

Abstract
HIV encodes four essential enzymes: protease, integrase, reverse transcriptase (RT) associated DNA polymerase, and RT-associated ribonuclease H (RNase H). Current clinically approved anti-AIDS drugs target all HIV enzymatic activities except RNase H, which has proven to be a very difficult target for HIV drug discovery. Our high-throughput screening activities identified the dihydroxycoumarin compound F3284-8495 as a specific inhibitor of RT RNase H, with low micromolar potency in vitro. Optimization of inhibitory potency can be facilitated by structural information about inhibitor-target binding. Here, we report the crystal structure of F3284-8495 bound to the active site of an isolated RNase H domain of HIV-1 RT at a resolution limit of 1.71 Å. From predictions based on this structure, compounds were obtained that showed improved inhibitory activity. Computational analysis suggested structural alterations that could provide additional interactions with RT and thus improve inhibitory potency. These studies established proof-of-concept that F3284-8495 could be used as a favorable chemical scaffold for development of HIV RNase H inhibitors.
Keywords: RNase H Inhibitors, dihydroxybenzopyrone derivatives, HIV ribonuclease H, protein-inhibitor complex, structure-based drug design
INTRODUCTION
Drugs that target HIV reverse transcriptase (RT) are critical components of highly active antiretroviral therapy (HAART) used to treat HIV infection1; 2. HIV RT converts the single-stranded viral genomic RNA to the double-stranded viral DNA form that is then integrated into the infected host cell genome, an essential process in the HIV replication cycle. The conversion of HIV genomic RNA to DNA is a complex process entirely catalyzed by reverse transcriptase. RT RNA-dependent DNA polymerase (RDDP) activity synthesizes a complementary DNA copy of the HIV RNA template, ribonuclease H (RNase H) activity degrades the RNA strand in the DNA:RNA heteroduplex formed by the RT RDDP activity, and RT DNA-dependent DNA polymerase activity (DDDP) converts the newly synthesized single-stranded DNA into double-stranded viral DNA. This process requires RT to be multifunctional, with both DNA polymerase and RNase H activities. In HIV RT, the active sites for these two different activities are located in different protein domains and are separated by over 50 Å (Figure 1). Both activities are essential for HIV replication3; 4; yet, all current RT-directed therapeutics target only RT DNA polymerase activity. The continued emergence of HIV variants resistant to these and all other clinically used drugs5; 6; 7; 8; 9 underscores the need to develop drugs against new targets such as RNase H that may be effective against resistant viruses. Despite considerable effort, no clinically useful drugs targeting RT RNase H have been developed.
Figure 1. Ribbon diagram of the structure of full-length RT.

RT is a heterodimer that consists of p66 (color) and p51 (gray) subunits. The two enzymatic active sites and the subdomains of the p66 subunit are labelled. Right inset: A closer view of the isolated RNase H domain of the current structure, with F3284-8495 bound at the active site. Selected alpha-helices and beta-sheets are labelled based on ref. 52. The chemical structure of F3284-8495 is shown at the bottom center of the figure.
RT RNase H has proven to be a difficult target for drug discovery and development. Structural information about the interaction of RNase H inhibitors (RNHIs) identified in screening activities with RT may facilitate the development and optimization of RNHIs with potential clinical utility. X-ray crystal structures have been reported for a number of RNHIs in complex with full-length RT and/or the isolated HIV-1 RNase H domain, including one allosteric inhibitor10 and several RNase H active site inhibitors11; 12; 13; 14; 15; 16. However, none of these RNase H active site directed inhibitors show substantial antiviral activity, and the search continues for chemical scaffolds that readily facilitate synthesis of compounds that can be tested for inhibitory activity. Compound F3284-8495 [IUPAC name: (7,8-dihydroxy-2-oxo-2H-chromen-4-yl)acetic acid] is a dihydroxycoumarin that exhibits low micromolar inhibition of the RNase H activity of RT without significant inhibition of RT polymerase activity. We here report an X-ray crystal structure at a 1.71 Å resolution limit for F3284-8495 in complex with the isolated RNase H domain of HIV-1 RT (Figures 1 and 2). The inhibitor has a chemically-modifiable ethanoic acid substituent positioned in the crystal complex in such a manner as to present opportunities for structural analogs that can form additional protein contacts. Several such analogs were obtained and evaluated for inhibitory potency in vitro. We also present a computational docking analysis of some of these analogs, based on the crystal structure, to deduce the binding mode that most likely accounts for their improved inhibitory activities and to suggest strategies for developing inhibitors based on the dihydroxycoumarin scaffold.
Figure 2. Inhibitor electron density at the RNase H active site.

Two Fo-Fc simulated annealing omit maps are shown at the RNase H active site. In one (light blue, contoured at 2.8σ), the inhibitor has been excluded from the phase calculation, and in the other (violet, contoured at 11σ), both the Mn2+ cations and the inhibitor have been omitted from phasing. F3284-8495 (yellow carbon atoms) and two Mn2+ cations (black orbs) are shown modeled into the electron density. The cations are designated A and B. Two vantage points are shown rotated 90° from each other about the vertical axis.
