein to form insoluble aggregates.
It should also be noted that a significant difference in the level of Aβ42-GFP relative to Aβ40-GFP and AβEP-GFP was observed in the insoluble fraction. Because all the Aβ-GFP fusions used were expressed from otherwise identical plasmids/promoters, reduced levels of Aβ42-GFP in cells likely stemmed from increased degradation of the insoluble aggregates of Aβ42-GFP in cells relative to Aβ40-GFP and AβEP-GFP. This hypothesis is consistent with other findings in yeast using Aβ fused to fluorescent reporters (Hamada et al., 2008; Morell et al., 2011; Villar-Pique and Ventura, 2013). The length of the linker sequence between an aggregation-prone domain and GFP influences the degree to which aggregation or misfolding inhibits the appearance of fluorescence. Fusion proteins containing longer linker sequences or where the aggregation-prone region of a multidomain protein is distal to GFP may display robust fluorescence despite forming aggregates (Hamada et al., 2008). Indeed, D'Angelo et al. (2013) introduced an expanded glycine-alanine linker into their Aβ-GFP expression construct specifically to ensure that Aβ-GFP fluorescence could be observed in the endoplasmic reticulum, after failing to detect a GFP signal from a shorter linker form. Because our aim was to create a screening platform for which fluorescence was only observed under conditions in which Aβ solubility is increased, we used a very short (four amino acids) linker region between the C-terminus of Aβ and the N-terminus of GFP.
Expression of each of the respective Aβ-GFP fusions (and GFP alone) was not associated with any discernible change in growth rate of wild-type cells (unpublished data). These findings are consistent with previous findings in which amyloid was also expressed cytosolically. In yeast, cytosolic expression of wild-type Aβ42-GFP, as well as of a comprehensive set of Aβ peptide variants (fused to GFP), was not associated with any observable cytotoxicity (Morell et al., 2011; Villar-Pique and Ventura, 2013). Caine et al. (2007) reported a minor reduction (5%) in growth of yeast cells (at ∼10 h) expressing cytosolically localized Aβ42-GFP. In contrast, substantial toxicity was reported when Aβ was expressed in the endoplasmic reticulum (Treusch et al., 2011; D'Angelo et al., 2013).
Aβ42 seeds formation of punctate aggregates of Aβ40
Aβ toxicity has been shown to correlate with the presence of fibrils or β-sheet structures (Howlett et al., 1995; Simmons et al., 1994; Seilheimer et al., 1997). However, gaps remain in understanding the mechanisms by which Aβ aggregation mediates neuronal death. Aβ aggregation proceeds by a multistep, nucleation-dependent process (Jarrett and Lansbury, 1993). Formation of nucleation seeds is rate limiting, and in the absence of preformed seed fibrils, there is a significant lag period for the formation of Aβ fibrils, followed by a rapid fibril elongation phase once seed fibrils have been generated. The lag time for fibril formation can be dramatically shortened by adding preformed fibril seeds to Aβ monomer (Jarrett and Lansbury, 1993). The rate of Aβ fibril formation is controlled by both fibril seed and monomer concentrations (Naiki and Nakakuki, 1996). To examine whether the more-aggregation-prone AB42-GFP affected fluorescence produced by the less-aggregation-prone AB40-GFP form, we undertook parallel expression in wild-type cells of Aβ42 plus Aβ40, Aβ42 plus AβEP, or Aβ40 plus AβEP.
Coinduction of Aβ42-GFP and Aβ40-GFP gave rise to more fluorescent cells (∼22%) than did expression of the Aβ42-GFP alone (5%) but significantly fewer than cells expressing Aβ40-GFP alone (∼40%; Figure 2). Cells expressing both Aβ42-GFP and Aβ40-GFP exhibited trace cytosolic fluorescence, with intense large puncta, and in some cells there were elongated structures (Figure 2). Coinduction of Aβ42-GFP and AβEP-GFP in wild-type cells also gave rise to more fluorescent cells (∼28%; exhibiting cytosolic fluorescence with small, intense puncta) compared with those expressing Aβ42-GFP alone but significantly fewer than cells expressing AβEP-GFP alone (∼70%). Coinduction of Aβ40-GFP and AβEP-GFP in wild-type cells gave rise to intense cytosolic fluorescent cells (∼60%) comparable to wild-type cells expressing AβEP alone, and 30% of the fluorescent cells contained small puncta (Figure 2). The increased presence of puncta and lower levels of cytosolic fluorescence in wild-type cells coexpressing either Aβ42-GFP and Aβ40-GFP or Aβ42-GFP and AβEP-GFP indicated that the more-aggregation-prone Aβ42-GFP can act as a seed for aggregation. Preformed Aβ42-GFP aggregates formed in the cytosol may therefore accelerate nucleation and act as seeds for further formation of intracellular aggregates and fibrils.
FIGURE 2.
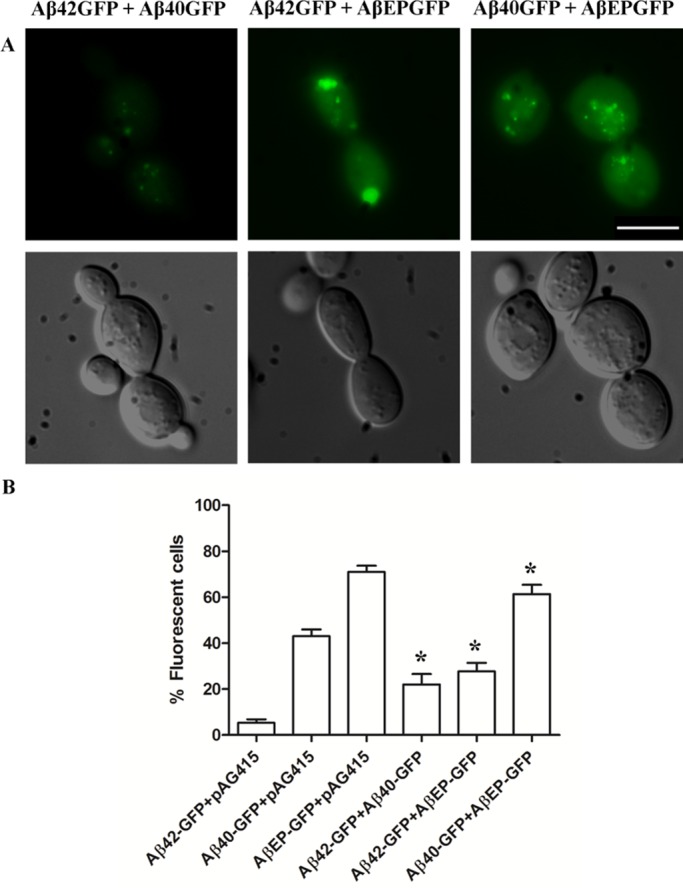
(A) Fluorescence microscope images of wild-type cells coexpressing Aβ42-GFP and Aβ40-GFP; Aβ42-GFP and AβEP-GFP; or Aβ40-GFP and AβEP-GFP. Wild-type cells expressing these constructs were induced in SC-galactose medium, and fluorescence was analyzed at OD600 of 1.5. Bar, 5 μm. (B) Proportion of fluorescent wild-type cells expressing GFP, Aβ42-GFP, Aβ40-GFP, or AβEP-GFP. Nine hundred cells were counted per sample, and data shown are averages of three independent experiments. *p < 0.01.
Together these data highlight the inverse correlation between the relative aggregation propensity of the Aβ moiety fused to GFP and the level of fluorescence intensity of the Aβ-GFP fusion protein in yeast cells.
Cellular processes affecting Aβ42-GFP–associated fluorescence
To identify processes affecting intracellular Aβ42-GFP aggregation, individual homozygous diploid strains of the genome-wide S. cerevisiae deletion library (Winzeler et al., 1999) were transformed with the Aβ42-GFP construct and screened using fluorescence microscopy to identify mutants exhibiting increased Aβ42-GFP–associated fluorescence and/or altered localization relative to wild-type cells. Expression of the Aβ42-GFP fusion in each strain was induced by growth in SC galactose medium (–Ura), and Aβ42-GFP–associated fluorescence was analyzed 12–18 h postinduction. Rescreening, in duplicate, of mutants that exhibited altered fluorescence during the primary screen led to identification of 344 mutants that exhibited fluorescence reproducibly different from that of the wild type. These mutants fell into two broad sets according to the intensity of fluorescence and/or percentage of fluorescent cells. The first set of 110 mutants contained those exhibiting strong fluorescence in ≥15% of cells (Table 1). The other 234 mutants exhibited moderate to weak fluorescence in 5–10% of cells (Supplemental Table S3). Of note, 50 of the 110 S. cerevisiae genes in Table 1 have orthologues in humans.
