

TβRIII is basolaterally localized in polarized breast epithelial cells. The disruption of TβRIII targeting by mutation of proline 826 results in global loss of cell polarity through enhanced EMT. The mistargeting of TβRIII results in enhanced proliferation, migration, and invasion in vitro and enhanced tumor formation and invasion in vivo.
Abstract
The majority of breast cancers originate from the highly polarized luminal epithelial cells lining the breast ducts. However, cell polarity is often lost during breast cancer progression. The type III transforming growth factor-β cell surface receptor (TβRIII) functions as a suppressor of breast cancer progression and also regulates the process of epithelial-to-mesenchymal transition (EMT), a consequence of which is the loss of cell polarity. Many cell surface proteins exhibit polarized expression, being targeted specifically to the apical or basolateral domains. Here we demonstrate that TβRIII is basolaterally localized in polarized breast epithelial cells and that disruption of the basolateral targeting of TβRIII through a single amino acid mutation of proline 826 in the cytosolic domain results in global loss of cell polarity through enhanced EMT. In addition, the mistargeting of TβRIII results in enhanced proliferation, migration, and invasion in vitro and enhanced tumor formation and invasion in an in vivo mouse model of breast carcinoma. These results suggest that proper localization of TβRIII is critical for maintenance of epithelial cell polarity and phenotype and expand the mechanisms by which TβRIII prevents breast cancer initiation and progression.
INTRODUCTION
Apical–basolateral cell polarity refers to the asymmetric cellular distribution of proteins and lipids by which the apical membrane domain faces the lumen of the duct and the basolateral domain forms cell–cell contacts and interacts with the extracellular matrix and basement membrane (Feigin and Muthuswamy, 2009). Apical–basolateral cell polarity is a characteristic of many epithelial cells, including the luminal cells that line the breast duct. The apical and basolateral membranes are separated from one another by tight junctions, which prevent the movement of proteins and lipids between the two domains (Shin et al., 2006). In addition, adherens junctions, of which the E-cadherin and β-catenin proteins are key components, use the forces generated by the actin cytoskeleton to maintain close cell–cell contact (Halbleib and Nelson, 2006; Shapiro and Weis, 2009). The formation and maintenance of apical–basolateral cell polarity depends on the functions of three separate groups of proteins. These include the Crumbs, PALS, and PATJ proteins, which are important for apical membrane development (Bazellieres et al., 2009); Par3, Par6, and aPKC, which control tight junction formation (Izumi et al., 1998; Joberty et al., 2000; Macara, 2004); and Scribble, Dlg, and Lgl, which define basolateral membrane identity (Yamanaka and Ohno, 2008).
Epithelial cells rely on cell polarity to maintain the cell and tissue architecture required for organ function. Of interest, loss of cell polarity is a consequence of epithelial-to-mesenchymal transition (EMT), which is believed to be of critical importance during cancer metastasis (Thiery, 2003; Thiery and Sleeman, 2006; Gupta and Massague, 2006; Heldin et al., 2012; Creighton et al., 2013). Cancer-associated EMT involves the initiation of a transcriptional program that uses specific transcription factors, including Snail, Slug (also known as Snail 2), Zeb1/2, and Twist (Peinado et al., 2007; Heldin et al., 2012). These transcription factors induce the loss of proteins that are characteristic of epithelial cells, including E-cadherin, Crumbs, Lgl2, and PATJ (Batlle et al., 2000; Bolos et al., 2003; Kang and Massague, 2004; Yang et al., 2004; Vanderwalle et al., 2005; Aigner et al., 2007; Spaderna et al., 2008; Whiteman et al., 2008), and the gain of proteins characteristic of mesenchymal cells, including fibronectin, vimentin, and α-smooth muscle actin (α-SMA; Thiery and Sleeman, 2006; Peinado et al., 2007). As a result of this transcriptional program, EMT commonly results in cell morphology changes (cobblestone to fibroblastic/spindle shaped), alterations to the actin cytoskeleton and extracellular matrix, increased cell proliferation, migration, and invasion, and loss of cell adhesion and polarity (Conacci-Sorrell et al., 2002; Cowin et al., 2005; Hugo et al., 2007; Moreno-Bueno et al., 2008).
One of the key drivers of EMT, and thus the loss of cell polarity, is transforming growth factor-β (TGF-β; Zavadil and Bottinger, 2005; Heldin et al., 2012). Canonically, TGF-β signals via the type I and type II TGF-β receptors, resulting in the phosphorylation and activation of the Smad2 or Smad3 transcription factors (Massague, 2008; Moustakas and Heldin, 2009; Heldin et al., 2012). These factors then associate with Smad4, enter the nucleus, and regulate gene transcription (Heldin et al., 2012). TGF-β can also signal through several noncanonical pathways, including NFκB, p38, PI3K/Akt, and Erk1/2 (Yamashita et al., 2008; Parvani et al., 2011). Of importance, TGF-β induces the expression of Snail, Slug, and other key EMT mediators to decrease E-cadherin expression (Heldin et al., 2012).
In addition to the type I and type II receptors, the type III TGF-β receptor (TβRIII, or betaglycan) is a critical regulator of TGF-β signaling. In the context of breast cancer, the loss of TβRIII results in increased cell growth, invasion, and migration in vitro and increased tumor size, angiogenesis, and metastasis in vivo (Dong et al., 2007; Lee et al., 2010). Conversely, the overexpression of TβRIII decreases metastasis (Dong et al., 2007; Lee et al., 2010). The type I and type II receptors have been localized to tight junctions in polarized breast epithelial cells (NMuMG; Ozdamar et al., 2005) and to the basolateral membrane in canine kidney epithelial cells (MDCK; Murphy et al., 2004, 2007). Because the loss of cell polarity is a key feature of ductal carcinoma in situ and invasive ductal carcinoma (Debnath et al., 2003; Debnath and Brugge, 2005; Lee and Vasioukhin, 2008; Zhan et al., 2008), here we investigate the localization of TβRIII and the effect of this localization on polarity, EMT, TGF-β signaling, and breast cancer initiation and progression.
RESULTS
TβRIII is basolaterally localized in polarized epithelial cells
TβRIII is expressed on the cell surface of most epithelial cells. To determine whether TβRIII is specifically localized to the apical or basolateral membrane domain, we plated NMuMG murine mammary epithelial cells on Transwells, transfected them with TβRIII, and allowed them to form a polarized monolayer. Polarization was confirmed by the absence of fluorescein-dextran leakage from the apical chamber into the basolateral chamber, with the polarity model cell line MDCK serving as control (Figure 1A). TβRIII expression was detected by immunofluorescence, revealing a honeycomb staining pattern, indicating that TβRIII is localized to cell–cell junctions (Figure 1B, left). Sections perpendicular to the monolayer (XZ) demonstrate that TβRIII is primarily basolaterally localized, with little staining observed at the apical membrane (Figure 1B, left).
FIGURE 1.
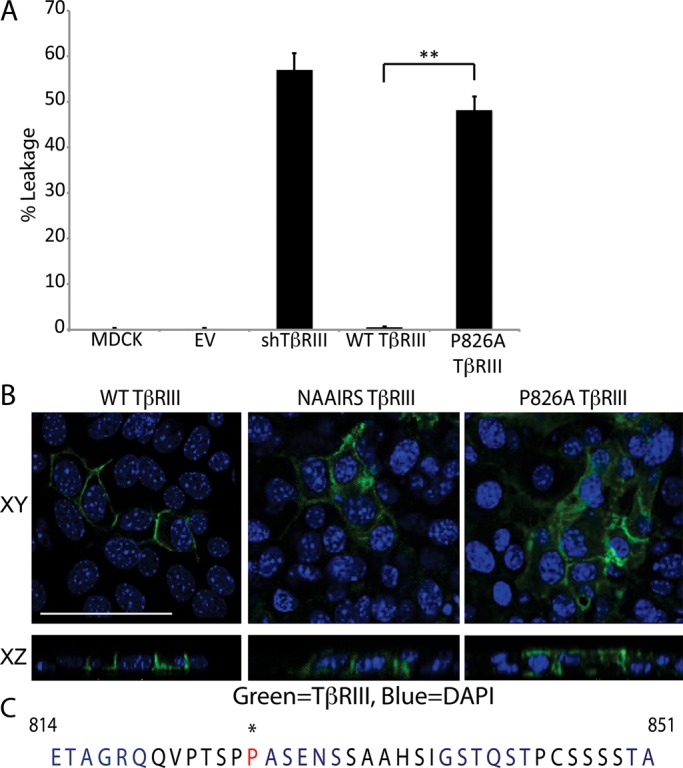
TβRIII is basolaterally localized. (A) We plated 2.5 × 105 NMuMG or MDCK cells on Transwells and grew them for 5 d to allow for polarization. A FITC-dextran tracer was placed in the upper chamber, and the cells were incubated at room temperature for 30 min. Apical and basolateral media samples were excited at a wavelength of 490 nm, and the absorbance at 520 nm was measured. Percentage leakage = basolateral signal/apical signal × 100. **p < 0.01 (Student's t test). (B) Cells were plated as in A and transfected with WT TβRIII, NAAIRS mutant TβRIII, or P826A TβRIII. Two days posttransfection, the cells were fixed and stained with primary antibody against TβRIII and an Alexa 488 secondary (green). Nuclei (blue) were stained with DAPI. Images were collected at a magnification of 400× and show the localization of TβRIII to cell junctions in the flat sections (XY, top). The cross-sectional images (XZ, bottom) show the basolateral localization of WT TβRIII and the apical–basolateral localization of NAAIRS and P826A TβRIII. Bar, 25 μm. (B) The 42–amino acid sequence of the C-terminus of human TβRIII. Each 6–amino acid–long group altered via NAAIRS mutagenesis is shown in alternating blue and black. Residue P826, which is critical for TβRIII's basolateral localization, is starred and shown in red.