RESULTS
F3284-8495 Binds at the RNase H Active Site
The RNase H domain crystallized in the space group P31, with two RNase H molecules per asymmetric unit (Table 1). Several highly conserved residues in the RNase H active site are required for catalysis, including four carboxylic acids (Asp443, Glu478, Asp498, and Asp549) and His5393; 17; 18. The four residues with carboxylate side-chains have been shown to coordinate one or two divalent cations11; 12; 13; 14; 15; 16; 19; 20. Our data provide unambiguous difference Fourier (Fo-Fc) electron density for F3284-8495 at the RNase H active site, along with strong electron density peaks for two Mn2+ cations separated by a distance of about 3.7 Å (Figures 2 and 3). The cations are coordinated both by the inhibitor and by the four active site carboxylates (Asp443, Glu478, Asp498, and Asp549). According to the two-cation mechanism hypothesis, the Mn2+ cations are conventionally designated as cations "A" and "B" 21; 22. Both divalent cations have approximately octahedral coordination geometry, including six to seven oxygen atoms from the inhibitor, active site side-chains, and two water molecules (Figure 3). For cation A, one octahedral axis is formed by the two water molecules. The four other coordinating oxygen atoms comprise the two coumarin ring hydroxyls, and a carboxyl oxygen atom from each of Asp549 and Asp443, with a coordinating distance ranging from 2.0 to 2.2 Å. The remaining Asp443 side-chain oxygen and the coumarin ether oxygen of F3284-8495 form an octahedral axis for cation B. The four other coordinating atoms for cation B are one coumarin hydroxyl, both side-chain carboxyl oxygens of Asp498, and at least one side-chain carboxyl oxygen of Asp478. The coordinating distances for cation B range from 2.0 to 2.6 Å. The octahedral coordination sphere about cation B is non-ideal. A second Asp478 side-chain carboxyl oxygen forms a longer distance contact with cation B of 3.2 to 3.3 Å (Figure 3). The cation coordination geometry observed in this structure agrees well with the coordination geometry observed in several structures of HIV RNase H ligand complexes reported by Lansdon et al.16 at resolutions ranging from 1.4 Å to 2.1 Å, including the positions of the Mn2+ cations and contact distances. In the Lansdon et al. structures, however, only one of the side-chain carboxyl oxygens of Asp498 coordinates cation B. As a result, Lansdon et al. count only five coordinating oxygen atoms about cation B, whereas we count six coordinating oxygen atoms. In addition to coordinating the cations, residues Glu478, Asp498, Asp549, and His539 form hydrophobic or hydrogen-bonding contacts directly with the inhibitor. In some previous structures for RNHIs in complex with the isolated RNase H domain, the side-chain of Arg557 formed a salt bridge with Asp54913; 16. This interaction is not seen, however, in the F3284-8495/RNase H domain complex. Compared to RT structures without an inhibitor bound at the RNase H active site, the current structure shows no overall change in the conformation of the RNase H domain (Figure 4a). The conformation of the RNase H active site is similar with and without bound ligand except that the side-chain of Glu478 points away from the active site if divalent cation B is not present (Figure 4b).
Table 1.
Crystallographic data collection and refinement statistics.
| Unit Cell and Space Group a, b = 51.2, c = 112.1 Å α, β = 90.0° γ = 120° P31 | |||
|---|---|---|---|
| PDB ID Code: 4QAG | Refinement | ||
| Resolution Range Used (Å) | 28.6 – 1.71 | ||
| Data Collection | Completeness in Range (%) | 98.3 | |
| Resolution Limit (Å) | 40 – 1.71 | Sigma Cutoff | 0.0 |
| Unique Reflections | 34856 | R-factor/Rfree(%) Cross-validated Coordinate |
18.2/19.0 |
| Completeness (%) / Multiplicity: | Error (Å)48;49;50 | 0.23 | |
| All data | 98.5 / 7.8 | No. of Protein/Solvent Atoms | 2076/174 |
| 2.03–2.15 Å Shell: | 99.7 / 7.2 | No. of Inhibitor/Cation Atoms | 34/4 |
| 1.93–1.84 Å Shell: | 99.3 / 5.3 | RMS Bond Lengths (Å)51 | 0.008 |
| 1.84–1.77 Å Shell: | 97.6 / 4.2 | RMS Bond Angles (°)51 | 0.94 |
| 1.77–1.71 Å Shell: | 89.5 / 2.9 | ||
| Average B-factors (Å2) | |||
| Rsym (%) / Average I/σ: | Protein/Solvent | 55.5/70.3 | |
| All Data | 8.8 / 27.1 | Inhibitor/cations | 61.5/46.0 |
| 2.03–2.15 Å Shell: | 16.3 / 9.3 | ||
| 1.93–1.84 Å Shell: | 31.0 / 4.8 | Ramachandran Regions | |
| 1.84–1.77 Å Shell: | 42.5 / 3.1 | Most favored | 98.1% |
| 1.77–1.71 Å Shell: | 45.1 / 2.4 | Additional Allowed | 1.9% |
| Generous or disallowed | 0% | ||
Figure 3. Interactions between the protein, cations, and inhibitor.

Coordination about the cations (light blue orbs) is shown as solid red lines. Two water molecules participate in this coordination (red orbs). The separation distance between the cations is shown in blue. Black dashed lines designate protein-inhibitor contacts. Distances are in Å. Wherever the distances differ for each of the two protein-inhibitor complexes in the asymmetric unit, both distances are given. Two vantage points are given, rotated about 70° about the vertical axis.
Figure 4. Effect of F3284-8495 and cations on RNase H conformation.

Shown is the superposition of the current structure (orange, inhibitor not shown) with the RNase H domain of several RT structures without inhibitors bound at the RNase H active site (referenced here by Protein Data Bank accession numbers): 3DLK53 (cyan), with no ligands bound; 4G1Q54 (violet), with Mg2+ bound at the Cation A position; and 2BE255 (gray), with Mn2+ bound at the Cation B position. Superposition is based on main-chain atoms for residues 441 to 448 of the RNase H domain. a) A view of the entire RNase H domain. The overall conformation of the RNase H domain is highly similar whether or not ligands are bound. b) A closer view of the active site, rotated toward the viewer by about 60 degrees. Glu478 tends to point away from the active site if there is no Cation B to coordinate (3DLK and 4G1Q). His539 may adjust its position and side-chain conformation to interact with a ligand such as F3284-8495 at the active site.
Binding and Inhibitory Properties
F3284-8495 inhibited the RNase H activity of the p66/p51 reverse transcriptase heterodimer in vitro with an IC50 of 4.8 µM when assayed under putative physiological conditions (pH 7.4, 1 mM Mg2+) (Table 2). The compound showed higher inhibitory activity (IC50 1.1 µM) when assayed under conditions optimal for RNase H activity (pH 8.0, 10 mM Mg2+). F3284-8495 exhibited no detectable inhibition of RT DNA polymerase activity at 10 µM, the highest concentration tested. Surprisingly, this compound was a poor inhibitor of a catalytically active HIV RNase H domain fragment (IC50 >10 µM). This enzyme construct, termed p15-EC, was created by replacing a small loop segment of HIV-1 RT RNase H with a 24 residue α-helical substrate-binding loop derived from E. coli RNase HI, and has been widely used to screen RNase H inhibitors and characterize protein-inhibitor interactions23; 24; 25; 26.
Table 2.