TABLE 1:
S. cerevisiae genes whose deletion led to a strong increase in Aβ42-GFP–associated fluorescence, together with the respective localization of fluorescence.
| Open reading frame/gene name | Aβ42-GFP localization pattern | Respiratory deficiency | Human orthologue |
|---|---|---|---|
| Chromatin remodeling/histone exchange | |||
| CHD1 | Cytosolic | No | CHD2 |
| HIR1 | Single small punctate | No | HIRA |
| HTA2 | Single small punctate | No | H2AFX |
| SWC5 | Single small punctate | No | CFDP1 |
| SWR1 | Multiple puncta | No | |
| VPS71 | Multiple small puncta | No | |
| VPS72 | Cytosolic with single large punctate | No | |
| YDL041W | Multiple small puncta | No | |
| Lipid metabolism/transport | |||
| CHO2 | Cytosolic with one or two small puncta | No | |
| DET1 | Cytosolic | No | |
| INO2 | ER-associated | No | |
| INO4 | Punctate | No | |
| IPK1 | Cytosolic with single large punctate | No | |
| OPI3 | ER-associated | No | PEMT |
| PDX3 | Cytosolic with punctate and nuclear | Yes | PNPO |
| PSD1 | ER-associated | No | PISD |
| SCS2 | Single large punctate | No | VAPA |
| UME6 | Single small punctate | No | |
| YER119C-A | Single small punctate | No | |
| Mitochondrial functions | |||
| ACO1 | Cytosolic with one or two small puncta | Yes | ACO2 |
| ACO2 | Cytosolic | No | |
| AIM4 | Single small punctate | Yes | |
| ATP11 | Cytosolic with one or two puncta | Yes | ATPAF1 |
| CBP3 | Cytosolic with puncta | Yes | UQCC |
| CIT1 | Cytosolic with one or two small puncta | Yes | |
| CIT3 | Cytosolic with one or two small puncta | No | |
| COX16 | Cytosolic | Yes | |
| COX20 | Cytosolic | Yes | |
| CYM1 | Single small punctate | No | PITRM1 |
| FUM1 | Cytosolic with puncta | Yes | FH |
| HAP2 | Cytosolic | Yes | NFYA |
| HAP3 | Single small punctate | Yes | NFYB |
| IDH1 | Cytosolic with small puncta | Yes | IDH3B |
| IDH2 | Cytosolic with small puncta | Yes | IDH3A |
| IDP1 | Cytosolic with small puncta | No | IDH1 |
| KGD1 | Cytosolic | Yes | OGDH |
| KGD2 | Cytosolic | Yes | DLST |
| LPD1 | Cytosolic | Yes | DLD |
| LSC1 | Cytosolic | Yes | SUCLG1 |
| LSC2 | Cytosolic | No | |
| MDH1 | Cytosolic | No | MDH2 |
| MIC14 | Cytosolic | No | |
| MRPL35 | Cytosolic | Yes | MRPL35 |
| MRPL7 | Cytosolic with small puncta | Yes | |
| PET112 | Single small punctate | Yes | PET112L |
| PET117 | Cytosolic | Yes | |
| PYC1 | Cytosolic with small puncta | No | PC |
| PYC2 | Cytosolic | No | PC |
| RIM1 | Cytosolic | Yes | |
| RRG8 | Large punctate and nuclear-diffused | Yes | |
| RSM18 | Single small punctate | Yes | |
| RSM7 | Cytosolic | Yes | |
| SDH1 | Cytosolic | No | SDHA |
| SDH2 | Cytosolic | Yes | SDHB |
| SDH4 | Cytosolic with puncta | Yes | SDHD |
| STF2 | Cytosolic with single large punctate | No | |
| TUF1 | Single small punctate | Yes | TUFM |
| YDR230W | Cytosolic | Yes | |
| Gene expression/regulation | |||
| CTK1 | One or two small puncta | No | |
| DEG1 | Single large punctate | No | PUS3 |
| ELC1 | Single small punctate | No | |
| GAT1 | Single small punctate | No | |
| GDT1 | Single small punctate | Yes | TMEM165 |
| HFI1 | Cytosolic | Yes | |
| LSM7 | Multiple small puncta | Yes | LSM7 |
| MED1 | Cytosolic with one or two small puncta | No | MED1 |
| MOT2 | Single small punctate | Yes | |
| NCL1 | Single small punctate | No | NSUN2 |
| SNT309 | Cytosolic with one or two small puncta | Yes | |
| SRB8 | One or two small puncta | Yes | MED12L |
| SSN2 | Cytosolic with single large punctate | No | MED13L |
| SSN3 | Multiple small puncta | Yes | CDK8 |
| SYC1 | Single small punctate | No | |
| TIF4631 | Multiple small puncta | Yes | EIF4G1 |
| Mitotic cell cycle | |||
| CTS1 | Cytosolic | No | |
| DCC1 | Single small punctate | Yes | DSCC1 |
| POG1 | Single small punctate | No | |
| SWI4 | Cytosolic with single small punctate | No | |
| Methionine metabolism | |||
| MET16 | Single small punctate | No | |
| MET8 | Single small punctate | No | |
| MXR1 | Single small punctate | No | MSRA |
| Purine metabolism | |||
| ADE12 | Single small punctate | No | ADSSL1 |
| ADK1 | Single small punctate | No | AK2 |
| Spindle pole body | |||
| BFA1 | Single small punctate | No | |
| BIM1 | Single small punctate | No | MAPRE1 |
| Ubiquitin/proteasome | |||
| SAN1 | Cytosolic | No | |
| SHP1 | Cytosolic with puncta | Yes | NSFL1C |
| UBR1 | Cytosol with large punctate | No | UBR1 |
| Bud-site selection | |||
| BUD23 | Cytosolic with punctate | Yes | WBSCR22 |
| BUD31 | Cytosolic with puncta | Yes | BUD31 |
| MAP kinase activity | |||
| PBS2 | Single small punctate | No | MAP2K4 |
| SLG1 | Single small punctate | No | |
| SOK1 | Multiple small puncta | No | |
| Others/unknown | |||
| APJ1 | Single small punctate | No | |
| ASM4 | Cytosolic with large punctate | No | |
| EMI2 | Cytosolic with small puncta | No | |
| GTT3 | Single small punctate | No | |
| ICE2 | ER-associated | No | |
| ICY2 | Single small punctate | No | |
| PAU11 | Cytosolic | No | |
| PHM6 | Cytosolic puncta | No | |
| RAD61 | Cytosolic punctate | No | |
| RIB1 | Multiple puncta | Yes | |
| RKM4 | Single small punctate | No | |
| SNX41 | Cytosolic with one or two small puncta | No | |
| YDL242W | Cytosolic | No | |
| YDR015C | Cytosolic | No | |
| YEL008W | Single small punctate | No | |
| YIM2 | Cytosolic | No | |
| YOR364W | Single small Punctate | No | |
Because many S. cerevisiae genes have human orthologues, identification of these may help to identify cellular processes in humans that play a role in Aβ42 aggregation. Of the 110 S. cerevisiae genes in Table 1, 50 have human orthologues identified using the National Center for Biotechnology Information database, HomoloGene. These genes may provide a point for more targeted studies in mammalian AD model systems.
Five distinct Aβ42-GFP localization patterns were observed among the 110 mutants identified by the genome-wide screen (Figure 3): single punctate (29%), multiple puncta (9%), cytosolic-diffuse (22%), distinct arc-shaped (5%), and a combination of punctate and cytosolic-diffuse fluorescence (35%). The percentages are based on the number of mutants in each group. An example of each of these localization patterns is given in Figure 3.
FIGURE 3.

Fluorescence microscope images of representative mutants exhibiting various Aβ42-GFP localization patterns. Strains expressing Aβ42EGFP were induced in SC-galactose medium, and Aβ42-GFP–associated fluorescence was analyzed between 12 and 18 h postinduction (OD600 of ∼1.5). The Aβ42-GFP–associated localization patterns were classified as (A) fluorescent punctate, (B) multiple fluorescent puncta, (C) cytosolic-diffuse fluorescence, (D) arc-shaped perinuclear fluorescence, and (E) combination of puncta and cytosolic fluorescence. Arrows indicate the specific type of localization in each of the mutants. Bar, 5 μm. (F) Graphic representation of cellular processes identified affecting Aβ42-GFP–associated fluorescence in S. cerevisiae. Percentages reflect the number of mutants identified in a particular cellular process relative to the total number of mutants identified by the genome-wide screen.
Functional categories overrepresented in the group of 110 genes indicated the main cellular processes likely to affect intracellular Aβ42 aggregation. Manual inspection of the list identified processes including phospholipid metabolism, mitochondrial function, and chromatin remodeling. In addition, histone exchange (p = 2.12 × 10−5), DNA-dependent transcription (p = 0.0009), chromatin remodeling (p = 0.009) and modification (p = 0.001), and tricarboxylic acid (TCA) cycle (p > 1 × 10−14) functions were significantly overrepresented using SGD FunCat GO Term Finder. Analysis using Munich Information Centre for Protein Sequences (MIPS) FunCatDB yielded a very similar set of functional categories but also identified phosphatidylcholine (PC) biosynthesis/phospholipid metabolism (p = 0.001), regulation of lipid, fatty acid, and isoprenoid metabolism (p = 0.008), sulfate assimilation (p = 0.007), and transcriptional control (p = 0.0005) to be overrepresented (Figure 3).