The majority of basolateral proteins are targeted to this membrane domain by a unique signal sequence that is typically found within the cytoplasmic domain (Aroeti et al., 1998; Ikonen and Simons, 1998; Rodriguez-Boulan et al., 2005; Murphy et al., 2007). To determine whether a basolateral targeting sequence is present within the cytoplasmic domain of TβRIII, we used a mutagenesis strategy in which every six amino acids in the 42–amino acid–long cytosolic domain of TβRIII was changed to the NAAIRS protein sequence (Figure 1C). The NAAIRS sequence is capable of adopting multiple secondary structures. Therefore NAAIRS sequence mutation minimizes the potential for structural changes (Wilson et al., 1984). Whereas alteration of most of the cytoplasmic domain had no effect on TβRIII localization, mutation of residues 826–831 (PASENS) in the cytosolic domain of TβRIII to the NAAIRS sequence resulted in both apical and basolateral localization (Figure 1, B, middle, and C). Such nonspecific localization is consistent with a disruption of a basolateral targeting sequence (Murphy et al., 2007). To further refine the motif regulating TβRIII localization, we changed each individual amino acid in the PASENS sequence to alanine and assessed the localization. A single amino acid alteration of proline 826 to alanine fully recapitulated the nonspecific targeting observed in the NAAIRS mutant (Figure 1, B, right, and C). All other mutations had no effect on, or only mildly disrupted, TβRIII localization (unpublished data).
Cells expressing P826A TβRIII have disrupted epithelial cell polarity
To determine whether the mislocalization of TβRIII affects cell signaling or biology, we first generated NMuMG cell lines in which endogenous TβRIII was stably silenced. Whereas endogenous TβRIII expression is low in NMuMG cells, we were able to observe ∼60% knockdown of expression by retroviral transduction (Figure 2A, left). Stable rescue of TβRIII expression was then performed by using transduction of human wild-type (WT) TβRIII or P826A TβRIII, with empty vector (EV) lentivirus serving as a control. Although the cell surface expression level of P826A TβRIII was slightly greater than that of WT TβRIII, neither protein was expressed to the extent of endogenous TβRIII in the MDA-MB-231 breast cancer cell line (Figure 2A, right).
FIGURE 2.

P826A TβRIII expression results in a loss of cell polarity. (A) Left, binding and cross-linking of surface TβRIII showing a lower level of TβRIII in the stable shTβRIII cells compared with the EV cells. β-Actin Western blotting was used as a loading control. Right, binding and cross-linking, showing the relative surface levels of TβRIII in the EV and shTβRIII lines, as well as in the WT TβRIII and P826A TβRIII rescued lines. The endogenous TβRIII level in the MDA-MB-231 breast cancer cell line is shown for comparison. Note that with this lighter exposure, the EV and shTβRIII signals are not observable. (B) Caco-2 and NMuMG WT and P826A TβRIII cells were grown in Transwells for 5 d. Cells were incubated with fresh medium on the apical and basal sides for a further 24 h. Media were collected, and the level of shed TβRIII in each compartment was detected via ELISA. The percentage of signal in the apical vs. basolateral chamber was calculated and graphed. **p < 0.01 (Student's t test). (C) Light images taken at 100× magnification show the morphological differences between the cell lines. Bar, 200 μm. (D) Cells were grown on coverslips to confluency, allowed to polarize for 5 d, and fixed and stained with an anti-Scribble primary antibody, followed by an Alexa 488–labeled secondary antibody (green). Nuclei were stained with DAPI (blue). Images were obtained at 400× magnification. Right, enlarged images. Bar, 200 μm.
Because the levels of TβRIII in each stable cell line were too low to detect by immunofluorescence, we followed TβRIII localization by assessing the constitutive ectodomain shedding and release of soluble TβRIII into the media in a Transwell format. Consistent with the results observed with transient expression, the majority of soluble TβRIII was detected in the basal media in the WT TβRIII cell line (∼64%; Figure 2B). However, only ∼33% of soluble TβRIII was detected in the basal media in the P826A TβRIII cell line (Figure 2B). We also examined the localization of endogenous soluble TβRIII in Caco-2 cells, which are a well-characterized epithelial cell model of polarity. Consistent with our observations in NMuMG cells, the majority of soluble TβRIII was detected in the basal media of Caco-2 cells (Figure 2B). Of interest, no apical TβRIII was detectable in WT TβRIII cells by immunofluorescence (Figure 1B), yet a percentage of the signal was detected in the apical media by the enzyme-linked immunosorbent assay (ELISA) (Figure 2B). Because ELISA is a more sensitive and quantitative method than immunofluorescence, this indicates that a fraction of endogenous TβRIII is delivered apically in NMuMG and Caco-2 cells. Alternatively, some basal-to-apical transcytosis may occur. Collectively these data suggest that the majority of TβRIII is basolaterally localized in polarized epithelial cells. Of interest, the type I and type II TGF-β receptors have also been localized at or near the basolateral membrane in NMuMG and MDCK cells (Murphy et al., 2004, 2007), suggesting that most TGF-β signaling occurs from the basolateral membrane in polarized cells.
Whereas EV and WT TβRIII-expressing cells exhibited the typical cobblestone-like epithelial cell morphology of parental NMuMG cells, P826A TβRIII-expressing cells displayed a more mesenchymal phenotype, with elongated, spindle-shaped cells and fewer cell–cell contacts (Figure 2C). shTβRIII cells also displayed this mesenchymal phenotype, although to a lesser degree (Figure 2C). The mesenchymal phenotype suggested that P826A TβRIII and shTβRIII cells might have disrupted epithelial cell polarity. To investigate this possibility, we used immunofluorescence to examine the organization of the Scribble protein, which is critical for the proper formation of apical–basolateral cell polarity (Yamanaka and Ohno, 2008). Whereas Scribble staining was well organized and localized to cell–cell junctions in EV and WT TβRIII-expressing cells, this pattern was clearly disrupted in P826A TβRIII cells and was partially disrupted in shTβRIII cells (Figure 2D). Similar results were observed with PALS, another critical regulator of apical–basolateral cell polarity (Supplemental Figure S1). Collectively these data indicate that shTβRIII and P826A TβRIII NMuMG cells have undergone a loss of epithelial cell apical–basolateral polarity.
The observed mislocalization of P826A TβRIII could result from either apical misdelivery or disruption of normal cell polarity. To determine whether P826A TβRIII is targeted properly, we used a lentiviral Tet-inducible system to acutely induce WT or P826A TβRIII expression. Both WT and P826A TβRIII were readily induced by 12 h of doxycycline treatment (unpublished data). E-cadherin staining of doxycycline-treated WT and P826A TβRIII Transwell filters revealed that cell polarity was still intact after 12 h of induction (Figure 3A). The majority of soluble TβRIII was secreted basolaterally in induced WT TβRIII cells (60%; Figure 3B). In contrast, no above-baseline signal for soluble TβRIII was detectable in the basolateral media from P826A TβRIII cells (Figure 3B), with 100% of the detectable signal occurring in the apical media. These results were verified in Caco-2 cells, in which significantly more soluble TβRIII was found within the apical chamber in Tet-induced P826A TβRIII cells in comparison to WT TβRIII cells, confirming the importance of residue P826 in the targeting of TβRIII. Collectively these results suggest that the P826A mutation results in apical misdelivery (Figure 3B), with this apical localization occurring before, and perhaps mediating, the subsequent loss of polarity.
FIGURE 3.