In vitro inhibitory properties of F3284-8495 and selected analogs
| Compound | Structure | In vitro IC50 (µM)a | |||
|---|---|---|---|---|---|
| RT-RNase H | p15-EC RNase Hb |
RT DNA polb |
|||
| pH 7.4 1 mM Mg2+ |
pH 8.0 10 mM Mg2+ |
||||
| F3284-8495 | 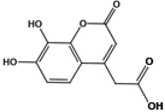 |
4.8 | 1.1 | >10 | >10 |
| F3385-2581 | 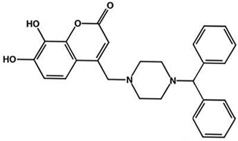 |
2.2 | 1.9 | 0.1 | 2.2 |
| F3385-2588 | 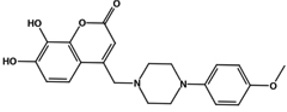 |
2.1 | 0.7 | 1.5 | >10 |
| F3385-2590 | 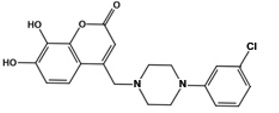 |
0.8 | 0.4 | 0.2 | 2.5 |
Data are the averages of two separate determinations, each carried out over a series of at least eight different concentrations
Assayed at pH 7.4, 1 mM Mg2+
Based on the structure reported here, we obtained a series of analogs of F3284-8495 in which the ethanoic acid substituent was replaced with a variety of bulkier substituents anticipating that these might form additional contacts with the RNase H protein and thus improve inhibitory potency. Most of the analogs showed similar or only slightly increased potency for inhibition of RT-associated RNase H activity compared to F3284-8495 (Table S1). However, the most potent of the analogs, F3385-2590, inhibited RT RNase H activity under physiological assay conditions with an in vitro IC50 of about 0.8 µM, a six-fold increase in inhibitory potency compared to the parent compound. This compound also inhibited the DNA polymerase activity of RT to a lesser extent. Compound F3385-2588 inhibited RT RNase H activity with an in vitro IC50 of 2.1 µM at pH 7.4 and 0.7 µM at pH 8 but did not inhibit the DNA polymerase activity of RT. Unlike the parent compound F3284-8495, most of the analogs inhibited the catalytically active p15-EC RT RNase H domain fragment (Table S1). Two of the compounds, F3385-2581 and F3385-2590, showed nanomolar inhibitory potency against p15-EC RNH (Table 2). The increased inhibitory activity of these analogs against the RNase H domain fragment is consistent with the structural predictions arising from the RNase H/F3284-8495 crystal structure reported in the present work, because the analogs can form contacts with additional residues in the RT RNase H domain.
Docking Experiments
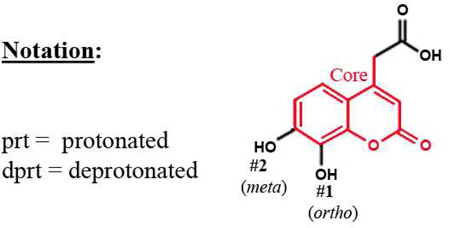
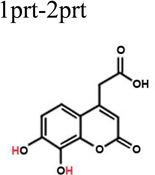
To better understand the binding of F3284-8495 and the improved activity of some of its derivatives, computational docking experiments were conducted using the Glide module of the Schrödinger Software suite. Since the two hydroxyls on the coumarin ring system of F3284-8495 are acidic, all possible coumarin ring protonation states of the inhibitor were generated for completeness and docked to the active site. The resulting Glide docking scores were compared, and the docked models were superimposed on the crystal structure for comparison (Table 3). Deprotonation of the ortho hydroxyl alone led to good docking scores and the best agreement with the crystal structure, with a coumarin core root-mean-square (RMS) deviation ranging from 0.17 to 0.26 Å, and excellent superposition on visual inspection. The best docking scores were obtained with deprotonation of both hydroxyls, but the predicted binding positions did not agree as well with the crystal structure, giving a RMS deviation of 0.21 to 0.31, and good (but not excellent) superposition on visual inspection. By contrast, protonation of either the ortho hydroxyl alone or both hydroxyls led to poor predictions of binding. Thus, it appears that the ortho hydroxyl of the inhibitor is likely to be deprotonated in order to approximate the binding mode observed in the crystal structure. Deprotonation of the second (meta) hydroxyl may occur less frequently at physiological pH.
Table 3.
Protonation effect on the docking of F3284-8495 to the RNase H active site
 | |||
|---|---|---|---|
| Protonation State |
Best Dockingbr/>Scores |
Core RMS Difference from Crystal Structurea |
Quality of Core Superposition By Visual Inspection |
| 1dprt-2dprt | −8.69 | 0.309 | Good superposition |
 |
−8.64 | 0.249 | " |
| −8.63 | 0.268 | " | |
| −8.57 | 0.212 | Very good superposition. | |
| 2prt-1dprt | −8.18 | 0.260 | Very good superposition. |
 |
−8.09 | 0.184 | Excellent superposition. |
| −8.08 | 0.189 | " | |
| −8.07 | 0.172 | " | |
| 1prt-2dprt | −6.77 | 0.414 | Fair superposition. |
 |
−6.09 | 0.330 | " |
| −6.08 | 0.338 | " | |
| −5.83 | 0.294 | " | |
| 1prt-2prt | −3.43 | 0.927 | Poor superposition. |
 |
−3.35 | 0.919 | " |
| −3.32 | 0.458 | Fair superposition. | |
| −3.25 | 0.908 | Poor superposition. | |
Superposition for RMS calculations was based on the main-chain atoms for residues 441 to 444.
The ethanoic acid group of F3284-8495 is attached to the coumarin ring system by a freely rotatable single bond. As a result, for a number of F3284-8495 derivatives reported here, the substructures that replace the ethanoic acid substituent of F3284-8495 are long enough (8 – 10 Å) to bring them within reach of additional residue contacts near the RNase H active site. These include Gln500 (which has recently been proposed as a possible binding site for allosteric RNHIs27), Trp535, Arg448, and, importantly, active site residue His539. The most potent derivatives F3385-2581, F3385-2588 and F3385-2590 (Table 2) were selected for induced-fit docking analysis. In all of these structures, the ethanoic acid substituent of F3284-8495 is replaced by a piperazine ring linked to one or more aromatic rings. The conformers with the top Glide scores and coumarin cores that superimposed well on that of the crystal structure are shown in Figure 5 through 7.
Figure 5. Docking result for F3385-2581.

The docking experiment resulted in one pose. Predicted electrostatic interactions (defined as hydrogen bonds, ionic-ionic, ionic-dipole, and dipole-dipole) are depicted by red dashes, and hydrophobic interactions by black dashes. Two additional hydrophobic contacts between residue Pro537 and the analog are not shown. An inset of the chemical structure is shown on the upper left side of the figure. Predicted protein-analog contacts were dominated by electrostatic interactions. An induced fit Schrödinger Glide score of −10.25 was computed for this docking result.