Of the 110 mutants identified in the genome-wide screen, 35% (39 mutants from 110) were annotated in the Saccharomyces Genome Database as exhibiting disrupted mitochondrial respiratory function. Analysis of process ontology identified acetyl-CoA catabolism and enzymes of the TCA cycle, including isocitrate dehydrogenase (Idp1p), α-ketoglutarate dehydrogenase (Kgd2p), succinate dehydrogenase (Sdh4p), aconitase (Aco1p), and fumarase (Fum1p) to be significantly overrepresented (p = 7.71 × 10−22). Fluorescent Aβ42-GFP in TCA cycle mutants was cytosolic-diffuse, or cytosolic-diffuse with single to multiple small puncta (Figure 4), which did not appear to occur in a distinct structure in the cell. Mitochondrial mutants that had strongly affected Aβ42-GFP–associated fluorescence were mainly defective in metabolism of pyruvate to oxaloacetate and disruption of the TCA cycle. It was previously demonstrated that 341 nuclear genes affect respiratory growth capacity and/or mitochondrial morphology, including numerous genes encoding subunits of the mitochondrial electron transport chain complexes and factors required for their assembly, enzymes of the TCA cycle, mitochondrial ribosome function and maintenance, and inheritance of the mitochondrial genome (Dimmer et al., 2002). In the event that some mutants may have been missed in the initial screen, a comprehensive rescreening of representative mutants affected in mitochondrial functions was undertaken and Aβ42-GFP–associated fluorescence reassessed in each strain. These analyses not only validated the 39 mitochondrial mutants identified in the initial screen as having strongly affected Aβ42-GFP–associated fluorescence, but they also confirmed that loss of many other genes involved in mitochondrial functions, including respiratory energy production, mitochondrial ribosome function, and mitochondrial genome maintenance, did not affect Aβ42-GFP fluorescence in the same manner. Of those ∼30 non-TCA cycle mutants that were affected in Aβ42-GFP fluorescence, the effect was comparatively weak in terms of fluorescence intensity and the proportion of affected cells in a given population (Supplemental Table S3).
FIGURE 4.

Fluorescence microscope images of mutants expressing Aβ42-GFP induced in galactose medium, and analysis of Aβ42-GFP–associated fluorescence (OD600 of ∼1.5). (A) Δaco1, (B) Δfum1, (C) Δidp1, (D) Δkgd2, and (E) Δsdh4 predominantly exhibit cytosolic-diffuse Aβ42-GFP–associated fluorescence, with some mutants exhibiting fluorescent puncta. Phospholipid mutants (F) Δopi3, (G) Δpsd1, (H) Δcho2, and (I) Δino2. Aβ42-GFP-associated fluorescence in wild-type cells overexpressing (J) INM1 cytosolic diffuse, (K) INM2 cytosolic diffuse, or (L) CDS1 exhibiting perinuclear Aβ42-GFP localization. Arrows point to a distinct localization pattern. Bar, 5 μm
The mitochondrial genome (mtDNA) of S. cerevisiae encodes eight proteins that are essential for oxidative phosphorylation (Tzagoloff and Dieckmann, 1990), and deletion of mtDNA leads to respiratory incompetence due to disruption of the mitochondrial electron transport chain. S. cerevisiae is a facultative aerobe, and respiratory-incompetent cells can grow on carbon sources such as glucose or galactose. To further examine whether the disruption of respiratory function per se affected Aβ42-GFP fluorescence, we grew rho-zero (rho0) cells lacking mtDNA derived from wild-type (BY4743) S. cerevisiae in SC galactose (inducing) medium and analyzed the effect on Aβ42-GFP. Respiratory-incompetent rho0 cells yielded similar trace levels of Aβ42-GFP fluorescence (∼5%), comparable to wild-type grande cells, indicating that loss of respiratory function per se does not affect Aβ42-GFP–associated fluorescence.
Many mutants affected in mitochondrial function, including those affected in operation of the TCA cycle, exhibit a reduced growth rate (Giaever et al., 2002; McCammon et al., 2003), and this may have indirectly influenced Aβ42-GFP fluorescence in the present screen. However, this explanation is unlikely, since there was complete lack of correlation between the genes identified in the present study and ∼720 yeast genes whose deletion was previously shown to cause slow growth (Giaever et al., 2002; Hillenmeyer et al., 2008), and, more broadly, the ∼340 genes identified previously that are involved in mitochondrial morphology and/or respiration. Mitochondrial dysfunction in rho0 cells has been shown to promote increased Gal4-dependent transcription (Jelicic et al., 2005). Loss of mitochondrial function was not a primary cause of increased Aβ42-GFP fluorescence in the mitochondrial mutants, since rho0 cells exhibited the same Aβ42-GFP fluorescence as the wild-type parent. These data support the hypothesis that loss of distinct mitochondrial functions, including those involved in TCA cycle function, metabolism of pyruvate to oxaloacetate, and cytochrome c oxidase activity, leads to increased Aβ42-GFP–associated fluorescence and thus influences Aβ aggregation.
Disruption of genes involved in the TCA cycle leads to altered expression of ∼400 genes, including decreased expression of genes involved in glucose uptake (HXT7, HXT6), glycolysis (PFK26, FBP26), and glycogen/trehalose production (GDB1, GPH1, GSY1, TPS1, TSL1), and, conversely, induction of genes involved in inositol or phosphatidylcholine biosynthesis biosynthesis (INO1, OPI3; McCammon et al., 2003). Induction of INO1 and OPI3 does not occur in rho0 cells (Epstein et al., 2001; Traven et a1., 2001). This raised the possibility that increased inositol synthesis may lead to increased Aβ42-GFP fluorescence. To examine this hypothesis more directly, we assessed the effect on Aβ42-GFP fluorescence in wild-type BG1805 cells grown to exponential phase (SC galactose –URA–HIS) of overexpressing genes encoding Ino1p (inositol-3-phosphate synthetase) or Inm1p or Inm2p (inositol monophosphatase), which perform subsequent steps in inositol biosynthesis. Strikingly, overexpression of either INM1 or INM2 led to intense diffuse fluorescence in ∼18% of cells compared with the empty vector control and cells overexpressing INO1, in which only trace fluorescence was observed (Figure 4). Although these data do not establish a direct causal link between increased Aβ42-GFP fluorescence in mutants disrupted in TCA function and altered inositol synthesis, it is worth noting that both situations led to the appearance of intense diffuse Aβ42-GFP fluorescence in cells. The broader implications of these data are addressed further in the Discussion.
Phospholipid homeostasis plays an important role in intracellular aggregation of Aβ42-GFP
Of the Aβ42-GFP localization patterns identified, the arc-shaped fluorescence localization in mutants affected in phospholipid homeostasis was distinctive, and these mutants were selected for further investigation. Five deletion mutants expressing Aβ42-GFP exhibited structured arc-shaped Aβ42-GFP fluorescence (Figure 4). Of the five genes involved, three encode consecutive steps in the conversion of phosphatidylserine to phosphatidylcholine (phosphatidylserine decarboxylase, PSD1; and phosphatidylethanolamine methyltransferases, CHO2 and OPI3) and two (INO2 and INO4) encode a heterodimeric transcriptional regulator of phospholipid and inositol biosynthesis (Ambroziak and Henry, 1994). From Figure 5, Aβ42-GFP or Aβ40-GFP were localized in a perinuclear location in the Δopi3 mutant. These data indicate that the perinuclear localization of Aβ42-GFP and Aβ40-GFP did not depend on the last two residues of the Aβ moiety in the former. High-resolution confocal microscopy indicated that in Δopi3 cells the perinuclear fluorescence consisted of numerous intense puncta localized around the nucleus (Figure 5). The elongated cell morphology observed with the Δopi3 mutant was not due to induction of AβGFP fusion proteins, since it was also observed in otherwise genetically identical cells expressing the empty vector control (pUG35GAL1; see Supplemental Figure S1).
FIGURE 5.

Aβ-GFP localization in the Δopi3 mutant. (A) Δopi3 cells expressing Aβ42-GFP or Aβ40-GFP (green) were grown in galactose medium, and fluorescence was analyzed. Cells were costained with DAPI (blue) to visualize the nucleus, indicating the perinuclear localization of Aβ42-GFP or Aβ40-GFP in Δopi3 cells. (B) Confocal microscopic image of a Δopi3 cell expressing Aβ42-GFP (green) stained with DAPI (blue). Bar, 5 μm. (C) Western blot analysis, using the 6E10 antibody, of subcellular fractions of the ER and mitochondria from wild-type and Δopi3 cells expressing Aβ42-GFP. Aβ42-GFP band, ∼31 kDa. Antibodies against Por1p and Wbp1p were used as controls to validate lack of cross-contamination between mitochondria and ER fractions, respectively. Dotted lines delineate individual lanes on gels.