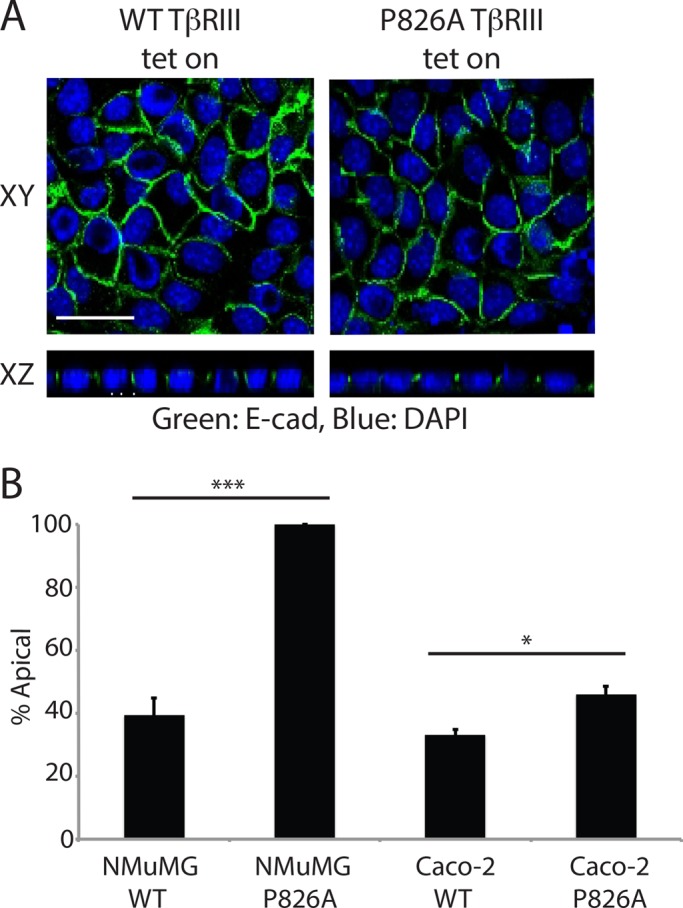
P826A TβRIII is mistargeted. (A) Tet-inducible NMuMG WT and P826A TβRIII cells were grown on Transwells for 5 d. Cells were treated with 1.5 μg/ml doxycycline for 12 h and fixed and stained with an anti-TβRIII primary antibody, followed by an Alexa 488–labeled secondary (green). Nuclei were stained with DAPI (blue). Images were taken at a magnification of 400× and are shown as flat sections (XY) above cross-sectional images (XZ). Bar, 25 μm. (B) The apical and basal media were collected from Tet-induced NMuMG and Caco-2 Tet-inducible cells and analyzed for soluble TβRIII by ELISA. The percentage of signal in the apical vs. basolateral chamber was calculated, and the percentage of signal in the apical chamber was graphed. ***p < 0.001, *p < 0.05 (Student's t test).
P826A TβRIII induces EMT
The loss of polarity and change in cell morphology observed with the stable loss of TβRIII or P826A TβRIII expression in NMuMG cells are consistent with an epithelial-to-mesenchymal transition (EMT). Because TGF-β is a known inducer of EMT, we used immunofluorescence, Western blotting, and quantitative PCR (qPCR) to follow the expression and localization of several epithelial and mesenchymal markers over a time course of TGF-β treatment to examine the effect of P826A TβRIII expression on EMT.
Polarized NMuMG cells typically exhibit cortical actin staining and a junctional localization of the epithelial markers E-cadherin and β-catenin. Consistent with this, actin staining was mainly cortical, and minimal stress-fiber formation was observed in untreated EV, shTβRIII, and WT TβRIII NMuMG cells (Figure 4A). Treatment with TGF-β induced stress fiber formation in EV, shTβRIII, and WT TβRIII cells by 24 h (Figure 4A). In contrast, stress fiber formation was already evident in untreated P826A TβRIII-expressing cells (Figure 4A, right), and stress fibers continued to accumulate after TGF-β ligand stimulation (Figure 4A).
FIGURE 4.

P826A TβRIII cells have reduced epithelial marker expression. (A) Cells were grown on coverslips and treated with 100 pM TGF-β1 for 0, 6, 24, or 48 h. Cells were subsequently stained with Alexa 488–conjugated phalloidin for visualization of actin (green). Enlarged images show the basal difference between actin staining in the WT and P826A TβRIII cells. Bar, 200 μm. (B) Cells were grown and treated as in A and stained with a primary antibody against E-cadherin, followed by an Alexa 594–conjugated secondary antibody (red). Nuclei were stained with DAPI (blue). Enlarged images show the basal difference in E-cadherin staining between WT and P826A TβRIII cells. All images were taken at 400× magnification. Bar, 200 μm. (C) Cells were grown in six-well dishes and treated with 100 pM TGF-β1 for 0, 6, 12, 24, 48, or 72 h. Cell lysates were analyzed by Western blotting for the epithelial marker E-cadherin. β-Actin was used as a loading control. The ratios of E-cadherin to actin are noted beneath each lane. (D) Cells were grown in six-well format and treated with 100 pM TGF-β1 for 0, 24, and 72 h. To analyze the differences in E-cadherin mRNA levels, total RNA was harvested, and qPCR for E-cadherin was performed. A representative graph shows the fold expression change for each cell line and time of treatment in comparison to the EV 0-h time point. Data were normalized to GAPDH expression. The experiment was repeated three times.
Similar results were observed with E-cadherin and β-catenin staining, for which, in the absence of TGF-β ligand stimulation, P826A TβRIII cells lacked significant E-cadherin staining and displayed disrupted β-catenin staining (Figure 4B, right; and unpublished data). In contrast, all other cell lines required 48 h of TGF-β treatment to observe the same effects on E-cadherin expression (Figure 4B). In addition, E-cadherin protein levels (Figure 4C) were decreased more rapidly and more efficiently in P826A TβRIII cells relative to EV, shTβRIII, and WT TβRIII cells (Figure 4C). Further, the mRNA level of E-cadherin was basally reduced in P826A TβRIII cells and was rapidly lost upon TGF-β–mediated EMT induction (Figure 4D). shTβRIII and P826A TβRIII cells had similar basal levels of E-cadherin mRNA, consistent with their mesenchymal morphology.
We also examined the expression of several mesenchymal markers. shTβRIII and P826A TβRIII cells showed an increase in fibronectin accumulation in response to TGF-β in comparison to EV and WT TβRIII cells (Figure 5A). Fibronectin accumulation was detectable in P826A TβRIII cells before TGF-β treatment (Figure 5A). We also measured the protein and mRNA levels of the mesenchymal marker α-SMA. Similar to fibronectin, the protein levels of α-SMA accumulated more rapidly in the P826A TβRIII cells than in the other cell lines (Figure 5B). Of interest, no α-SMA protein accumulation was detected in the WT TβRIII cells, suggesting that WT TβRIII may inhibit some aspects of EMT (Figure 4B). α-SMA mRNA expression was also dramatically elevated in P826A TβRIII cells compared with the other cell lines after 72 h of treatment (Figure 5C).
FIGURE 5.

P826A TβRIII cells have enhanced mesenchymal marker expression. (A) Cells were grown on coverslips, treated with 100 pM TGF-β1 for 0, 6, 24, or 48 h, and stained with an anti-fibronectin primary antibody, followed by an Alexa 488–conjugated secondary antibody (green). DAPI was used for nuclear staining (blue). Enlarged images show the difference in fibronectin staining between WT and P826A TβRIII cells at the 48-h time point. All images were taken at 400× magnification. Bar, 200 μm. (B) Cells were grown in six-well dishes and treated with 100 pM TGF-β1 for 0, 6, 12, 24, 48, or 72 h. Cell lysates were analyzed by Western blotting for the mesenchymal marker α-SMA. β-Actin was used as a loading control. The ratios of α-SMA to β-actin are noted beneath each lane. (C) Cells were grown in six-well dishes and treated with 100 pM TGF-β1 for 0, 24, and 72 h. To analyze the differences in α-SMA mRNA levels, total RNA was harvested, and qPCR for α-SMA was performed. A representative graph shows the fold expression change for each cell line and time of treatment in comparison to the EV 0-h time point. Data were normalized to GAPDH expression. Each experiment was repeated three times. (D) Cells were grown in six-well format and left untreated to analyze the basal levels of Snail and Slug mRNA. Total RNA was harvested, and qPCR for Snail (left) or Slug (right) was performed. The representative graphs show the fold expression change for each cell line in comparison to EV cells. Data were normalized to GAPDH expression. Each experiment was repeated a minimum of three times.
To explore which transcription factors might be driving the EMT process in P826A TβRIII cells, we examined the mRNA levels of the key EMT regulators Snail and Slug (also known as Snail2) by qPCR. Although no differences were observed with TGF-β treatment (unpublished data), the basal mRNA levels of these two transcription factors were higher in the P826A TβRIII and shTβRIII cells than in the EV and WT TβRIII cell lines (Figure 5D), providing further evidence that the EMT transcriptional program is basally activated in the P826A TβRIII cell line. Collectively these data suggest that the P826A TβRIII mutation partially induces EMT in the absence of exogenous ligand and primes cells to TGF-β–mediated EMT relative to EV or WT TβRIII cells.
P826A TβRIII-expressing cells exhibit increased proliferation
In addition to affecting EMT, TβRIII and TGF-β regulate cell proliferation. Using thymidine incorporation assays, we observed no significant differences between the EV, shTβRIII, WT, and P826A TβRIII cell lines over a 24-h time period, either in the presence or absence of TGF-β (Figure 6A). TGF-β inhibited proliferation in all cases, suggesting that the P826A mutation or loss of TβRIII does not alter the ability of TGF-β to suppress cell growth in this time frame.
FIGURE 6.