The piperazine ring of F3385-2581 is linked via a branch point to two phenyl rings. For this compound, a single high-scoring docking conformation (pose) was obtained (Figure 5). In that pose, the two phenyl rings formed hydrophobic side-chain interactions with Trp535, Ala538, and Lys540, main- and side-chain interactions with Pro537, as well as side-chain contacts with p51 subunit residue Leu422. Electrostatic interactions with the side-chain of Gln500, the main-chain of Ala538, and the side-chain of p51 subunit residue Asn265 were also noted. The total free energy contributions from the electrostatic interactions were predicted to be energetically slightly more favorable (by about 5 kcal/mol) than the combined free energy contributions of the hydrophobic contacts. An induced fit Glide docking score of −10.25 kcal/mol was obtained for this binding mode.
Compounds F3385-2588 and F3385-2590 both gave better docking scores for their top-ranked results. In compound F3385-2588 (Figure 6), the piperazine ring is linked to a para-methoxyphenyl ring. Compound F3385-2590 is similar, except that the para-methoxyphenyl ring is replaced with a meta-chlorophenyl ring (Figure 7). Both of these compounds gave similar docking results. In the best scoring pose (Figure 6 and 7, pose group 1), the combined free energy contribution from hydrophobic contacts was equal or greater than that of the electrostatic interactions. The docked structures predicted interactions of the piperazine-phenyl ring system with RNase H residues Trp535, Pro537, and Ala538, as well as residue Asn265 of the p51 subunit of RT. F3385-2588 (pose group 1) also interacted Lys540 and with p51 residue Gly262 (Figure 6). The docking study also predicted alternative ligand conformations (Figure 6 and 7, pose groups 2 and 3) that could interact with residues such as Gln475 or Tyr501, and residues that help position the scissile phosphate of the substrate RNA strand at the RNase H active site, such as Arg448 or Asn47428. Gln475 or Tyr501 are part of the so-called "RNase H primer grip", which in RT also includes p66 residues Gly359, Ala360, His361, Thr473, Lys476, Ile505, as well as p51 residues Lys395 and Glu396. The RNase H primer grip residues interact with the DNA primer strand of an RNA:DNA substrate and may play a role in catalysis and RNA cleavage specificity28. Most of these predicted conformations featured both main-chain and side-chain contacts with the inhibitor, including both hydrophobic and electrostatic interactions. The electrostatic contacts had a larger energetic component than the hydrophobic contacts in pose groups 2 and 3.
Figure 6. Docking results for F3385-2588.

An electrostatic potential surface diagram (calculated in PyMol) is shown of RT with analog F3385-2588 docked to the RNase H active site. An inset of the chemical structure is shown on the upper right side of the figure. The induced-fit docking experiments resulted in multiple poses that could be categorized into several pose groups based on contacts formed with the protein. The most energetically favorable pose obtained for each of these groups is shown. Moving clockwise about the potential surface diagram, these pose groups interacted primarily with either His539 and Ala538 (pink pose), Trp535 and Pro537 (violet "Pose Group 1"), Gln500 and Trp535 (yellow pose), Gln475 and Tyr501 (gold "Pose Group 3"), or Arg448 and Asn474 (cyan, "Pose Group 2"). For the top three docking results (Pose Groups 1 −3), the Schrödinger Glide score is given (more negative = more energetically favorable), and insets are shown giving representative protein-inhibitor interactions predicted by the docking experiment (interactions color scheme as in Figure 5). Residues of the RNase H primer grip are indicated by an asterisk (*), and residues that interact with the scissile phosphate of the substrate's RNA strand (or with adjacent nucleotides) are indicated by a caret (^)28.
Figure 7. Docking results for F3385-2590.

An electrostatic potential surface diagram (calculated in PyMol) is shown of RT with analog F3385-2590 docked to the RNase H active site. An inset of the chemical structure is shown on the upper right side of the figure. The resulting poses are similar to those obtained for F3385-2588 and are grouped as described in Figure 6. The Schrödinger Glide scores are given for the top three scoring pose groups, and insets are shown giving representative protein-inhibitor interactions predicted by the docking experiment (interactions color scheme as in Figure 5). Residues of the RNase H primer grip are indicated by an asterisk (*), and residues that interact with the scissile phosphate of the substrate's RNA strand (or with adjacent nucleotides) are indicated by a caret (^)28
DISCUSSION AND CONCLUSIONS
The RNase H active site is a shallow, solvent-exposed binding site that presents few binding opportunities for a ligand other than hydrophilic interactions. As a result, the site scores low as a "druggable" binding site16; 27 using the Schrödinger Suite program SiteMap29; 30, which evaluates a binding pocket based on such factors as site enclosure, and potential for hydrophobic and electrostatic interactions. In developing a highly effective RNase H active site inhibitor, one strategy is to exploit nearby regions of the RNase H domain to form favorable interactions, or at least to optimize interactions with conserved active site residues. Crystal structures have recently been reported for RNHIs with submicromolar inhibitory activity (a pyrimidinol carboxylic acid and an N-hydroxy quinazolinedione) that both coordinate two Mn2+ cations at the active site and simultaneously form π-π interactions with the imidazole side-chain of active site residue His53916. In addition, it is possible that two manicol-derivatives with submicromolar inhibitory activity that coordinate Mn2+ cations at the RNase H active site also form favorable interactions with His53915.
We were interested in understanding in greater detail the structural basis for the difference between the inhibitory activity of F3284-8495 and other more active HIV RNase H inhibitors. We compared F3284-8495 to pyrimidinol carboxylic acid compound 1 ("3QIN #1")16, pyrimidinol carboxylic acid compound 2 ("3QIP #2")16, N-hydroxy-quinazolinedione ("3QIO #3)16, MK114, MK214, β-thujaplicinol13; 31, and manicol15 (Table 4). Structures for all of these compounds exhibited very similar cation coordination geometry, although the inhibitory activities differed significantly. In particular, the cation coordination geometry and contact distances observed in the RNase H/F3284-8495 structure are practically identical (within 0.2 Å, except for one of the Asp498 carboxyl oxygens) to the geometry and distances observed in 3QIO #316 and MK214. In spite of this similarity, the F3284-8495 inhibition of the RT RNase H activity is 20- to 40-fold weaker than the latter two compounds. The distance between cation A and cation B ranges from 3.5 Å (manicol) to 3.8 Å (3QIP #2, 3QIO #3) for all compounds examined here (including F3284-8495), with no linear relationship between this distance and inhibitory activity. In summary, it does not appear that any features of cation geometry and contact distances can be correlated consistently with differences in activities for the various inhibitors analyzed here.
Table 4.