To investigate more precisely the localization of the fluorescent Aβ42-GFP in the Δopi3 cells, wild-type and Δopi3 cells expressing the Aβ42-GFP fusion construct were grown to exponential phase, and subcellular fractionation of endoplasmic reticulum (ER) and mitochondria was performed. Aβ42-GFP was detected via Western blot using the 6E10 antibody. In wild-type cells, a ∼31-kDa Aβ42-GFP band was mainly observed in the mitochondrial fraction, with a very faint band also visible in the ER fraction (Figure 5). In contrast, Δopi3 cells expressing Aβ42-GFP exhibited intense ∼31-kDa bands corresponding to Aβ42-GFP in both the ER and mitochondrial fractions. The Aβ42-GFP identified in the mitochondrial fraction by cell fractionation and immunoblotting was therefore nonfluorescent aggregated Aβ42-GFP, since Aβ42-GFP–associated fluorescence was not observed in the mitochondria in whole cells via microscopic analysis. Owing to the appearance of an intense band of Aβ42-GFP in the mitochondrial fraction in both the wild type and the Δopi3 mutant, the possibility that aggregated Aβ42-GFP may have simply cosedimented with the mitochondrial fraction cannot be ruled out. Taken together with the data from Western blotting, which would detect both fluorescent and aggregated nonfluorescent Aβ42-GFP, these data support the proposal that fluorescent Aβ42-GFP in Δopi3 cells may be localized to the ER/ER membrane. Because nonaggregated/soluble Aβ-GFP correlated with fluorescence intensity (Figure 1), these data also support the proposal that it was the soluble form of Aβ42-GFP that had interacted with the ER/ER membrane. These data strongly implicate phospholipid metabolism/homeostasis as an effector of Aβ42 aggregation. Given the role of phospholipids as structural components of cell membranes, disruption of phospholipid homeostasis—for example, as in Δopi3 cells, influences the interaction of “soluble” Aβ42 (and probably Aβ40) with the ER membrane, influencing Aβ aggregation. These data are of particular interest, given the localization of many steps of phospholipid synthesis to the ER.
An alternative route of PC synthesis, via the Kennedy salvage pathway, depends on the availability of precursor molecules, including ethanolamine (EA), monomethylethanolamine, dimethylethanolamine (DMEA), or choline (Summers et al., 1988; McGraw and Henry, 1989). Because the growth medium used in mutant screening did not contain any of these substrates, it is likely that the membranes of Δpsd1, Δopi3 mutants were depleted in PC and/or additional phospholipids and that this was associated with altered Aβ42-GFP–associated fluorescence. To test this, Δopi3 cells expressing Aβ42-GFP or GFP alone were cultured in media supplemented with 1 mM EA, DMEA, or choline. Media supplementation with 1 mM DMEA or choline (Figure 6) resulted in reversal to a wild-type phenotype (Figures 6 and 1) for Δopi3 cells, with a comparable level of fluorescence to the wild-type strain. In contrast, treatment of Δopi3 cells with EA did not lead to altered fluorescence relative to untreated cells. This is consistent with the capacity of Opi3p to methylate either phosphatidyldimethylethanolamine or phosphatidylmonomethylethanolamine but not phosphatidylethanolamine, leading to PC formation. Treatment of cells expressing GFP alone in a comparable manner did not lead to any difference in fluorescence. The perinuclear localization of Aβ42-GFP fluorescence in Δopi3 cells therefore appears to be related to their capacity to maintain PC homeostasis.
FIGURE 6.

Fluorescence microscopic images of Δopi3 cells expressing GFP or Aβ42-GFP grown in galactose (induction) medium either lacking or supplemented with 1 mM ethanolamine (EA), dimethylethanolamine (DMEA), or choline. Cells were analyzed for fluorescence in exponential phase (OD600 of ∼1.5). Bar, 5 μm.
Overexpression of CDS1, encoding cytidine diphosphate–diacylglycerol synthase, alters Aβ42-GFP–associated fluorescence
To obtain further insight into the link between altered phospholipid homeostasis and Aβ42-GFP fluorescence, we assessed the effect of overexpressing genes involved in phospholipid metabolism on Aβ42-GFP. Wild-type cells were transformed with the plasmids carrying one of each of numerous genes involved in lipid metabolism (listed in Supplemental Table S4) and the cells examined to identify those exhibiting increased Aβ42-GFP–associated fluorescence and/or altered fluorescence morphology. Cells with the plasmids that encode proteins involved in lipid synthesis/regulation were cotransformed with the pAβ42-GFP construct in corresponding deletion mutant strains, as well as in wild-type cells. Expression of the Aβ42-GFP fusion protein and the lipid-related gene in each strain was induced by growth in galactose medium, and Aβ42-GFP–associated fluorescence was analyzed 12–18 h postinduction.
Cells overexpressing CDS1 exhibited ordered localization of intensely fluorescent puncta of Aβ42-GFP in a perinuclear arrangement analogous to that seen in the Δpsd1, Δcho2, and Δopi3 mutants (Figure 4). CDS1 encodes CDP-DAG synthase, which catalyzes CDP-DAG–dependent synthesis of phospholipids from phosphatidic acid (Homann et al., 1985). These data further support that Aβ42-GFP aggregation is strongly influenced by altered phospholipid homeostasis in S. cerevisiae cells.
DISCUSSION
The finding that fluorescence of an Aβ-GFP fusion protein is inversely proportional to its propensity to aggregate in E. coli (Kim and Hecht, 2005, 2008) provides an elegant and adaptable approach for exploring the intracellular properties of Aβ. The present study demonstrates that when expressed in the cytosol of the yeast S. cerevisiae, the inverse relationship between Aβ-GFP fluorescence and aggregation potential is preserved. This system forms the foundation of a genetically tractable eukaryotic platform for identifying processes that affect intracellular Aβ aggregation, an important feature in the pathology of Alzheimer's disease. We transformed an Aβ42-GFP expression plasmid into the entire S. cerevisiae genome deletion mutant collection and examined individual mutants for changes in Aβ42-GFP fluorescence. Conversely, we also overexpressed a targeted panel of genes in wild-type cells expressing Aβ42-GFP and examined the effect on fluorescence.
This approach identified 110 deletion mutants exhibiting strong Aβ42-GFP–associated fluorescence and 234 deletion mutants that exhibited weak fluorescence. After clustering mutants into broad functional categories, three emerged as being significantly overrepresented: mitochondrial function, phospholipid metabolism, and transcriptional/translational regulation. Overexpression of the genes CDS1, INM1, and INM2 significantly increased fluorescence in Aβ42-GFP–expressing wild-type cells, additionally implicating inositol biosynthesis as a fourth functional class.
These functional categories parallel some of those affected in Alzheimer's disease patients, and several genes identified in our screen have human homologues that have been found to be directly related to Aβ and Alzheimer's pathology. Mutants affected in mitochondrial function and tricarboxylic acid cycle function exhibited the highest changes in fluorescence identified in the screen (both intensity and proportion of cells), mirroring the long association of mitochondrial dysfunction with Alzheimer's pathology (de Leon et al., 1983; Atamna and Frey, 2007). Human homologues of the yeast TCA cycle screen hits α-ketoglutarate dehydrogenase (KGD2) and isocitrate dehydrogenase (IDP1) show reduced activity in AD patient brains, with the extent of reduction correlating closely with decreased mental performance (Gibson et al., 2000, 2005; Ko et al., 2001; Bubber et al., 2005; Chaturvedi and Beal, 2013). Decreased activity of α-ketoglutarate dehydrogenase leads to sensitivity to oxidative damage, which is strongly associated with AD (Shi et al., 2011). Given that rho0 respiratory-deficient cells and a range of other mitochondrial mutants displayed wild-type Aβ42-GFP fluorescence, altered Aβ aggregation appears to be specifically linked to TCA cycle and oxaloacetate-to-pyruvate defects.