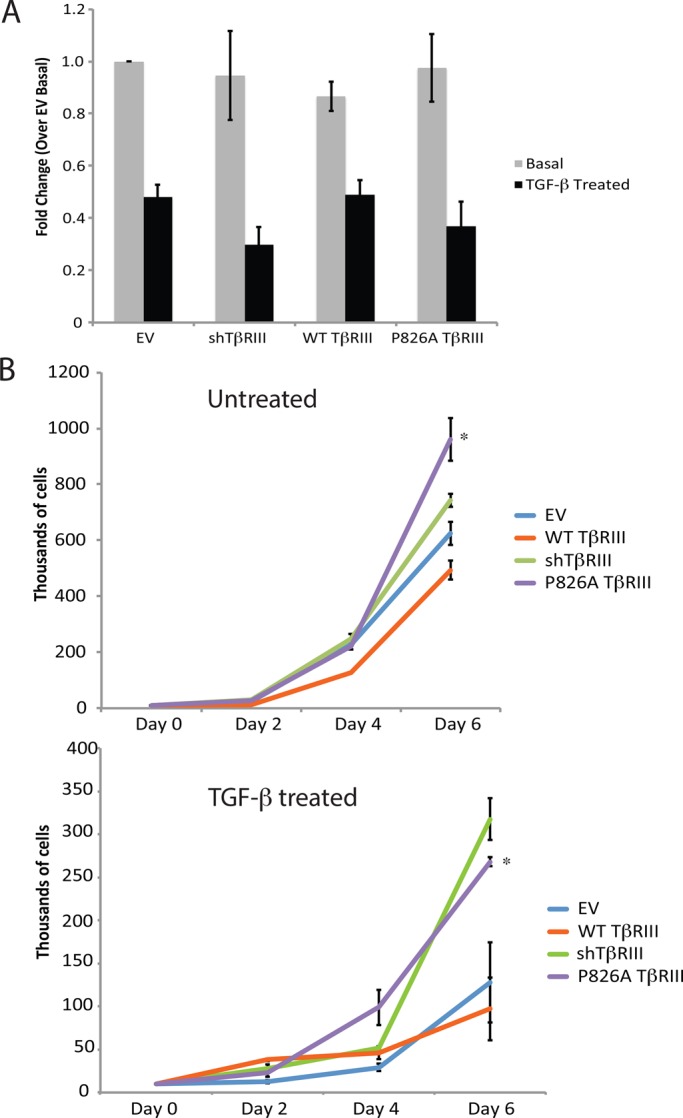
P826A TβRIII cells exhibit an enhanced rate of proliferation. (A) We plated 1 × 104 cells in triplicate in 96-well format and grew them for 24 h in the absence or presence of 100 pM TGF-β1. Cells were then labeled with 1 μCi of 3H for 4 h, and the level of incorporation was measured by scintillation counting. The graph depicts the fold change for each cell line over the untreated EV cells. Data were analyzed using Student's t test. No significant differences were found within each group (treated or untreated). The experiment was repeated three times. (B) We plated 1 × 104 cells in a six-well dish. Cells were grown in the presence (top) or absence (bottom) of 50 pM TGF-β1, with fresh media and TGF-β1 added every other day. Cells were trypsinized and counted 2, 4, and 6 d after plating, using a hemocytometer. Dead cells were excluded via staining with trypan blue. The graphs show the number of cells counted for each day (thousands) on the y-axis. Each cell line was plated in duplicate, and the experiments were performed three separate times. *p < 0.05 for P826A TβRIII compared with WT TβRIII cells at day 6 (Student's t test).
To determine whether TβRIII affected proliferation over a longer period of time, we used cell counting. No differences were noted between the untreated cell lines until day 4, when WT TβRIII-expressing cells exhibited decreased cell numbers (Figure 6B, top). By day 6, significant differences were observed among all cell lines, with cells expressing WT TβRIII having the lowest number of cells (Figure 6B, top). Compared to EV, shTβRIII and P826A TβRIII had more cells, with P826A TβRIII having more cells than shTβRIII (Figure 6B, top), suggesting that the P826A TβRIII mutation had more pronounced effects than the loss of TβRIII expression. In the presence of TGF-β, P826A TβRIII cells again had increased cell numbers compared with EV or WT TβRIII, with differences apparent at day 6 (Figure 6B, bottom). Of interest, shTβRIII and P826A TβRIII had similar cell numbers in the presence of ligand, suggesting that, although shTβRIII and P826A TβRIII cells are growth inhibited by TGF-β, they are less affected by its cytostatic actions over time relative to the other cell lines (Figure 6B; compare top and bottom). We also examined apoptosis in the absence or presence of TGF-β by Western blotting for cleaved caspase 3 at days 2, 4, and 6. However, cleavage levels remained low, and no significant differences were observed (unpublished data), supporting the idea the observed changes in cell number were due to changes in proliferation.
P826A TβRIII-expressing cells exhibit increased migration and invasion
We previously demonstrated that TβRIII regulates single-cell migration and invasion (Dong et al., 2007; Hempel et al., 2007; Turley et al., 2007; Gordon et al., 2008; Mythreye and Blobe, 2009; Lee et al., 2010). Therefore we examined the basal migratory and invasive capacities of the EV, shTβRIII, WT TβRIII, and P826A TβRIII cell lines. Consistent with previous reports, EV cells exhibited only a modest degree of migration (Figure 7A) or invasion (Figure 7B). However, shTβRIII cells exhibited a significant increase in both migration (Figure 7A) and invasion (Figure 7B). Consistent with prior reports, WT TβRIII expression rescued the shTβRIII-mediated increase in migration, further suppressing migration to a level lower than that observed for the EV cells (Figure 7A). For invasion, a similar suppressive trend was observed for the WT TβRIII cells; however, the invasive capacity of these cells was not significantly lower than that of the shTβRIII or EV cells (Figure 7B). In contrast, P826A TβRIII was unable to rescue the shTβRIII-mediated increases in migration (Figure 7A) or invasion (Figure 7B). Instead, P826A TβRIII cells exhibited twofold and threefold increases in the levels of migration and invasion, respectively, compared with shTβRIII cells (Figure 7, A and B).
FIGURE 7.

P826A TβRIII cells have enhanced migration and invasion. (A) We plated 25 × 103 (EV and WT TβRIII) or 15 × 103 (shTβRIII and P826A TβRIII) cells in 200 μl of serum free media and allowed them to migrate for 24 h toward 500 μl of medium containing 10% FBS. The number of migrated cells in three independent fields of view (100×) was counted for each cell line. Data are graphed as the number of migrating cells/the total plated cell number (percentage migration). (B) We plated 75 × 103 (EV and WT TβRIII) or 50 × 103 (shTβRIII and P826A TβRIII) cells in 500 μl of serum-free media and allowed them to invade for 24 h toward 600 μl of media containing 10% FBS. The number of invasive cells in three independent fields of view (100×) was counted for each cell line. Data are graphed as the number of invasive cells/the total plated cell number (percentage invasion). (C) WT and P826A TβRIII cells were plated and analyzed as in A and B, with the exception that dimethyl sulfoxide (DMSO; control [C]), 10 μg/ml TGF-β1–neutralizing antibody (Ab), or 10 μg/ml TβRI kinase inhibitor SB435142 (SB) was added to the upper chamber. N.S., not significant. ***p < 0.001, **p < 0.01, *p < 0.05 compared with DMSO (C) or EV (A,B) control (Student's t test).
Enhanced migration and invasion in P826A TβRIII cells is independent of TGF-β ligand and TβRI kinase activity
Because P826A TβRIII cells exhibited higher levels of basal invasion and migration than the other cell lines, to determine whether autocrine TGF-β production or signaling was involved, we performed the migration and invasion assays in the presence of a TGF-β–neutralizing antibody. Surprisingly, treatment with this antibody, although effective at inhibiting TGF-β–mediated Smad2 signaling (Supplemental Figure S2A), did not significantly decrease the migratory or invasive capacities of the WT or P826A TβRIII cell lines (Figure 7C). Further, WT and P826A TβRIII cells secreted similar levels of TGF-β over a 24-h time period (Supplemental Figure S2B). These data suggest that the secretion of TGF-β by P826A or WT TβRIII cells is not responsible for the observed differences in migration and invasion between these cell lines.
We next investigated whether TGF-β signaling was important for the enhanced migration and invasion of the P826A TβRIII cells. Of interest, phospho-Smad3 levels were slightly lower in shTβRIII and P826A TβRIII cells than in EV or WT TβRIII cells (Supplemental Figure S2C), suggesting that increased Smad3 activation is not responsible for the enhanced migration or invasion in P826A TβRIII cells. Although we observed enhanced TGF-β–induced Smad2 activation in P826A TβRIII cells compared with the other cell lines (Supplemental Figure S2D), the type I TGF-β receptor inhibitor SB431542 did not significantly decrease migration or invasion in P826A TβRIII cells, despite effectively inhibiting pSmad2 activation (Figure 7C and Supplemental Figure S2E). Collectively these data suggest that neither the TGF-β ligand nor TβRI kinase activity is responsible for the enhanced basal level of migration and invasion in P826A TβRIII cells.
TGF-β can also signal through a number of noncanonical pathways, including the p38 and Erk1/2 MAP kinases, NFκB, and PI3K/Akt. We examined the EV, shTβRIII, WT TβRIII, and P826A TβRIII cell lines for differences in basal and TGF-β–stimulated noncanonical signaling; however, no clear differences were observed by Western blotting (Supplemental Figure S3A). In addition, we tested whether small-molecule inhibitors of these noncanonical TGF-β signaling pathways could affect cell migration or invasion. However, no single inhibitor was able to specifically affect the invasion or migration capacities of the P826A TβRIII cells (Supplemental Figure S3B). Although some reduction in invasion was observed with the NFκB pathway inhibitor (IKK inhibitor IV) or the Src inhibitor (PP2), this was not unique to the P826A TβRIII cell line. Indeed, the WT TβRIII cell line was similarly affected (Supplemental Figure S3C).