Comparison of F3284-8495 and some other RNHIs
| Compound | PDB Code |
Resolution Limit (Å) |
IC50, RT- RNase H (µM) |
Number of Ligand His539 Contacts (< 4.0Å) |
|---|---|---|---|---|
| F3284-8495 | 4JE2 | 1.65 | 4.8 | 8 |
| 3QIN #1 | 3QIN | 1.70 | 1.18 | 12 |
| 3QIP #2 | 3QIP | 2.10 | 0.80 | 5 |
| Manicol | 3QLH | 2.70 | 0.60 | 20 |
| 3QIO #3 | 3QIO | 1.40 | 0.23 | 22 |
| β-Thujaplicinol | 3K2P | 2.00 | 0.21 | 7 |
| MK2 | 3LP1 | 2.20 | 0.12 | 8 |
| MK1 | 3LP0 | 2.80 | 0.11 | 11 |
On the other hand, a distinguishing feature of most of the more active inhibitors is the ability to form significantly more contacts with conserved residue His539 or other nearby amino acid residues than F3284-8495. F3284-8495 forms only eight contacts (<4.0 Å) with His539. By contrast, 3QIO #3 forms 22 interactions with His539 alone, and manicol forms twenty His539 contacts. MK1 has eleven contacts with His539. In addition, MK1, MK2, and 3QIP #3 form other amino acid contacts (such as Glu444, Ser499, Ala538, and Val552) not seen with the RNase H/F3284-8495 structure, including main-chain contacts.
Both the RNase H/F3284-8495 (Figure 3) and the RT/ β-thujaplicinol13 structures indicate a similar number of contacts with His539 and a similar number of protein-ligand contacts overall. Yet, of the two compounds only β-thujaplicinol inhibits an active RNase H domain and exhibits greater than 20-fold greater activity than F3284-8495 against the RNase H of full-length RT. One possible explanation for this difference could be that β-thujaplicinol may form a tropylium ion (in which it would carry a partial positive charge on its tropolone ring and a corresponding partial negative charge shared by its carbonyl and hydroxyl oxygen atoms), leading to a more productive interaction with His539 and tighter (more energetically favorable) binding to the RNase H active site than F3284-849513. In addition, it has previously been suggested13 that β-thujaplicinol might form favorable interactions with the nucleic acid substrate that further stabilize inhibitor binding. In like fashion, MK2 also appears capable of extensive interactions with the RNA strand of a nucleic acid substrate, based on superposition with an RT/RNA:DNA structure28, but F3284-8495 does not appear capable of forming as many favorable interactions with substrate. Structures containing RT in complex with both the RNHI and nucleic acid substrate would be required to confirm this assessment. However, full-length RT binds nucleic acid more tightly than an RNase H domain fragment, and nucleic acid bound to RT could trap F3284-8495 at the RNase H binding site, so that the RNHI becomes active against the RNase H activity of full-length RT. In summary so far, good ligand efficiency at the RNase H active site may enhance inhibitory activity, including contacts with conserved active site residues such as His539 and favorable interactions with the nucleic acid substrate that the ligand dislodges from the active site, whereas the exact geometry of divalent cation coordination does not appear to be as critical a factor to inhibitory potency. The advantage of F3284-8495 is that it is readily amenable to chemical modification and can be the basis for synthesizing compounds that interact more productively with the RNase H domain.
The computational analysis presented here suggests an alternative strategy. Although the most active F3284-8495 analogs have a derivative substructure long enough to reach key regions of the RNase H domain (such as the RNase H primer grip), the docking results favor a different possibility; namely, certain poses of these compounds assume binding modes that form contacts with residues of both the p66 and p51 subunits of RT along the cleft between the RNase H domain and the p51 subunit. This alternative is attractive, because it presents a way to develop inhibitors that are selective for the RNase H domain of HIV-1 RT instead of interacting with human RNase H. For example, the scaffold of analog F3385-2588 suggests that building onto the methoxyphenyl substituent might enable exploitation of features on the p51 subunit, unique to HIV-1 RT.
Caution is warranted in the interpretation of these docking results. It is noteworthy that the highest-scoring docking results for F3284-8495 (Table 3) were not the best predictors of the observed crystal structure. Structures having slightly lower scores provided better agreement with the crystal structure and favored more plausible protonation states. This discrepancy between the crystal structure and the docking analysis may be due in part to the fact that Glide does not take into consideration either molecular strain or the energetics of water hydrogen-bonding networks before and after binding of the ligand. A newer version of Glide, which was not yet available during this work, attempts to address this shortcoming in part by incorporating a module called WaterMap32. Thus, secondary docking results may provide a better representation of ligand interactions and should therefore be considered in addition to the highest scoring conformations. As an example, pose groups 2 and 3 for F3385-2588 (Figure 6) and F3385-2590 (Figure 7) suggest that the inhibitors could interact with residues of the RNase H primer grip region that are vital for positioning the RNA:DNA substrate at the RNase H active site. All three analogs in Table 2 showed greater inhibition against the activity of the p15-EC RNase H than against the full-length RT-RNase H, suggesting that the analog substructures do not interact as productively with full-length RT. This result suggests that all three analogs bind in a fashion that bring them in contact with residues outside of the RNase H domain, when binding to full-length RT, and indicates that these interactions would require considerable optimization. For the docking experiments with both F3385-2588 and F3385-2590, only pose 1 predicts contacts with residues outside of the RNase H domain (Figures 6 and 7). It is unclear from the current analysis why interactions outside the RNase H domain would decrease inhibitory activity relative to p15-EC RNase H, but it is possible that conformational changes may occur across the cleft between the RNase H domain and the p51 subunit. Protein mutagenesis studies may clarify whether these ligand poses contribute to the observed inhibitory activities.