One potential explanation for this may relate to inositol metabolism. In yeast, disruption of TCA function increases expression of inositol-3-phosphate synthase (encoded by INO1), a key enzyme in inositol synthesis (McCammon et al., 2003). We demonstrated that overexpression of either of the myo-inositol biosynthetic genes INM1 or INM2 led to the appearance of similar intense, diffuse Aβ42-GFP fluorescence as observed in the TCA cycle mutants, suggesting a link between inositol metabolism, the TCA cycle, and Aβ aggregation. In human brain, myo-inositol is the most abundant stereoisomer (Haris et al., 2011) and forms small stable micelles with the Aβ peptide (McLaurin et al., 1998). Amyloid fibril formation is inhibited by myo-inositol (Gellermann et al., 2007), and complexation of Aβ with inositol protects neuronal cultures from Aβ toxicity (McLaurin et al., 2000). Similarly, the scyllo-inositol stereoisomer inhibits amyloid formation (Li et al., 2012; Ma et al., 2012), and treatment of animal AD models with scyllo-inositol reduces plaque formation and improves memory performance and hippocampal synaptic plasticity (McLaurin et al., 2006; Aytan et al., 2013). Of note, the activity of the human myo-inositol monophosphatase homologue of the yeast INM1/INM2 genes identified in this study is raised in the brains of AD patients, possibly reflecting a compensatory change to altered phospholipid metabolism (Shimohama et al., 1998). Furthermore, scyllo-inositol was previously identified as an inhibitor of Aβ oligomerization in yeast (Park et al., 2011). Our results, together with those previously reported, suggest that inositol metabolism modulates the aggregation of the Aβ peptide and offer a potential link to the increased Aβ42-GFP fluorescence observed in TCA cycle mutants. Increased inositol levels resulting in formation of soluble inositol/Aβ42-GFP micelles and inhibition of Aβ42-GFP oligomerization offers a potential mechanistic explanation for the significant fluorescence observed in INM1/INM2-overexpressing cells.
In addition to alteration of inositol production, TCA cycle dysfunction would also affect NAD+/NADH homeostasis. NAD+ availability may modulate sirtuin-2 activity, leading to altered Aβ deposition, tubulin deacetylation, and potentiation of tau hyperphosphorylation (Silva et al., 2011). Given the role of sirtuins in chromatin remodeling, it is interesting to note that our screen identified numerous mutants affected in similar functions, particularly those affecting the Swr1 chromatin remodeling complex, suggesting the existence of a potential link between Aβ aggregation, TCA metabolite levels, and chromatin dynamics.
Several mutants affecting phospholipid synthesis exhibited increased Aβ42-GFP fluorescence in our screen. There is strong evidence of perturbed phospholipid metabolism in AD patients (Hung et al., 2008; Frisardi et al., 2011; Grimm et al., 2011). Phosphatidylcholine levels are reduced in cortical membranes (Nitsch et al., 1992) and erythrocytes (Selley, 2007) of AD patients, and studies of the global brain and plasma lipidomes in mouse AD models revealed changes in several sterol, sphingolipid, and phospholipid species, each occurring at specific stages of AD progression (Tajima et al., 2013). Deletion mutants identified in our screen related to phospholipid metabolism included enzymes involved in the de novo synthesis of phosphatidylcholine from phosphatidylserine and phosphatidylethanolamine (PE; PSD1, CHO2 and OPI3; Kodaki and Yamashita, 1987; Summers et al., 1988; McGraw and Henry, 1989) and both members of a heterodimeric transcription complex positively regulating the transcription of a large family of phospholipid biosynthetic genes (including OPI3; Loewy and Henry, 1984; Hirsch and Henry, 1986; Bailis et al., 1987; Jesch et al., 2005).
Defects in these PC biosynthetic pathways lead to accumulation of PC precursors, such as PE and phosphatidylmonomethylethanolamine and altered membrane composition (Kennedy and Weiss, 1956). Supplementation with choline or dimethylethanolamine restores normal synthesis of PC via the Kennedy salvage pathway (Kennedy and Weiss, 1956; McGraw and Henry, 1989) and, in this study, significantly reduced Aβ42-GFP fluorescence in Δopi3 cells. Of interest, mutations in PEMT, the human homologue of yeast OPI3, are associated with increased risk of developing AD (Bi et al., 2011). SCS2 encodes a key ER regulator of inositol and phospholipid metabolism (Greenberg et al., 1982; Kagiwada et al., 1998; Gavin et al., 2002; Loewen et al., 2003), with Δscs2 mutants accumulating 10% more PC than wild-type cells (Kagiwada and Zen, 2003). Thus, the intense Aβ42-GFP fluorescence in Δscs2 mutants indicates that PC depletion alone is unlikely to be the underlying cause of altered Aβ42-GFP fluorescence in Δopi3 and Δcho2 cells. We demonstrated that overexpression of CDP-diacylglycerol synthase (CDS1), responsible for synthesis of the global phospholipid precursor CDP-diacylglycerol, also resulted in increased Aβ42-GFP fluorescence. Together these results indicate overall perturbation of normal phospholipid homeostasis and membrane composition rather than changes in a single lipid species as being an important regulator of Aβ aggregation.
Aβ40/42 peptides may be absorbed onto phospholipid membranes (Terzi et al., 1997; Kanfer et al., 1999; Ege and Lee, 2004; Ege et al., 2005), and there is strong evidence that lipid composition modulates the association, dissociation, and aggregation of Aβ on membranes (Maltseva and Brezesinski, 2004; Maltseva et al., 2005; Chi et al, 2008; Hane et al., 2011; Lemkul and Bevan, 2011). The influence of membrane composition on Aβ dynamics suggests a mechanism for the changes in Aβ42-GFP fluorescence observed in Δino2, Δino4, Δpsd1, Δopi3, and Δcho2 mutants and CDS1-overexpressing cells. In these strains, disrupted lipid homeostasis alters normal membrane composition, including incorporation of intermediates of phospholipid biosynthetic pathways. Aβ42-GFP interaction with these membranes may be altered, favoring reduced aggregation of Aβ and increased fluorescence. The distinctive perinuclear ER membrane–like localization of Aβ42-GFP fluorescence in these strains indicates that such compositional changes are localized to specific organelles or subcellular regions. A detailed subcellular lipidomic analysis of organelle-specific membranes may provide data to further test this hypothesis, allowing in vitro measurement of Aβ aggregation on synthetic membranes, with compositions matched to those of the mutants.
This study identifies specific genes and broader functional classes that influence the aggregation of an Aβ42-GFP fusion protein in the cytosol of yeast, complementing recent screens focusing on intracellular Aβ toxicity (Treusch et al., 2011; D'Angelo et al., 2013. We found that deletion of PBS2, encoding a mitogen-activated protein (MAP) kinase of the high osmolarity glycerol (HOG) pathway, resulted in the appearance of a single intense, punctate fluorescent Aβ42-GFP structure in cells, and Treusch et al. (2011) determined that overexpression of PBS2 enhanced toxicity of Aβ expressed in the yeast endoplasmic reticulum and that expression of the human PBS2 homologue, MAP2K4, in a Caenorhabditis elegans neuronal model resulted in enhanced cell death. These complementary results together suggest that altered intracellular Aβ aggregation may contribute to Aβ-induced cell toxicity in which signaling in the HOG pathway is compromised. The high concordance of functional classes identified in our screen with those affected in animal AD models and AD patients, particularly related to the TCA cycle, inositol metabolism, and phospholipid homeostasis, hints at the involvement of novel modulators of intracellular Aβ aggregation in the development of the disease in humans.
MATERIALS AND METHODS
Strains and culture conditions
S. cerevisiae homozygous diploid deletants for all nonessential genes were obtained from the European Saccharomyces cerevisiae Archive for Functional Analysis (Winzeler et al., 1999). These deletants were produced in the BY4743 diploid strain background (MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 LYS2/lys2Δ0 ura3Δ0/ura3Δ0), which was used as the wild-type reference strain. Standard yeast growth media and techniques were used throughout this study. Yeast strains were grown in YEPD medium (2% [wt/vol] glucose, 2% [wt/vol] peptone, 1% yeast extract) or synthetic defined complete medium (SC; 2% [wt/vol] glucose, 0.17% yeast nitrogen base without amino acids [Becton Dickinson, Franklin Lakes, NJ], 5% [wt/vol] ammonium sulfate [Sigma, St. Louis, MO]) supplemented with appropriate amino acids and bases as indicated in Supplemental Table S1. Expression of genes under the control of the GAL1 promoter was induced by growth in induction medium (SC-galactose; SC medium with 2% [wt/vol] galactose instead of glucose). Where required, filter-sterilized phospholipid precursors were added to SC or induction media at 1 mM final concentration. Respiratory-incompetent (rho0) cells, lacking mitochondrial DNA, were generated by ethidium bromide treatment of the wild-type strain (Fox et al., 1991), identified by their inability to grow on YEPG medium and confirmed by a lack of mitochondrial DNA, visualized using 4,6-diamidino-2-phenylindole dihydrochloride (DAPI [Sigma]). At least five independent rho0 cells generated were examined for each experiment. Unless otherwise stated, all other reagents were purchased from Sigma.