P826A TβRIII cells form invasive tumors in athymic nude mice
To determine whether the localization of TβRIII affected tumorigenicity in vitro, we assessed colony formation in soft agar. A greater number of colonies was observed with the P826A TβRIII cells than in all of the other cell lines (Figure 8A), suggesting that, consistent with its EMT phenotype, P826A TβRIII increases tumorigenicity in vitro.
FIGURE 8.

P826A TβRIII cells form invasive tumors in mice. (A) For the soft colony assay, six-well dishes were coated with 0.8% agar, and 1 × 105 NMuMG cells in 0.4% agar were plated on top. Cells were plated in triplicate and were grown for 4 wk. The number of colonies per well was determined using crystal violet staining. Data were analyzed for significance by Student's t test. The p value for P826A TβRIII vs. WT TβRIII cells is indicated. (B) We injected 1 × 106 cells into the right #4 mammary gland of 6-wk-old female athymic nude mice. Tumors were measured with calipers weekly for a total of 14 wk. Tumor volume was calculated using volume = length × (width2) × 0.5. Ten mice were analyzed for each cell line. *p < 0.05 for P826A TβRIII mice compared with EV mice (Mann–Whitney test). (C) Primary tumors were excised and are shown to demonstrate size differences. The black lines indicate intervening samples that were removed for clarity. (D) Photos taken at 100× magnification show the overall tumor morphology as moderately to poorly differentiated and stroma rich. Bar, 400 μm. (E) Representative images (100×) showing localized invasion of the tumor into the muscle wall in an shTβRIII and a P826A TβRIII sample. Bar, 400 μm. (F) IHC was performed to detect incorporated BrdU. The percentage of BrdU-positive cells per field of view (100×) was determined for each cell line. No BrdU+ cells were observed in the single EV sample that was large enough for analysis. N = 3 for WT TβRIII, N = 8 for shTβRIII, and N = 10 for P826A TβRIII. Data were analyzed by the Mann–Whitney test. The p value for P826A TβRIII vs. WT TβRIII cells is noted above the graph. (G) Tumor sections were stained for E-cadherin (brown), which was absent from P826A and shTβRIII tumors but present around ductal structures in EV and WT TβRIII tumors (images are at 400× magnification). Bar, 100 μm.
Whereas parental NMuMG cells are not largely tumorigenic in vivo, stable NMuMG shTβRIII cells have been reported to form tumors in an orthotopic mouse model of breast cancer (Criswell and Arteaga, 2007). To determine whether P826A TβRIII altered the tumorigenicity of NMuMG cells, we orthotopically implanted EV, shTβRIII, WT TβRIII, or P826A TβRIII-expressing cells into athymic nude mice. Whereas a small number of EV (3 of 10) and WT TβRIII (4 of 10) mice developed tumors, the tumors remained small and were slow growing (Figure 8B). In contrast, nine of 10 shTβRIII and nine of 10 P826A TβRIII mice formed palpable tumors, which grew rapidly relative to EV and WT TβRIII tumors (Figure 8B). On comparing the P826A TβRIII and EV groups, these differences became significant at week 6 and remained significant until the date the mice were killed. P826A TβRIII mice also developed tumors earlier than EV and WT TβRIII mice. Tumors were confirmed in several shTβRIII and P826A TβRIII mice as early as week 4 but were not confirmed in EV or WT TβRIII mice until week 8 (Figure 8B).
After killing at week 14, the primary tumors, lungs, and mammary fat pads were harvested. The primary tumor size was noticeably larger for the shTβRIII and P826A TβRIII mice compared with the EV and WT TβRIII mice (Figure 8C). All tumors were stroma-rich, moderately to poorly differentiated carcinomas (Figure 8D). No macrometastases or micrometastases to the lung or noninjected fat pads were observed in any group. However, microscopic evaluation did confirm the presence of one additional primary tumor for the P826A group, bringing the total number of tumors in this group to 10. Of interest, we observed local invasion of the tumor into the chest wall in three of nine shTβRIII and three of 10 P826A TβRIII samples, whereas no local invasion was noted for tumors from EV or WT TβRIII mice (Figure 8E).
The WT and P826A TβRIII tumors retained TβRIII expression, as verified by immunohistochemistry (IHC; Supplemental Figure S4A). Before killing, the mice were intraperitoneally injected with bromodeoxyuridine (BrdU) to allow for the analysis of tumor cell proliferation. We observed a trend toward increased proliferation with TβRIII silencing or the expression of P826A TβRIII, consistent with our in vitro results (Figure 8F). We also examined apoptosis in the tumor sections by terminal deoxynucleotidyl transferase dUTP nick end labeling; however, we noted no differences (Supplemental Figure S4B). This suggests that the larger tumor size in the P826A TβRIII and shTβRIII groups is likely due to earlier tumor initiation coupled with an increase in tumor cell proliferation. In addition, consistent with the EMT phenotype of P826A TβRIII and shTβRIII cells in vitro, P826A TβRIII and shTβRIII tumors lacked E-cadherin expression in vivo, whereas E-cadherin was readily detectable in WT TβRIII tumors and in the single EV tumor specimen large enough for analysis, particularly around partially or completely filled-in ductal structures (Figure 8G).
DISCUSSION
Apical–basolateral cell polarity is essential for tissue and organ function and is often lost during carcinogenesis. Here we demonstrate that the mistargeting of a single protein, TβRIII, from predominantly basolateral to basolateral and apical membranes in breast epithelial cells results in a global disruption of cell polarity, an EMT phenotype, and increased proliferation, migration, and invasion. Further, mislocalization of TβRIII results in increased tumor growth and invasion in an in vivo mouse model of breast cancer.
TβRIII influences several aspects of cancer cell biology, including inhibiting migration and invasion in multiple cancer types (Dong et al., 2007; Turley et al., 2007; Finger et al., 2008; Gordon et al., 2008; Bilandzic et al., 2009; Mythreye and Blobe, 2009; Lee et al., 2010; Lambert et al., 2011; Mythreye et al., 2013), promoting fibroblast growth factor 2–mediated neuronal differentiation in neuroblastoma (Knelson et al., 2013), regulating immune system tolerance to breast cancer and melanoma (Hanks et al., 2013), and proliferation in breast cancer, kidney cancer, and multiple myeloma (Sun and Chen, 1997; Copland et al., 2003; Margulis et al., 2008; Lambert et al., 2011). These complex and diverse TβRIII functions underscore the importance of this cell surface receptor in cancer development and progression. Here we have identified a novel role for TβRIII in the regulation of breast epithelial cell polarity. Although TβRIII expression is typically lost during breast cancer progression (Dong et al., 2007), the loss of cell polarity can occur quite early in carcinogenesis (Debnath et al., 2003; Lee and Vasioukhin, 2008; Zhan et al., 2008). Therefore the proper regulation of cell polarity by TβRIII may be of critical importance for normal breast epithelial cell homeostasis.
Consistent with our data, Criswell and Arteaga (2007) demonstrated that loss of TβRIII expression in NMuMG cells results in increased EMT, proliferation, migration, and invasion. In our in vitro experiments, the P826A mutation resulted in more-severe phenotypes than did the loss of TβRIII expression, suggesting that the P826A alteration does not simply abolish TβRIII function. Although our in vitro experiments highlight the differences between shTβRIII and P826A TβRIII cells, these cells behaved similarly in our in vivo assay. Indeed, P826A and shTβRIII tumors grew at similar rates, lacked E-cadherin expression, and were locally invasive (Figure 8). One explanation for these results is that the tumor cells were exposed to TGF-β superfamily ligands in vivo. In support of this, whereas the P826A and shTβRIII cell lines grew at different rates in vitro in the absence of TGF-β, these two lines grew at similar rates in its presence (Figure 6B). Exposure to TGF-β in vivo would also drive EMT, resulting in a more complete loss of E-cadherin. In addition, although both P826A and TβRIII cells exhibited localized invasion, the time course of our in vivo experiment was too short to allow for the formation of metastases. It is possible that differences in metastasis, due to differences in invasion/migration between P826A and shTβRIII cells, would be observable with additional time or with a more aggressive model of breast cancer. Although only a small percentage of cells were migratory or invasive in our in vitro assay, this is consistent with the idea that metastases arise from a subpopulation of cells within a primary tumor (Fidler and Kripke, 1977; Wang et al., 2004, 2007; Plaks et al., 2013). Indeed, recent studies suggest that EMT may contribute to the formation of “cancer stem cells” or “tumor-initiating cells,” which exhibit increased migratory and invasive capacities yet comprise only a small percentage of the primary tumor (Chaffer and Weinberg, 2011). Therefore future studies will investigate the role of TβRIII localization in metastasis.