The piperazine ring of most analogs analyzed here give the inhibitor the ability to add some modest interactions with the protein, which could include hydrophobic interactions with Ala538. Functional groups attached to the distal nitrogen of the piperazine ring are capable of interacting with other RT residues, with the result that inhibitory activity improves compared to F3284-8495. For example, the docking results indicate that the chlorine atom on the terminal phenyl ring of F3385-2590 could form important electrostatic interactions with Trp535 or alternatively with Asn474 and Gln475 (Figure 7). Based on pose 1, the chlorine on F3385-2590 leads to considerably more productive interactions with Trp535 than the terminal methoxyl group on F3385-2588. In the crystal structure, the side-chain of Trp535 forms hydrophobic stacking interactions with the side-chain of Pro537. The docking results suggest that compound F3385-2588 or F3385-2590 could replace Trp535 in contacts with Pro537, and Trp535 would become available for interactions with appropriately situated substituents on the terminal phenyl ring of the compound. Only in F3385-2590 is a halogen substituent present to take advantage of this opportunity. It may be possible to build a more bulky substituent in place of the chlorine atom to optimize hydrophobic interactions with Trp535 and spatially adjacent residues Leu422 and Trp426. Alternatively, it is possible that a derivative substructure would have additional interaction opportunities if it were linked to the F3284-8495 core by a two-methylene linker instead of the single methylene present in the analogs tested here. In that case, it might be possible to develop a branched substructure that could interact simultaneously with His539 and more distant residues, such as Trp535 and the p51 subunit of RT. F3284-8495 showed substantially increased inhibitory potency when assayed at pH 8.0 compared to pH 7.4. Our docking analysis is consistent with these biochemical data. The coumarin ring hydroxyls have pKa values of about 8.9. The best agreement between a docked structure and the crystal structure of the RNase H/F3284-8495 complex occurred when the ortho hydroxyl of F3284-8495 was deprotonated.
Conclusions
F3284-8495 is a low micromolar specific inhibitor of the RNase H activity of HIV-1 RT. We used X-ray crystallography to determine the structure of this compound bound to the active site of an RNase H domain derived from HIV-1 RT. This structure was used to predict analogs with potentially improved binding; these analogs were obtained and many showed significantly improved inhibitory potency. This inhibition was more potent at pH values greater than physiological pH 7.4. Docking analyses suggested that this is due to the need for deprotonation of the coumarin ring ortho hydroxyl to provide an optimal binding mode. Additional docking studies with analogs of F3284-8495 provided intriguing possibilities for ligand optimization involving interactions with Trp535 and residues in the p51 subunit of RT. This investigation provided proof-of-concept that synthesis based on the F3284-8495 scaffold could be used for development of new and possibly more potent RNase H inhibitors.
MATERIALS AND METHODS
Materials
Compound F3284-8495 and analogs were purchased from Life Chemicals (Burlington, ON, Canada). All compounds were greater than 95% purity according to the supplier. Wild-type p66/p51 heterodimeric HIV-1 RT was expressed from plasmid p6HRT (a gift from Dr. S. Le Grice, NCI-Frederick, Frederick, MD) and purified essentially as previously described33. Plasmid pCSR231 encoding a codon-optimized chimeric HIV-1 RNase H domain fragment protein containing an i-helical substrate-binding loop derived from E. coli RNase HI23; 24; 25 was a generous gift from Dr. Daria Hazuda (Merck, West Point, PA). This protein, termed p15-EC, was overexpressed and purified as described26 . RT RNA-dependent DNA polymerase activity was measured as described34. Ribonuclease H activity of the RT heterodimer and the p15-EC RNase H domain fragment was determined using a rapid fluorescence assay35.
Expression and Purification of HIV-1 RT RNase H Domain for Structural Studies
A plasmid encoding the HIV-1 RT RNase H domain (427–560) bearing an N-terminal 6x-His tag (a kind gift from Karen Maegley-Pfizer) was transformed into the E. coli BL21 (DE3) pLysS bacterial strain and grown at 37°C in 1 L Luria Broth with constant shaking at 280 rpm. When cells reached an OD600 of 0.7-0.8, they were induced by addition of 1 mM IPTG and then left to grow for four hours before they were harvested by centrifugation and stored at −20°C.
Cells were resolubilized in a buffer consisting of 20 mM Tris pH 7.9, 300 mM NaCl, 20 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by sonication and the resulting lysate was centrifuged at 18000 rpm (38360 x g) for 30 min. The supernatant was recovered, filtered through a 0.45 µm membrane, and then applied to a His-trap nickel column (GE healthcare). The column was washed with at least fifty column volumes of lysis buffer and then eluted with 20 mM Tris pH 7.9, 300 mM NaCl, and 250 mM imidazole. The eluate was concentrated to 1 mL and incubated with a 1:10 ratio of His-tagged TEV protease overnight at 4°C. The mixture was then injected back onto a His-trap column to remove the cleaved His tag and the TEV protease. The flow-through was collected, concentrated to a volume of 1 mL, and injected onto a Superdex S200 size exclusion column pre-equilibrated with 10 mM Tris pH 8.0 and 75 mM NaCl. Fractions containing pure protein as assessed by SDS-PAGE gel were pooled, concentrated to 20 mg mL−1, and flash frozen at −80°C.
Crystallization and Data Collection
The RNase H domain was crystallized at 20°C (293K) by vapor diffusion in hanging drops containing 2.0 mL each of protein solution (see above) and precipitant solution [100 mM Bicine pH 8.2, 10 mM manganese sulfate, 1 mM sodium azide, and 9% (w/v) PEG 3350]. The drops were equilibrated over a reservoir containing 420 µL of the precipitant solution to which 288 µL of 50% (w/v) PEG 3350 were added. The chosen crystal was transferred stepwise to each of three soaking solutions that had in common 100 mM Bicine pH 8.2, 10 mM manganese sulfate, 1 mM sodium azide, and 26% PEG 3350. The first soaking solution also contained 1% (v/v) glycerol and 5 mM F3284-8495, and the soak time was 3.5 hours. The second and third soak solutions each contained 2.5 mM F3284-8495, as well as 13% and 27% glycerol, respectively. The soak time for the last two solutions was 15 – 20 seconds each. Finally, the crystal was flash-cooled and stored in liquid nitrogen. X-ray data were collected at 100K and a wavelength of 1.1 Å at the National Synchrotron Light Source at Brookhaven National Laboratories, Beamline X25. The data were processed using DENZO/SCALEPACK36; 37.
Structure Determination and Refinement
Phases for the HIV-1 RNase H/F3284-8495 X-ray data were determined by molecular replacement with the CCP4 program PHASER38; 39, using as an initial search model an RNase H domain model derived from coordinates of RT in complex with β-thujaplicinol (PDB accession number 3IG1)13. Stepwise structure refinement and model building were conducted using CNS 1.140 with a bulk solvent correction and the Coot graphics package (version 0.6)41. The inhibitor coordinates were constructed and energy-minimized using the Maestro™ graphics and Impact™ applications of the Schrödinger Software suite (Schrödinger, L.L.C.) and were subsequently built into electron density maps using Coot, with ligand restraints generated by Sketcher in CCP439. Water molecules were built into the structure both manually and using the "Find Waters" tool of Coot. In later stages of refinement, both Phenix42; 43; 44 and Schrödinger PrimeX45 were utilized to overcome local energy minima and improve the geometry and placement of the inhibitor and Mn2+ atoms. Finally, anisotropic B-factor refinement and TLS refinement were carried out in Phenix.