Plasmid construction
A derivative of the pUG35GFP fusion vector (http://mips.gsf.de/proj/yeast/info/tools/hegemann/gfp.html) containing the GAL1 promoter was created by excising the MET25 promoter from pUG35 by digestion with SacI and XbaI. The GAL1 promoter was amplified from a pESC-URA template (Stratagene), using primers ESC-URA-F and ESC-URA-R (Supplemental Table S2) containing SacI and XbaI restriction sites, respectively. The fragment containing GAL1 was ligated into pUG35 to produce pUG35GAL1. To generate Aβ-GFP fusion constructs for the deletion library screen, Aβ42 coding sequence was amplified using pAS1N.AβGFP plasmid DNA (Caine et al., 2007) as a template. The C-terminus of Aβ42 was fused to the N-terminus of GFP (including a four–amino acid linker) by amplifying the Aβ42 coding sequence, using primers ABETA-F and ABETA-R containing BamHI and SalI sites, respectively. After digestion with these enzymes, the Aβ42 fragment was ligated into pUG35GAL1 to produce plasmid pAβ42-GFP. Similarly, GFP fusions of the C-terminally truncated Aβ40-GFP and a mutant form of Aβ42 (with amino acid substitutions I41E and A42P) were created by amplification using ABETA-F with ABETA-40-R and ABETA-EP-R primers, respectively, to yield pAβ42-GFP and pAβEP-GFP. To generate Aβ-GFP fusion constructs for the overexpression screen, the respective PCR fragments were used for the BP reaction of the Gateway system (Invitrogen, San Diego, CA) with the destination vector pDonr221. The resulting plasmids, pDonr221-Aβ42-GFP/pDonr221-Aβ40-GFP/pDonr221-AβEP-GFP/pDonr221-EGFP, were used for the LR reaction using the destination vector pAG415GAL-ccdB and pAG416GAL-ccdB (Alberti et al., 2007). Plasmids were maintained and amplified in E. coli DH5-α cells. For gene overexpression experiments, high-copy galactose-inducible plasmids of the Yeast ORF Collection containing S. cerevisiae open reading frames in the BG1805 plasmid background were used (Gelperin et al., 2005; Thermo Scientific).
Protein extraction, SDS–PAGE, and Western blot analysis
Yeast cultures (50 ml) were grown to OD600 of 1.5 in induction medium (SC-galactose) lacking uracil at 30°C. Cells were harvested, washed with water, and lysed by shaking with glass beads in ice-cold lysis buffer (0.1 mM Tris-HCl, pH 8.0) supplemented with protease inhibitors (Complete; Roche). Intact cells and large debris were removed by centrifugation at 2000 rpm for 5 min at 4°C. Supernatant from this step was subject to ultracentrifugation at 100,000 × g for 1 h at 4°C to yield a supernatant fraction containing soluble proteins and an insoluble pellet. The pellet was solubilized by agitation in 2% SDS at 100°C. Total protein concentration for both supernatant and pellet fractions was determined by BCA Protein Assay (Pierce Biotechnology, Rockford, IL), which was used to normalize protein loading onto 4–12% gradient SDS–PAGE gels (20 μg/lane for supernatant fractions, 5 μg/lane for pellet fractions). Proteins were electroblotted onto nitrocellulose membranes for Western blotting and probed with mouse anti-Aβ (6E10; Covance, Sydney, Australia) antibodies overnight at 1:1000 dilution. Immunodetection was performed using horseradish peroxidase–conjugated anti-mouse secondary antibodies (Jackson ImmunoResearch, West Grove, PA), chemiluminescence reagents (Bio-Rad, Sydney, Australia), and ChemiDoc MP charge-coupled device imaging system (Bio-Rad). Analysis and quantitation of Western blot images was performed using ImageJ, version 1.38 (National Institutes of Health, Bethesda, MD).
High-throughput transformation of the S. cerevisiae genome knockout collection
Microtiter plates containing strains from the S. cerevisiae genome-wide deletion collection were replicated into 96-well plates containing 160 μl of YEPD medium/well and incubated at 30°C with shaking for 1 d. Cells were pelleted and resuspended in 20 μl of sterile MilliQ water before the addition of 160 μl of transformation mix/well (40% [wt/vol] PEG-3350, 100 mM lithium acetate, 1 mM EDTA, 10 mM Tris-HCl, pH 7.5, 20 ng/ml single-stranded herring sperm DNA, and 5 μg/well pAβ42-GFP plasmid DNA). Plates were incubated at 30°C overnight, followed by heat shock at 42°C for 30 min. Cells were pelleted, resuspended in 150 μl of SC medium (lacking uracil for selection of the pAβ42-GFP plasmid), and incubated at 30°C with shaking for 2 d. Further selection was performed by replicating cells into fresh 96-well plates (containing 150 μl/well of uracil-free SC media) and growing for 1 d. Strains were stored for later analysis by resuspension in 10% (vol/vol) glycerol and freezing at −80°C.
Epifluorescence and confocal microscopy, and the visualization of organelles
Fluorescence microscopy was performed using a Leica DM5500B microscope under 100× objective. Nuclear and mitochondrial DNA were visualized by staining cells with DAPI. Images were obtained and processed using the Leica Application Suite. Image overlays were performed using ImageJ, version 1.38 software. Confocal fluorescence microscopy was performed using an Olympus FV-1000 confocal microscope under 100× objective. GFP and DAPI images were visualized using 488- and 405-nm lasers, respectively. Three-dimensional confocal data analysis and image processing were performed using the Imaris 7.2 software package (Bitplane). The appearance of rod-like regions along the z-axis of Aβ42-GFP in the confocal image may be an artifact of limitation in imaging resolution along this z-axis.
Screening and analyzing knockout mutants for Aβ42-GFP fluorescence
Strains transformed with pAβ42-GFP were replicated and grown for 48 h with shaking at 30°C in microtiter plates containing 150 µl/well of induction medium. Strains were replicated in microtiter plates and grown for a further 24 h before Aβ42-GFP fluorescence was evaluated using fluorescence microscopy. A minimum of 900 cells per strain were examined for presence of Aβ42-GFP fluorescence. Mutant strains exhibiting altered fluorescence were subcultured in induction media and reexamined.
Positive strains were analyzed for overrepresentation of functional groups with Gene Ontology (GO) and MIPS databases using GO Biological Process, GO Molecular Function, and GO Cellular Component in the analysis. S. cerevisiae genes were mapped to human homologues using the HomoloGene database (www.ncbi.nlm.nih.gov/sites/entrez?db=homologene). All other analyses of S. cerevisiae genes and associated annotations were performed using the Saccharomyces Genome Database and associated tools (www.yeastgenome.org).
Subcellular fractionation
Subcellular fractions of S. cerevisiae cells were prepared as previously described by Rosenberger et al. (2009) and the cross-contamination of the ER and mitochondrial fractions verified by Western blot analysis using antibodies against Wbp1p and Por1p, respectively. Cells expressing Aβ42-GFP were grown to exponential phase (OD600 of 1.5) in galactose medium, harvested, washed in distilled water, and converted to spheroplasts (Daum et al., 1982). Preparation of spheroplasts was performed using 2 mg Zymolyase 20T/g cell wet weight and incubating for 1.5 h at 30°C shaking (600 rpm). Spheroplasts were homogenized on ice using a Dounce homogenizer with a tight-fitting pestle and centrifuged to remove unbroken cells and nuclei. For preparation of crude mitochondrial fraction, cell lysates were centrifuged at 30,000 × g (30 min, 4°C). To enrich for mitochondria, the resulting pellet was thrice resuspended in breakage buffer, rehomogenized, and centrifuged. For preparation of crude ER microsomal fraction, the remaining supernatant was centrifuged at 45,000 × g (45 min; 4°C). To enrich for ER, the resulting pellet was resuspended in breakage buffer, rehomogenized, and centrifuged. To validate lack of cross-contamination of subcellular fractions, proteins from ER and mitochondrial fractions were precipitated with 50% trichloroacetic acid for 1 h on ice, and Western blot analysis was subsequently performed as described.
Statistical analysis
Statistical analysis was performed using the unpaired Student's t test, using Prism 5 for Windows, version 5.02 (GraphPad Software). Data are presented as mean ± SD. Significant differences are indicated by a p value for data in the text and figures.
Supplementary Material
Acknowledgments
We thank Ian Macreadie for providing plasmid pAS1N.AβGFP; Phil Hogg for assistance with flow cytometry; Katharina Mack, Geoff Kornfeld, Joyce Chiu, and Shixiong Tan for technical advice; and Guenter Daum and Vid Vojko Flis for helpful discussion and techniques for subcellular fractionation. This research was supported by grants from the Australian Research Council (I.W.D.), Cancer Institute New South Wales (I.W.D., G.G.P.), and a University of New South Wales International Postgraduate Research Scholarship (S.N.).
Abbreviations used:
- Aβ
amyloid-β
- AD
Alzheimer's disease
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- TCA
tricarboxylic acid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-04-0216) on May 28, 2014.
*These authors contributed equally to this work.