P826A TβRIII expression likely results in the unique deregulation of one or more signaling pathways. With regard to the EMT phenotype, we observed a reduction in the mRNA levels of both Snail and Slug in P826A TβRIII cells treated with the TβRI kinase inhibitor and a suppression of TGF-β–mediated α-SMA induction with both the TβRI kinase inhibitor and the TGF-β–neutralizing antibody, suggesting that a TGF-β and/or TβRI kinase–mediated pathway is likely involved in this phenotype (unpublished data). Surprisingly, neither TGF-β nor TβRI kinase activity had a role in the enhanced migration and invasion observed in P826A TβRIII cells, as the kinase and TGF-β inhibitors were unable to decrease migration or invasion in this cell line despite inhibiting Smad2 activation (Figure 7C). In addition, WT and P826A TβRIII cells secreted similar levels of TGF-β1 ligand (Supplemental Figure S2B). Collectively these data suggest that a unique TGF-β/TβRI kinase–independent signaling pathway is responsible for the enhanced migration and invasion capacities of P826A TβRIII cells. To identify the signaling pathway involved, we screened a panel of chemical inhibitors for their effect on P826A TβRIII cell migration/invasion. However, no single inhibitor was able to selectively decrease P826A TβRIII-mediated migration (Supplemental Figure S3B). Although the NFκB and Src inhibitors decreased invasion in P826A TβRIII cells, they also did so in WT TβRIII cells, suggesting that these pathways globally regulate invasion in NMuMG cells (Supplemental Figure S3, B and C).
Collectively these data suggest that different signaling pathways are responsible for the EMT versus invasion and migration phenotypes observed upon P826A TβRIII expression, with TGF-β/TβRI signaling influencing EMT and a unique TGF-β/TβRI-independent pathway influencing migration and invasion. Although the specific signaling pathway(s) responsible for the enhanced migration and invasion phenotypes in P826A TβRIII cells is unknown, other growth factor signaling pathways, including epidermal, fibroblast, platelet-derived, or hepatocyte growth factors, may be involved. Integrin-mediated signaling might also have a role. Indeed, we recently demonstrated that TβRIII can modulate epithelial cell motility by regulating the function and localization of α5β1 integrin in a TGF-β–independent manner (Mythreye et al., 2013). Alternatively, a signaling pathway may have been transiently activated by early P826A TβRIII expression, setting up the EMT program and subsequent effects on migration and invasion. We attempted to examine cell signaling using a Tet-inducible P826A TβRIII cell line. However, the presence of endogenous TβRIII complicated data interpretation and, due to their loss of polarity and partial EMT, shTβRIII cells were not suitable for such experiments. The precise pathway by which P826A TβRIII regulates EMT and subsequent biology remains to be determined.
In NMuMG cells, the type I TGF-β receptor was localized to tight junctions (Ozdamar et al., 2005). The authors demonstrated that TβRI associates with the Par6 polarity protein. On TGF-β treatment, TβRII joins the complex and phosphorylates Par6, resulting in Smurf1 recruitment and RhoA degradation. This pathway was shown to be important for the dissolution of tight junctions during TGF-β–mediated EMT. Consistent with the activation of this pathway, the two cell lines with a basal EMT-like phenotype, shTβRIII and P826A TβRIII, exhibited lower levels of RhoA than did WT TβRIII or EV cells (unpublished data). In contrast to Ozdamar et al. (2005), Murphy et al. (2004, 2007) defined a basolateral localization of TβRI and TβRII in MDCK cells. Although the precise localization of the type I and II TGF-β receptors differs in these two reports, a localization of all three TGF-β receptors to cell junctions would allow for more precise control over junctional integrity. Indeed, both tight junction and adherens junction components have been shown to be associated with a number of signaling molecules that regulate gene transcription, proliferation, and apoptosis (Perez-Moreno and Fuchs, 2006; Hartsock and Nelson, 2008; Balda and Matter, 2009). Junction-associated signaling pathways also affect actin dynamics, which directly affect cell motility and invasion (Perez-Moreno and Fuchs, 2006; Hartsock and Nelson, 2008).
Collectively these studies suggest that the three TGF-β receptors serve key signaling roles at cell–cell junctions, controlling epithelial tissue organization and function. Disruption of receptor function through mistargeting or inappropriate EMT can have significant and lasting effects on cell and tissue function and contribute to tumor growth and invasion.
MATERIALS AND METHODS
Cell culture and reagents
NMuMG cells were purchased from the American Type Culture Collection and grown in DMEM (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Corning, Tewksbury, MA) and 10 μg/ml insulin (Life Technologies). EMT was induced with 100 pM TGF-β1 (R&D Systems, Minneapolis, MN). Caco-2 cells were purchased from the American Type Culture Collection and grown in MEM (Life Technologies) supplemented with 20% FBS. HEK 293FT cells (Invitrogen, Grand Island, NY) were grown in DMEM supplemented with 10% FBS, 0.1 mM nonessential amino acids, 6 mM l-glutamine, and 1 mM sodium pyruvate (Life Technologies). MDCK cells were purchased from the American Type Culture Collection and grown in DMEM supplemented with 10% FBS. All cell lines were grown at 37°C in a humidified incubator at 5% CO2. Fluorescein isothiocyanate (FITC)–dextran (anionic, molecular weight 40,000, D1844) was purchased from Life Technologies. For transfection of pcDNA 3.1 human WT TβRIII and variants, Lipofectamine 2000 reagent (Life Technologies) was used according the manufacturer's protocol. All restriction enzymes were purchased from New England Biolabs and were used as directed. The following antibodies were used in this study: TβRIII (AF-242-PB; R&D Systems), Scribble (sc-11049; Santa Cruz Biotechnology, Dallas, TX), α-SMA and fibronectin (A5228 and F3648; Sigma-Aldrich, St. Louis, MO), E-cadherin (610182; BD Transduction), and β-catenin (06-734, EMD Millipore, Billerica, MA). The TGF-β–neutralizing antibody 2G7 was supplied by Genentech. The TGF-β RI kinase inhibitor SB431542 was purchased from Sigma-Aldrich. Cells expressing Tet-inducible TβRIII were grown in media supplemented with 10% Tet System Approved FBS (Clontech) and induced with 1.5 μg/ml doxycycline (Enzo Life Sciences, Farmingdale, NY).
Apico-basolateral Transwell growth
We plated 2.5 × 105 cells in a total volume of 900 μl in the inner chamber of 0.4-μm pore-sized Transwells (Corning). We placed 1 ml of media into the bottom chamber. Cells were allowed to polarize for 5 d before further assays were performed. Cell polarization was measured using fluorescein-conjugated dextran. For this, 30 μl of 25 mg/mL FITC-dextran was placed into the upper chamber. After 30 min, the samples were diluted 1:1000. Excitation was carried out at 490 nm, and the absorbance for the upper and lower chambers was measured at 520 nm. Cells were considered polarized when <10% of the total fluorescence was detectable in the lower chamber.
Standard PCR, mutagenesis, and qPCR
NAAIRS mutants and single alanine alterations were made using pcDNA3.1 human WT TβRIII as a template. 5′-CCC CAC CTC CCC GAA TGC TGC TAT ACG ATC GAG TGC TGC CCA CAG C-3′ and its genetic complement were used to generate the PASENS → NAAIRS mutation, and 5′-GCA AGT CCC CAC CTC CCC GGC AGC CTC GGA AAA CAG CAG TGC-3′ and its genetic complement were used to generate the P826A TβRIII mutation. All mutations were made using the QuikChange Mutagenesis Kit (Agilent) according to the manufacturer's recommendations. All standard PCRs for the purpose of cloning were performed using Pfu Turbo (Agilent) in a Bio-Rad iCycler according to the manufacturer's recommendations.
For qPCR, total RNA was isolated from cells using the RNeasy Mini Kit (74106; Qiagen), and first-strand synthesis was performed using the iScript cDNA Synthesis Kit (170-8891; Bio-Rad). Quantitative real time PCRs were performed using the iQ SYBR Green SuperMix (170-882; Bio-Rad) and a Bio-Rad iCycler. Relative transcript levels were compared via the ΔΔCt method. All data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences used were as follows:
Mouse E-cadherin, forward: 5′-CCT TCC CCC AAC ACG TCC CCCC-3′
Mouse E-cadherin, reverse: 5′-TCT CCA CCT CCT TCT TCA TC-3′
Mouse α-SMA, forward: 5′-GGA CGT ACA ACT GGT ATT GTGC-3′
Mouse α-SMA, reverse: 5′-TCG GCA GTA GTC ACG AAG GA-3′
Mouse Snail, forward: 5′-GGA AGC CCA ACT ATA GCG AGC-3′
Mouse Snail, reverse: 5′-CAG TTG AAG ATC TTC CGC GAC-3′
Mouse Slug, forward: 5′-CTC ACC TCG GGA GCA TAC AGC-3′
Mouse Slug, reverse: 5′-TGA AGT GTC AGA GGA AGG CGG G-3′
Mouse GAPDH, forward: 5′-TTG ACC TCA ACT ACA TGG TCT A-3′
Mouse GAPDH, reverse: 5′-ACC AGT AGA CTC CAC GAC ATA C-3′
Retroviral and lentiviral production and stable cell line generation
To stably silence endogenous murine TβRIII, the complementary primers 5′-GAT CCC CGA AAT GAC ATC CCT TCC ACT TCA AGA GAG TGG AAG GGA TGT CAT TTC TTT TTC-3′ and 5′-TCG AGA AAA AGA AAT GAC ATC CCT TCC ACT CTC TTG AAG TGG AAG GGA TGT CAT TTC GGG-3′ were annealed and cloned into the BglII and XhoI sites of the pSuper.retro.puro vector (OligoEngine). For annealing, 1 μl of each oligo (3 mg/ml starting) was mixed with 48 μl of annealing buffer (100 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4). The mixture was incubated at 90°C for 4 min and 70°C for 10 min and then cooled in a stepwise manner to 37°C over a 20-min time period.