Computational Chemistry Analysis
Selected compounds were constructed from the RNase H/F3284-8495 crystal structure coordinates of one of the asymmetric unit molecules and energy-minimized using the Maestro™ graphics and Impact™ applications of the Schrödinger Software suite (Schrödinger, L.L.C.). The compounds were subjected to steepest descent and then conjugate gradient energy minimization. The protonation states of each compound was analyzed in the Schrödinger Epik™ application over a pH range of 5.5 to 9.5, and each of the protonation states was prepared for docking using Schrödinger LigPrep™. To prepare the protein receptor, RT subunit p51 residues 238 to 428 were taken from the coordinates of a 1.8 Å resolution RT structure (PDB code: 2ZD1)46 and were merged with the current RNase H/F3284-8495 coordinates, using p66 residues 505 to 540 as the basis of superposition. The p51/p66 interface was energy minimized using the Schrödinger Prime™ module. Water molecules were removed, except for the two water molecules that coordinate Mn2+ cation A (Figure 3). The receptor was merged with each compound to prepare a grid for docking.
To explore the conformational space of selected analogs of compound F3284-8495, it was assumed that the coumarin ring system of the analog interacts with the protein and Mn2+ cations exactly as observed in the crystal structure. The coumarin core (see Table 3) of each compound was aligned with that of the RNase H/F3284-8495 structure. Before preparing each grid, hydrogens were added and energy minimized with the Schrödinger "Protein Preparation Wizard Workflow" and Prime™. Docking was conducted using the Schrödinger Glide™ module, with imposition of coumarin core and metal coordination constraints to promote agreement with the crystal structure at the RNase H active site. Docking results with the most negative Glide scores and good core alignment with the crystal structure were subjected to induced fit energy minimization using Prime. New grids were generated for the receptor, and the compounds were re-docked in Glide. Structure figures were generated using PyMOL47.
Supplementary Material
Highlights.
Both the DNA polymerase and RNase H activities of reverse transcriptase are vital for HIV viability.
Only the DNA polymerase activity of this enzyme has been targeted by anti-AIDS drugs.
F3284-8495 is a micromolar inhibitor of HIV RNase H that is chemically modifiable.
A 1.71 Å crystal structure is presented of F3284-8495 bound to the RNase H active site and analysis of more active analogs.
Our analysis establishes proof-of-concept that the F3284-8495 scaffold can be the basis for development of more potent HIV RNase H inhibitors.
ACKNOWLEDGEMENTS
This study was supported in part by grants from the National Institutes of Health (AI073975 to M.A.P.; R37AI27690 to E.A.) and an unrestricted grant (to M.A.P.) from Johnson & Johnson. We are grateful to Ronald M. Levy and members of his laboratory for providing auxiliary computer resources and advice. We wish to thank Karen A. Maegley at Pfizer, Inc. for generously providing us with an expression clone for an isolated RNase H domain for crystallographic studies and Dr. Daria Hazuda at Merck, Inc. for kindly providing us with an expression clone for the p15-EC RNase H domain for biochemical studies. Crystallographic data were collected at the National Synchrotron Light Source, Brookhaven National Laboratory. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.
ABBREVIATIONS USED
- RNase H
ribonuclease H
- HIV
human immunodeficiency virus
- RT
reverse transcriptase
- AIDS
acquired immunodeficiency syndrome
- HAART
highly active antiretroviral therapy
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- PEG
polyethylene glycol
- PMSF
phenylmethylsulfonyl fluoride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 4QAG.
Contributor Information
Daniel M. Himmel, Email: himmel@cabm.rutgers.edu.
Nataliya S. Myshakina, Email: nsm11@pitt.edu.
Tatiana Ilina, Email: tai4@pitt.edu.
Alexander Van Ry, Email: avanry86@hotmail.com.
William C. Ho, Email: who@cabm.rutgers.edu.
Michael A. Parniak, Email: map167@pitt.edu.
Eddy Arnold, Email: arnold@cabm.rutgers.edu.
REFERENCES
- 1.De Clercq E. Antiretroviral Drugs. Curr. Opin. Pharmacol. 2010;10:507–515. doi: 10.1016/j.coph.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 2.Singh K, Marchand B, Kirby KA, Michailidis E, Sarafianos SG. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses. 2010;2:606–638. doi: 10.3390/v2020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tisdale M, Schulze T, Larder BA, Moelling K. Mutations Within the RNase H Domain of HIV-1 RT Abolish Virus Infectivity. J. Gen. Virol. 1991;72:59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- 4.Schatz O, Cromme F, Naas T, Lindermann D, Gruninger-Leitch F, Mous J, Le Grice SFJ. Inactivation of the RNase H domain of HIV-1 Reverse Transcriptase Blocks Viral Infectivity. In: Papas T, editor. Oncogenesis and AIDS. Houston, TX: Portfolio Publishing Company; 1990. pp. 55–68. [Google Scholar]
- 5.Ghosn J, Chaix ML, Delaugerre C. HIV-1 Resistance to First- and Second-Generation Non-Nucleoside Reverse Transcriptase Inhibitors. AIDS Rev. 2009;11:165–173. [PubMed] [Google Scholar]
- 6.Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. Nonnucleoside Reverse Transcriptase Inhibitor Resistance and the Role of the Second-Generation Agents. Ann. Pharmacother. 2010;44:157–165. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]
- 7.Gong J, Wang XQ, Tong X, Shen XH, Yang RG. Emerging Trends of Drug-Resistant HIV-1 Among Drug-Treated Patients in Former Blood Donors in Hubei, China: A Three-Year Surveillance from 2004 to 2006. Virol. Sin. 2011;26:386–392. doi: 10.1007/s12250-011-3210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menéndez-Arias L, Betancor G, Matamoros T. HIV-1 Reverse Transcriptase Connection Subdomain Mutations Involved in Resistance to Approved Non-Nucleoside Inhibitors. Antiviral Res. 2011;92:139–149. doi: 10.1016/j.antiviral.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Singh K, Marchand B, Rai DK, Sharma B, Michailidis E, Ryan EM, Matzek KB, Leslie MD, Hagedorn AN, Li Z, Norden PR, Hachiya A, Parniak MA, Xu HT, Wainberg MA, Sarafianos SG. Biochemical Mechanism of HIV-1 Resistance to Rilpivirine. J. Biol. Chem. 2012;287:38110–38123. doi: 10.1074/jbc.M112.398180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. HIV-1 Reverse Transcriptase Structure with RNase H Inhibitor Dihydroxy Benzoyl Naphthyl Hydrazone Bound at a Novel Site. ACS Chem. Biol. 2006;1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klumpp K, Mirzadegan T. Recent Progress in the Design of Small Molecule Inhibitors of HIV RNase H. Curr. Pharm. Des. 2006;12:1909–1922. doi: 10.2174/138161206776873653. [DOI] [PubMed] [Google Scholar]