REFERENCES
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroziak J, Henry SA. INO2 and INO4 gene products, positive regulators of phospholipid biosynthesis in Saccharomyces cerevisiae, form a complex that binds to the INO1 promoter. J Biol Chem. 1994;269:15344–15349. [PubMed] [Google Scholar]
- Atamna H, Frey WH. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer's disease. Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Aytan N, Choi JK, Carreras I, Kowall NW, Jenkins BG, Dedeoglu A. Combination therapy in a transgenic model of Alzheimer's disease. Exp Neurol. 2013;250C:228–238. doi: 10.1016/j.expneurol.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis AM, Poole MA, Carman GM, Henry SA. The membrane-associated enzyme phosphatidylserine synthase is regulated at the level of mRNA abundance. Mol Cell Biol. 1987;7:167–176. doi: 10.1128/mcb.7.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X-H, Zhao H-L, Zhang Z-X, Zhang J-W. PEMT G523A (V175M) is associated with sporadic Alzheimer's disease in a Chinese population. J Mol Neurosci. 2011;46:505–508. doi: 10.1007/s12031-011-9630-3. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Caine J, Sankovich S, Antony H, Waddington L, Macreadie P, Varghese J, Macreadie I. Alzheimer's Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007;7:1230–1236. doi: 10.1111/j.1567-1364.2007.00285.x. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson's and Huntington's diseases. Mol Cell Neurosci. 2013;55:101–114. doi: 10.1016/j.mcn.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Chi EY, Ege C, Winans A, Majewski J, Wu G, Kjaer K, Lee KYC. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer's disease amyloid-β peptide. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- Daum G, Bohni PC, Schatz G. Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- D'Angelo F, Vignaud H, Di Martino J, Salin B, Devin A, Cullin C, Marchal C. A yeast model for amyloid-beta aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis Model Mech. 2013;6:206–216. doi: 10.1242/dmm.010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, et al. Positron emission tomographic studies of aging and Alzheimer disease. Am J Neuroradiol. 1983;4:568–571. [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:847–853. doi: 10.1091/mbc.01-12-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege C, Lee KY. Insertion of Alzheimer's A beta 40 peptide into lipid monolayers. Biophys J. 2004;87:1732–1740. doi: 10.1529/biophysj.104.043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege C, Majewski J, Wu G, Kjaer K, Lee KY. Templating effect of lipid membranes on Alzheimer's amyloid beta peptide. Chemphyschem. 2005;6:226–229. doi: 10.1002/cphc.200400468. [DOI] [PubMed] [Google Scholar]
- Epstein CB, Waddle JA, Hale WT, Dave V, Thornton J, Macatee TL, Garner HR, Butow RA. Genome-wide responses to mitochondrial dysfunction. Mol Biol Cell. 2001;12:297–308. doi: 10.1091/mbc.12.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J Mol Biol. 2005;351:1081–1100. doi: 10.1016/j.jmb.2005.06.060. [DOI] [PubMed] [Google Scholar]
- Fox TD, Folley LS, Mulero JJ, McMullin TW, Thorsness PE, Hedin LO, Costanzo MC. Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol. 1991;194:149–165. doi: 10.1016/0076-6879(91)94013-3. [DOI] [PubMed] [Google Scholar]
- Frisardi V, Panza F, Seripa D, Farooqui T, Farooqui AA. Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer's disease pathology. Prog Lipid Res. 2011;50:313–330. doi: 10.1016/j.plipres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gellermann GP, Ullrich K, Unger C, Fandrich M, Sauter S, Diekmann S. Identification of molecular compounds critical to Alzheimer's-like plaque formation. J Neurosci Res. 2007;85:2037–2044. doi: 10.1002/jnr.21311. [DOI] [PubMed] [Google Scholar]
- Gelperin DM, et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Park LC, Sheu KF, Blass JP, Calingasan NY. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem Int. 2000;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gouras GK, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Greenberg ML, Reiner B, Henry SA. Regulatory mutations of inositol biosynthesis in yeast: isolation of inositol-excreting mutants. Genetics. 1982;100:19–33. doi: 10.1093/genetics/100.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MO, Grosgen S, Riemenschneider M, Tanila H, Grimm HS, Hartmann T. From brain to food: analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in Alzheimer's disease human post mortem brains and mice model via mass spectrometry. J Chromatogr. 2011;1218:7713–7722. doi: 10.1016/j.chroma.2011.07.073. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hamada D, Tsumoto K, Sawara M, Tanaka N, Nakahira K, Shiraki K, Yanagihara I. Effect of an amyloidogenic sequence attached to yellow fluorescent protein. Proteins. 2008;72:811–821. doi: 10.1002/prot.21971. [DOI] [PubMed] [Google Scholar]
- Hane F, Drolle E, Gaikwad R, Faught E, Leonenko Z. Amyloid-β aggregation on model lipid membranes: an atomic force microscopy study. J Alzheimers Dis. 2011;26:485–494. doi: 10.3233/JAD-2011-102112. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Haris M, Cai K, Singh A, Hariharan H, Reddy R. In vivo mapping of brain myo-inositol. Neuroimage. 2011;54:2079–2085. doi: 10.1016/j.neuroimage.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer ME, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JP, Henry SA. Expression of the Saccharomyces cerevisiae inositol-1-phosphate synthase (INO1) gene is regulated by factors that affect phospholipid synthesis. Mol Cell Biol. 1986;6:3320–3328. doi: 10.1128/mcb.6.10.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann MJ, Henry SA, Carman GM. Regulation of CDP-diacylglycerol synthase activity in Saccharomyces cerevisiae. J Bacteriol. 1985;163:1265–1266. doi: 10.1128/jb.163.3.1265-1266.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett DR, Jennings KH, Lee DC, Clark MS, Brown F, Wetzel R, Wood SJ, Camilleri P, Roberts GW. Aggregation state and neurotoxic properties of Alzheimer beta-amyloid peptide. Neurodegeneration. 1995;4:23–32. doi: 10.1006/neur.1995.0003. [DOI] [PubMed] [Google Scholar]
- Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Jelicic B, Traven A, Filic V, Sopta M. Mitochondrial dysfunction enhances Gal4-dependent transcription. FEMS Microbiol Lett. 2005;253:207–213. doi: 10.1016/j.femsle.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Jesch SA, Zhao X, Wells MT, Henry SA. Genome-wide analysis reveals inositol, not choline, as the major effector of Ino2p-Ino4p and unfolded protein response target gene expression in yeast. J Biol Chem. 2005;280:9106–9118. doi: 10.1074/jbc.M411770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S, Hosaka K, Murata M, Nikawa J, Takatsuki A. The Saccharomyces cerevisiae SCS2 gene product, a homolog of a synaptobrevin-associated protein, is an integral membrane protein of the endoplasmic reticulum and is required for inositol metabolism. J Bacteriol. 1998;180:1700–1708. doi: 10.1128/jb.180.7.1700-1708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S, Zen R. Role of the yeast VAP homolog, Scs2p, in INO1 expression and phospholipid metabolism. J Biochem. 2003;133:515–522. doi: 10.1093/jb/mvg068. [DOI] [PubMed] [Google Scholar]
- Kanfer JN, Sorrentino G, Sitar DS. Amyloid beta peptide membrane perturbation is the basis for its biological effects. Neurochem Res. 1999;24:1621–1630. doi: 10.1023/a:1021172620295. [DOI] [PubMed] [Google Scholar]
- Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipids. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- Kim W, Hecht MH. Sequence determinants of enhanced amyloidogenicity of Alzheimer Aβ42 peptide relative to Aβ40. J Biol Chem. 2005;280:35069–35076. doi: 10.1074/jbc.M505763200. [DOI] [PubMed] [Google Scholar]
- Kim W, Hecht MH. Mutations enhance the aggregation propensity of the Alzheimer's Abeta peptide. J Mol Biol. 2008;377:565–574. doi: 10.1016/j.jmb.2007.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LW, Sheu KF, Thaler HT, Markesbery WR, Blass JP. Selective loss of KGDHC-enriched neurons in Alzheimer temporal cortex: does mitochondrial variation contribute to selective vulnerability? J Mol Neurosci. 2001;17:361–369. doi: 10.1385/JMN:17:3:361. [DOI] [PubMed] [Google Scholar]
- Kodaki T, Yamashita S. Yeast phosphatidylethanolamine methylation pathway. Cloning and characterization of two distinct methyltransferase genes. J Biol Chem. 1987;262:15428–15435. [PubMed] [Google Scholar]