To generate pSMPUW (neo) lentiviral vectors, WT TβRIII and P826A TβRIII were PCR amplified and cloned into the EcoRV site in pSMPUW neo (Cell Biolabs). To generate Tet-on WT and P826A TβRIII, TβRIII was PCR amplified and cloned into the EcoRI site in the pLVX-Tight-Puro plasmid (Clontech). All constructs were screened by sequencing for proper orientation.
To generate retroviral particles, the pSuper.retro.puro shTβRIII (9 μg), gag/pol (15 μg), and VSVG (6 μg) plasmids were transfected into HEK 293FT cells. The medium was changed the day after transfection, and viral particles were harvested after a further 48 h of incubation. To generate lentiviral particles, the pMDL g/pRRE (3 μg), pRSV-Rev (2 μg), and pMD2.G (2 μg) packaging vectors and the appropriate lentiviral vector (pSMPUW TβRIII, pLVX-Tight-Puro TβRIII, or pLVX-Tet-On Advanced; 6 μg) were similarly transfected into HEK 293FT cells. Viral particles were harvested and stored as described.
To generate noninducible stable cell lines, NMuMG cells were infected with pSuper.retro.puro (EV) or pSuper.retro.puro shTβRIII in the presence of 6 μg/ml Polybrene (Sigma-Aldrich). Cells were selected in 2 μg/ml puromycin (Sigma-Aldrich). After confirmation of shTβRIII knockdown, cells were infected with pSMPUW-neo (EV), pSMPUW-WT TβRIII, or pSMPUW-P826A TβRIII lentiviral particles. Cells were selected in the presence of 500 μg/ml G418 (KSE Scientific). Stable cell lines were maintained in 1 μg/ml puromycin and 250 μg/ml G418. To generate Tet-inducible stable cell lines, NMuMG cells were infected with pLVX-Tet-On Advanced and either pLVX-Tight-Puro WT TβRIII or pLVX-Tight-Puro P826A TβRIII lentiviral particles. Cells were selected in 500 μg/ml G418 and 2 μg/ml puromycin.
Binding and cross-linking of [125I]TGF-β
Binding and cross-linking of 125I-labeled TGF-β ligand was performed as described previously (Jelinek et al., 2003; Dong et al., 2007; Gordon et al., 2008; Mythreye and Blobe, 2009). Immunoprecipitated samples were separated by SDS–PAGE and visualized using a Typhoon 9200 Variable Mode Phosphorimager (GE Healthcare).
ELISA
Cells were grown in Transwells for 5 d and then incubated with fresh media. Soluble TβRIII was allowed to accumulate for 24 h, with the exception of Tet-inducible cell lines, for which accumulation occurred over a 12-h time period. Media samples were collected, spun down to remove dead cells and debris, and frozen at −80°C.
For ELISA, the capture antibody (AF-242-PB; R&D Systems) was immobilized onto a Multi-Array 96-well plate (L15XA-3; Meso Scale Discovery [MSD]) overnight at 4°C. The plates were washed, the samples were loaded, and the plates were then incubated at room temperature for 2 h with shaking. The detection antibody (BAF-242; R&D Systems), coincubated with streptavidin-SULFO-TAG (R32AD-1; MSD) secondary antibody, was then applied, and the plates were incubated for 2 h with shaking. The plates were washed, and Read Buffer T (R92TC-3; MSD) was added. Images of the plates were taken within 10 min using an MSD Sector Imager 2400. The images were analyzed using MSD Sector Imager software, version 3.0. Analyte concentrations were calculated based on a standard curve derived by performing four serial dilutions of the corresponding protein standard on each plate. Samples were tested in duplicate, and the mean value was used for analysis.
Immunofluorescence and microscopy
Cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% TX-100 for 1 min (actin) or 10 min (all others). Blocking was done in 0.5% BSA in phosphate-buffered saline (PBS) for 1 h and was followed by a 20-min incubation with a 1:50 dilution of phalloidin-conjugated Alexa 488 for actin (Invitrogen) or a 1-h incubation with a 1:400 dilution of primary antibody (all others). Cells were washed three times with PBS and mounted (actin) or further incubated with a 1:500 dilution of Alexa 488– or 595–conjugated secondary antibody (Invitrogen) for 30 min. After washing, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Roche) and were mounted with ProLong Gold Antifade reagent (Life Technologies). Immunofluorescence imaging and z-stacking were performed using a Leica SP5 confocal microscope at a magnification of 400×.
Western blotting
Membranes were incubated with primary antibodies in 5% milk PBS/0.1% Tween overnight at 4°C. Membranes were then washed and incubated with horseradish peroxidase–conjugated secondary antibodies for 1 h at room temperature, and proteins were detected by the ECL Plus chemiluminescence system (GE Healthcare). Bands were visualized using Hyperfilm ECL film (GE Healthcare) and quantified by computer-assisted densitometry.
Thymidine incorporation and cell counting
For thymidine incorporation, 3000 cells were plated in each well of a 96-well plate in 100 μl of DMEM/10% FBS/insulin with or without TGF-β ligand (50 pM). Cells were pulsed with 1 μCi of 3H for 4 h at 37°C, washed in cold PBS and 10% trichloroacetic acid (TCA), and incubated for 1 h at 4°C in 10% TCA. Cells were washed with cold 10% TCA and lysed overnight in 0.2 M NaOH, and lysates were read using a scintillation counter.
For cell counting assays, 1 × 104 cells were plated in six-well dishes in the absence or presence of 50 pM TGF-β ligand. Cells were collected by trypsinization every other day, stained with trypan blue to exclude dead cells, and counted using a hemocytometer. All samples were plated in duplicate, and the assay was performed three separate times.
Invasion and migration assays
For invasion assays, Matrigel-coated Transwells (BD Biosciences, San Jose, CA) were preincubated with serum-free DMEM for 2 h at 37°C. We plated 75 × 103 (NMuMG EV and WT TβRIII) or 50 × 103 (NMuMG shTβRIII and P826A TβRIII) cells in 500 μl of serum-free media and allowed them to invade for 24 h toward 600 μl of media containing 10% FBS. Migration chambers (BD Biosciences) were coated with 50 μg/ml fibronectin (Calbiochem) in serum-free DMEM for 2 h at 37°C. We plated 25 × 103 (NMuMG EV and WT TβRIII) or 15 × 103 (NMuMG shTβRIII and P826A TβRIII) cells in 200 μl of serum-free media and allowed them to migrate for 24 h toward 500 μl of media containing 10% FBS. For both assays, cells remaining in the upper chamber were removed with a cotton swab, and the filters were stained using a Three-Step Stain Kit (Richard-Allan Scientific). Filters were mounted with VectaMount (Vector Laboratories). For each filter, the cells present in three random fields of view were counted. Images were collected at 100× magnification. Cells were plated in duplicate, and each experiment was conducted three times. When used, 10 μg/ml of the TGF-β–neutralizing antibody or the TGF-β RI kinase inhibitor was added simultaneously with cell plating. For Smad2-knockdown conditions, cells were first transfected with 2 μg of shSmad2 and incubated for 48 h before plating for migration or invasion.
Soft agar assay
Six-well dishes were coated with 0.8% agar in DMEM supplemented with 10% FBS and 10 μg/ml insulin. We plated 1 × 105 NMuMG cells/well in 0.4% agar/DMEM/10% FBS/insulin. Cells were incubated at 37°C for 4 wk and fed every 3 d. For colony detection, cells were fixed and stained with 0.005% crystal violet in 10% neutral buffered Formalin solution and washed with PBS. Colonies were counted and quantified using Bio-Rad Quantity One software.
In vivo xenograft assay
The Institutional Animal Care and Use Committee of Duke University approved all animal procedures. At 24 h before injection, NMuMG EV, shTβRIII, WT TβRIII, and P826A TβRIII cells were transferred to selection-free media. We resuspended 1 × 106 cells in 200 μl of PBS and injected them into the right #4 mammary gland of 6-wk-old female athymic nude mice (Harlan) using a 22-gauge needle. Tumors were measured with calipers weekly for a total of 14 wk. Tumor volume was calculated using volume = length × (width2) × 0.5. At 2 h before killing, mice were intraperitoneally injected with 0.01 ml/g of total body weight of a 7 mg/ml BrdU stock solution. After killing, the mammary glands, lungs, and primary tumors were removed and fixed in 4% paraformaldehyde (2 h), followed by 70% ethanol. We embedded 6-μm-thick sections in paraffin for further analysis. Sections of tumors, lungs, and mammary glands were stained with hematoxylin and eosin to examine histology.
Immunohistochemistry
For E-cadherin staining, tumor sections were deparaffinized in two changes of xylene for 10 min, each followed by 100, 95, 70, and 50% ethanol solutions for 5 min each. Sections were rinsed in deionized water for 5 min and boiled for 20 min in 10 mM citric acid, pH 6.0. After cooling (20 min), sections were quenched in 3% H2O2 in methanol for 30 min. After two washes in PBS (2 min each), tissues were blocked using the Avidin/Biotin Blocking Kit (SP-2001; Vector Laboratories) according to the manufacturer's instructions. Tissues were further processed for E-cadherin staining using the Mouse On Mouse (MOM) IHC kit (MMK-2202; Vector Laboratories) according to the manufacturer's instructions. A 1:200 dilution of the primary antibody (610182; BD Transduction) was used. Samples were further processed with the Vectastain mouse ABC kit (PK-4002; Vector Laboratories) for 20 min, and staining was visualized with DAB reagent (SK-4100; Vector Laboratories). After washing in PBS, samples were stained with hematoxylin and rehydrated in 50, 70, 95, and 100% ethanol, followed by xylene (5 min each). Sections were mounted using VectaMount reagent (H-5000; Vector Laboratories). For BrdU staining, the BrdU In-Situ Detection kit (550803; BD PharMingen) was used according to the manufacturer's instructions.