- Komano H, Seeger M, Gandy S, Wang GT, Krafft GA, Fuller RS. Involvement of cell surface glycosyl-phosphatidylinositol-linked aspartyl proteases in alpha-secretase-type cleavage and ectodomain solubilization of human Alzheimer beta-amyloid precursor protein in yeast. J Biol Chem. 1998;273:31648–31651. doi: 10.1074/jbc.273.48.31648. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C. Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul JA, Bevan DR. Lipid composition influences the release of Alzheimer's amyloid β-peptide from membranes. Protein Sci. 2011;20:1530–1545. doi: 10.1002/pro.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lesne S, Kotilinek L. Amyloid plaques and amyloid-beta oligomers: an ongoing debate. J Neurosci. 2005;25:9319–9320. doi: 10.1523/JNEUROSCI.3246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Rauscher S, Baud S, Pomes R. Binding of inositol stereoisomers to model amyloidogenic peptides. J Phys Chem B. 2012;116:1111–1119. doi: 10.1021/jp208567n. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Roy A, Levine TP. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22:2025–2035. doi: 10.1093/emboj/cdg201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewy BS, Henry SA. The INO2 and INO4 loci of Saccharomyces cerevisiae are pleiotropic regulatory genes. Mol Cell Biol. 1984;4:2479–2485. doi: 10.1128/mcb.4.11.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Thomason LA, McLaurin J. scyllo-Inositol, preclinical, and clinical data for Alzheimer's disease. Adv Pharmacol. 2012;64:177–212. doi: 10.1016/B978-0-12-394816-8.00006-4. [DOI] [PubMed] [Google Scholar]
- Macreadie I, Lotfi-Miri M, Mohotti S, Shapira D, Bennett L, Varghese J. Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer's amyloid-beta aggregation. J Alzheimers Dis. 2008;15:391–396. doi: 10.3233/jad-2008-15305. [DOI] [PubMed] [Google Scholar]
- Maltseva E, Brezesinski G. Adsorption of amyloid beta (1-40) peptide to phosphatidylethanolamine monolayers. Chemphyschem. 2004;5:1185–1190. doi: 10.1002/cphc.200400045. [DOI] [PubMed] [Google Scholar]
- Maltseva E, Kerth A, Blume A, Mohwald H, Brezesinski G. Adsorption of amyloid beta (1-40) peptide at phospholipid monolayers. Chembiochem. 2005;6:1817–1824. doi: 10.1002/cbic.200500116. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon MT, Epstein CB, Przybyla-Zawislak B, McAlister-Henn L, Butow RA. Global transcription analysis of Krebs tricarboxylic acid cycle mutants reveals an alternating pattern of gene expression and effects on hypoxic and oxidative genes. Mol Biol Cell. 2003;14:958–972. doi: 10.1091/mbc.E02-07-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw P, Henry SA. Mutations in the Saccharomyces cerevisiae OPI3 gene: effects on phospholipid methylation, growth and cross-pathway regulation of inositol synthesis. Genetics. 1989;122:317–330. doi: 10.1093/genetics/122.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J, Franklin T, Chakrabartty A, Fraser PE. Phosphatidylinositol and inositol involvement in Alzheimer amyloid-beta fibril growth and arrest. J Mol Biol. 1998;278:183–194. doi: 10.1006/jmbi.1998.1677. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta-induced toxicity. J Biol Chem. 2000;275:18495–18502. doi: 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- McLaurin J, et al. Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- Morell M, de Groot NS, Vendrell J, Aviles FX, Ventura S. Linking amyloid protein aggregation and yeast survival. Mol Biosyst. 2011;7:1121–1128. doi: 10.1039/c0mb00297f. [DOI] [PubMed] [Google Scholar]
- Naiki H, Nakakuki K. First-order kinetic model of Alzheimer's beta-amyloid fibril extension in vitro. Lab Invest. 1996;74:374–383. [PubMed] [Google Scholar]
- Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH, Wurtman RJ. Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci USA. 1992;89:1671–1675. doi: 10.1073/pnas.89.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev. 2005;126:1292–1299. doi: 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Pegan SD, Mesecar AD, Jungbauer LM, LaDu MJ, Liebman SW. Development and validation of a yeast high-throughput screen for inhibitors of Abeta(4)(2) oligomerization. Dis Model Mech. 2011;4:822–831. doi: 10.1242/dmm.007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger S, Connerth M, Zellnig G, Daum G. Phosphatidylethanolamine synthesized by three different pathways is supplied to peroxisomes of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2009;1791:379–387. doi: 10.1016/j.bbalip.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Seilheimer B, Bohrmann B, Bondolfi L, Muller F, Stuber D, Dobeli H. The toxicity of the Alzheimer's beta-amyloid peptide correlates with a distinct fiber morphology. J Struct Biol. 1997;119:59–71. doi: 10.1006/jsbi.1997.3859. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Amyloid protein and Alzheimer's disease. Sci Am. 1991;265:78. doi: 10.1038/scientificamerican1191-68. 68–71, 74–66. [DOI] [PubMed] [Google Scholar]
- Selley ML. A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer's disease. Neurobiol Aging. 2007;28:1834–1839. doi: 10.1016/j.neurobiolaging.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Shi Q, Xu H, Yu H, Zhang N, Ye Y, Estevez AG, Deng H, Gibson GE. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2011;286:17640–17648. doi: 10.1074/jbc.M110.203018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Sumida Y, Tsuda J, Fujimoto S. Alteration of myo-inositol monophosphatase in Alzheimer's disease brains. Neurosci Lett. 1998;245:159–162. doi: 10.1016/s0304-3940(98)00209-2. [DOI] [PubMed] [Google Scholar]
- Silva DF, Esteves AR, Oliveira CR, Cardoso SM. Mitochondria: the common upstream driver of amyloid-beta and tau pathology in Alzheimer's disease. Curr Alzheimer Res. 2011;8:563–572. doi: 10.2174/156720511796391872. [DOI] [PubMed] [Google Scholar]
- Simmons LK, et al. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- Summers EF, Letts VA, McGraw P, Henry SA. Saccharomyces cerevisiae cho2 mutants are deficient in phospholipid methylation and cross-pathway regulation of inositol synthesis. Genetics. 1988;120:909–922. doi: 10.1093/genetics/120.4.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima Y, et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer's disease. Lipids Health Dis. 2013;12:68. doi: 10.1186/1476-511X-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzi E, Holzemann G, Seelig J. Interaction of Alzheimer beta-amyloid peptide(1-40) with lipid membranes. Biochemistry. 1997;36:14845–14852. doi: 10.1021/bi971843e. [DOI] [PubMed] [Google Scholar]
- Traven A, Wong JM, Xu D, Sopta M, Ingles CJ. Interorganellar communication. Altered nuclear gene expression profiles in a yeast mitochondrial DNA mutant. J Biol Chem. 2001;276:4020–4027. doi: 10.1074/jbc.M006807200. [DOI] [PubMed] [Google Scholar]
- Treusch S, et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagoloff A, Dieckmann CL. PET genes of Saccharomyces cerevisiae. Microbiol Rev. 1990;54:211–225. doi: 10.1128/mr.54.3.211-225.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebroek T, Terwel D, Vanhelmont T, Gysemans M, Van Haesendonck C, Engelborghs Y, Winderickx J, Van Leuven F. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J Biol Chem. 2006;281:25388–25397. doi: 10.1074/jbc.M602792200. [DOI] [PubMed] [Google Scholar]
- Vandebroek T, Vanhelmont T, Terwel D, Borghgraef P, Lemaire K, Snauwaert J, Wera S, Van Leuven F, Winderickx J. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry. 2005;44:11466–11475. doi: 10.1021/bi0506775. [DOI] [PubMed] [Google Scholar]
- Vanhelmont T, Vandebroek T, De Vos A, Terwel D, Lemaire K, Anandhakumar J, Franssens V, Swinnen E, Van Leuven F, Winderickx J. Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 2010;10:992–1005. doi: 10.1111/j.1567-1364.2010.00662.x. [DOI] [PubMed] [Google Scholar]
- Villar-Pique A, Ventura S. Protein aggregation propensity is a crucial determinant of intracellular inclusion formation and quality control degradation. Biochim Biophys Acta. 2013;1833:2714–2724. doi: 10.1016/j.bbamcr.2013.06.023. [DOI] [PubMed] [Google Scholar]
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Winderickx J, Delay C, De Vos A, Klinger H, Pellens K, Vanhelmont T, Van Leuven F, Zabrocki P. Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim Biophys Acta. 2008;1783:1381–1395. doi: 10.1016/j.bbamcr.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer's Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–1290. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of Abeta 42 in Alzheimer's disease. J Physiol. 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- Zhang W, Espinoza D, Hines V, Innis M, Mehta P, Miller DL. Characterization of beta-amyloid peptide precursor processing by the yeast Yap3 and Mkc7 proteases. Biochim Biophys Acta. 1997;1359:110–122. doi: 10.1016/s0167-4889(97)00082-7. [DOI] [PubMed] [Google Scholar]
- Zhang H, Komano H, Fuller RS, Gandy SE, Frail DE. Proteolytic processing and secretion of human beta-amyloid precursor protein in yeast. Evidence for a yeast secretase activity. J Biol Chem. 1994;269:27799–27802. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.