Supplementary Material
Acknowledgments
We thank Duke University pathologists Michael B. Datto and Joseph Geradts for help in reviewing mouse tissue slides. We also thank fellow lab members for helpful suggestions and critical reading of the manuscript. This work was supported by National Institutes of Health Grants 5F32CA157030 (A.E.M.), R01-CA136786 (G.C.B.), and R01-CA135006 (G.C.B.) and Komen for the Cure Grant SAC100002 (G.C.B.).
Abbreviations used:
- α-SMA
α-smooth muscle actin
- BrdU
bromodeoxyuridine
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- EMT
epithelial-to-mesenchymal transition
- EV
empty vector
- IHC
immunohistochemistry
- TβRIII
type III transforming growth factor-β cell surface receptor
- TGF-β
transforming growth factor-β
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-03-0825) on May 28, 2014.
REFERENCES
- Aigner K, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial cell polarity. Oncogene. 2007;26:6979–6988. doi: 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroeti B, Okhrimenko H, Reich V, Orzech E. Polarized trafficking of plasma membrane proteins: emerging roles for coats, SNAREs, GTPases and their link to the cytoskeleton. Biochim Biophys Acta. 1998;1376:57–90. doi: 10.1016/s0304-4157(98)00005-7. [DOI] [PubMed] [Google Scholar]
- Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim Biophys Acta. 2009;1788:761–767. doi: 10.1016/j.bbamem.2008.11.024. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Gracia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Bazellieres E, Assemat E, Arsanto JP, Le Bivic A, Massey-Harroche D. Crumbs proteins in epithelial morphogenesis. Front Biosci (Landmark Ed) 2009;14:2149–2169. doi: 10.2741/3368. [DOI] [PubMed] [Google Scholar]
- Bilandzic M, Chu S, Farnworth PG, Harrison C, Nicholls P, Wang Y, Escalona RM, Fuller PJ, Findlay JK, Stenvers KL. Loss of betaglycan contributes to the malignant properties of human granulosa tumor cells. Mol Endocrinol. 2009;23:539–548. doi: 10.1210/me.2008-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Zhurinsky J, Ben-Ze'ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copland JA, Luxon BA, Ajani L, Maity T, Campagnaro E, Guo H, LeGrand SN, Tamboli P, Wood CG. Genomic profiling identifies alterations in TGFbeta signaling through loss of TGFbeta receptor expression in human renal cell carcinogenesis and progression. Oncogene. 2003;22:8053–8062. doi: 10.1038/sj.onc.1206835. [DOI] [PubMed] [Google Scholar]
- Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17:499–508. doi: 10.1016/j.ceb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Gibbons DL, Kurie JM. The role of epithelial-mesenchymal transition programming in invasion and metastasis: a clinical perspective. Cancer Manag Res. 2013;5:187–195. doi: 10.2147/CMAR.S35171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell TL, Arteaga CL. Modulation of NFkappaB activity and E-cadherin by the type III transforming growth factor beta receptor regulates cell growth and motility. J Biol Chem. 2007;282:32491–32500. doi: 10.1074/jbc.M704434200. [DOI] [PubMed] [Google Scholar]
- Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, Kelly P, Moeller BJ, Marks JR, Blobe GC. The type III TGF-beta receptor suppresses breast cancer progression. J Clin Invest. 2007;117:206–217. doi: 10.1172/JCI29293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin ME, Muthuswamy SK. Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Curr Opin Cell Biol. 2009;5:694–700. doi: 10.1016/j.ceb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. TbetaRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis. 2008;29:528–535. doi: 10.1093/carcin/bgm289. [DOI] [PubMed] [Google Scholar]
- Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis. 2008;29:252–262. doi: 10.1093/carcin/bgm249. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20:3199–3214. doi: 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- Hanks BA, et al. Type III TGF-b receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest. 2013;123:3925–3940. doi: 10.1172/JCI65745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGF-b in cancer. FEBS Lett. 2012;586:1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer Res. 2007;67:5231–5238. doi: 10.1158/0008-5472.CAN-07-0035. [DOI] [PubMed] [Google Scholar]
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- Ikonen E, Simons K. Protein and lipid sorting from the trans-Golgi network to the plasma membrane in polarized cells. Semin Cell Dev Biol. 1998;9:503–509. doi: 10.1006/scdb.1998.0258. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Hirose T, Tamai Y, Hirai S, Nagashima An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of Caenorhabditis elegans polarity protein PAR-3. J Cell Biol. 1998;143:95–106. doi: 10.1083/jcb.143.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek DF, Tschumper RC, Stolovitzky GA, Iturria SJ, Tu Y, Lepre J, Shah N, Kay NE. Identification of a global gene expression signature of B-chronic lymphocytic leukemia. Mol Cancer Res. 2003;1:346–361. [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Marcara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Knelson EH, Gaviglio AL, Tewari AK, Armstrong MB, Mythreye K, Blobe GC. Type III TGF-b receptor promotes FGF2-mediated neuronal differentiation in neuroblastoma. J Clin Invest. 2013;123:4788–4798. doi: 10.1172/JCI69657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert KE, Huang H, Mythreye K, Blobe GC. The type III transforming growth factor-beta receptor inhibits proliferation, migration, and adhesion in human myeloma cells. Mol Biol Cell. 2011;22:1463–1472. doi: 10.1091/mbc.E10-11-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Hempel N, Lee NY, Blobe GC. The type III TGF-beta receptor suppresses breast cancer progression through GIPC-mediated inhibition of TGF-beta signaling. Carcinogenesis. 2010;31:175–183. doi: 10.1093/carcin/bgp271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Vasioukhin V. Cell polarity and cancer-cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci. 2008;121:1141–1150. doi: 10.1242/jcs.016634. [DOI] [PubMed] [Google Scholar]
- Macara IG. Par proteins: partners in polarization. Curr Biol. 2004;14:R160–162. [PubMed] [Google Scholar]
- Margulis V, Maity T, Zhang XY, Cooper SJ, Copland JA, Wood CG. Type III transforming growth factor-beta (TGF-beta) receptor mediates apoptosis in renal cell carcinoma independent of the canonical TGF-beta signaling pathway. Clin Cancer Res. 2008;14:5722–5730. doi: 10.1158/1078-0432.CCR-08-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- Murphy SJ, Dore JJ, Edens M, Coffey RJ, Bernard JA, Mitchell H, Wilkes M, Leof EB. Differential trafficking of transforming growth factor-beta receptors and ligand in polarized epithelial cells. Mol Biol Cell. 2004;15:2853–2862. doi: 10.1091/mbc.E04-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SJ, Shapira KE, Henis YI, Leof EB. A unique element in the cytoplasmic tail of the type II transformation growth factor-beta receptor controls basolateral delivery. Mol Biol Cell. 2007;18:3788–3799. doi: 10.1091/mbc.E06-10-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mythreye K, Blobe GC. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci USA. 2009;106:8221–8226. doi: 10.1073/pnas.0812879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mythreye K, Knelson EH, Gatza CE, Gatza ML, Blobe GC. TbetaRIII/beta-arrestin2 regulates integrin alpha5beta1 trafficking, function, and localization in epithelial cells. Oncogene. 2013;32:1416–1427. doi: 10.1038/onc.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-b signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2011;16:127–146. doi: 10.1007/s10911-011-9207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Olemeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaks V, Koopman CD, Werb Z. Circulating tumor cells. Science. 2013;341:1186–1188. doi: 10.1126/science.1235226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Kreitzer G, Musch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005;6:233–247. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:a003053. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- Spaderna S, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537–544. doi: 10.1158/0008-5472.CAN-07-5682. [DOI] [PubMed] [Google Scholar]
- Sun L, Chen C. Expression of transforming growth factor beta type III receptor suppresses tumorigenicity of human breast cancer MDA-MB-231 cells. J Biol Chem. 1997;272:25367–25372. doi: 10.1074/jbc.272.40.25367. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007;67:1090–1098. doi: 10.1158/0008-5472.CAN-06-3117. [DOI] [PubMed] [Google Scholar]
- Vanderwalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Anderson H, Tulchinsky E, Van Roy F, Berx G. SIP/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005;33:6566–6578. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, Singer RH, Segall JE, Condeelis JS. Identification and testing of a gene expression signature of invasive carcinoma cells within primary metastatic tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- Whiteman EL, Liu CJ, Fearon ER, Margolis B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene. 2008;27:3875–3879. doi: 10.1038/onc.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Yamanaka T, Ohno S. Role of Lgl/Dlg/Scribble in the regulation of epithelial junction, polarity and growth. Front Biosci. 2008;13:6693–6707. doi: 10.2741/3182. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.