- 12.Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. RNase H Active Site Inhibitors of Human Immunodeficiency Virus Type 1 Reverse Transcriptase: Design, Biochemical Activity, and Structural Information. J. Med. Chem. 2009;52:5781–5784. doi: 10.1021/jm900597q. [DOI] [PubMed] [Google Scholar]
- 13.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD, Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. Structure of HIV-1 Reverse Transcriptase with the Inhibitor beta-Thujaplicinol Bound at the RNase H Active Site. Structure. 2009;17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural Basis for the Inhibition of RNase H activity of HIV-1 Reverse Transcriptase by RNase H Active Site-directed Inhibitors. J. Virol. 2010;84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. Synthesis, Activity, and Structural Analysis of Novel alpha-Hydroxytropolone Inhibitors of Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H. J. Med. Chem. 2011;54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lansdon EB, Liu Q, Leavitt SA, Balakrishnan M, Perry JK, Lancaster-Moyer C, Kutty N, Liu X, Squires NH, Watkins WJ, Kirschberg TA. Structural and Binding Analysis of Pyrimidinol Carboxylic Acid and N-Hydroxy Quinazolinedione HIV-1 RNase H Inhibitors. Antimicrob. Agents Chemother. 2011;55:2905–2915. doi: 10.1128/AAC.01594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schatz O, Cromme FV, Gruninger-Leitch F, Le Grice SFJ. Point Mutations in Conserved Amino Acid Residues within the C-Terminal Domain of HIV-1 Reverse Transcriptase Specifically Repress RNase H Function. FEBS Lett. 1989;257:311–314. doi: 10.1016/0014-5793(89)81559-5. [DOI] [PubMed] [Google Scholar]
- 18.Davies JF, 2nd, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal Structure of the Ribonuclease H Domain of HIV-1 Reverse Transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 19.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal Structures of RNase H Bound to an RNA/DNA Hybrid: Substrate Specificity and Metal-Dependent Catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of Human RNase H1 Complexed with an RNA/DNA Hybrid: Insight into HIV Reverse Transcription. Mol. Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of Ribonuclease H Phased at 2 A Resolution by MAD Analysis of the Selenomethionyl Protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 22.Steitz TA, Steitz JA. A General Two-Metal-Ion Mechanism for Catalytic RNA. Proc. Natl. Acad. Sci. U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keck JL, Marqusee S. Substitution of a Highly Basic Helix/Loop Sequence into the RNase H Domain of Human Immunodeficiency Virus Reverse Transcriptase Restores its Mn(2+)-Dependent RNase H Activity. Proc. Natl. Acad. Sci. U S A. 1995;92:2740–2744. doi: 10.1073/pnas.92.7.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahl SJ, Kaufman JD, Vikic-Topic S, Crouch RJ, Wingfield PT. Construction of an Enzymatically Active Ribonuclease H Domain of Human Immunodeficiency Virus Type 1 Reverse Transcriptase. Protein Eng. 1994;7:1103–1108. doi: 10.1093/protein/7.9.1103. [DOI] [PubMed] [Google Scholar]
- 25.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. Inhibition of HIV-1 Ribonuclease H by a Novel Diketo Acid, 4-[5-(benzoylamino)thien-2-Yl]-2,4-dioxobutanoic Acid. J. Biol. Chem. 2003;278:2777–2780. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- 26.Gong Q, Menon L, Ilina T, Miller LG, Ahn J, Parniak MA, Ishima R. Interaction of HIV-1 Reverse Transcriptase Ribonuclease H with an Acylhydrazone Inhibitor. Chem. Biol. Drug Des. 2011;77:39–47. doi: 10.1111/j.1747-0285.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felts AK, Labarge K, Bauman JD, Patel DV, Himmel DM, Arnold E, Parniak MA, Levy RM. Identification of Alternative Binding Sites for Inhibitors of HIV-1 Ribonuclease H Through Comparative Analysis of Virtual Enrichment Studies. J. Chem. Inf. Model. 2011;51:1986–1998. doi: 10.1021/ci200194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarafianos SG, Das K, Tantillo C, Clark AD, Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal Structure of HIV-1 Reverse Transcriptase in Complex with a Polypurine Tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halgren T. New Method for Fast and Accurate Binding-Site Identification and Analysis. Chem. Biol. Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 30.Halgren TA. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 31.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective Inhibition of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity by Hydroxylated Tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Repasky MP, Murphy RB, Banks JL, Greenwood JR, Tubert-Brohman I, Bhat S, Friesner RA. Docking Performance of the Glide Program as Evaluated on the Astex and DUD Datasets: A Complete Set of Glide SP Results and Selected Results for a New Scoring Function Integrating Watermap and Glide. J. Comput. Aided Mol. Des. 2012;26:787–799. doi: 10.1007/s10822-012-9575-9. [DOI] [PubMed] [Google Scholar]
- 33.Fletcher RS, Holleschak G, Nagy E, Arion D, Borkow G, Gu Z, Wainberg MA, Parniak MA. Single-step Purification of Recombinant Wild-Type and Mutant HIV-1 Reverse Transcriptase. Protein Expr. Purif. 1996;7:27–32. doi: 10.1006/prep.1996.0004. [DOI] [PubMed] [Google Scholar]
- 34.Borkow G, Fletcher RS, Barnard J, Arion D, Motakis D, Dmitrienko GI, Parniak MA. Inhibition of the Ribonuclease H and DNA Polymerase Activities of HIV-1 Reverse Transcriptase by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde Hydrazone. Biochem. 1997;36:3179–3185. doi: 10.1021/bi9624696. [DOI] [PubMed] [Google Scholar]
- 35.Parniak MA, Min KL, Budihas SR, Le Grice SF, Beutler JA. A Fluorescence-Based High-Throughput Screening Assay for Inhibitors of Human Immunodeficiency Virus-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Anal. Biochem. 2003;322:33–39. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W, Borek D, Cymborowski M. DENZO and SCALEPACK. In: Arnold E, Himmel DM, Rossmann MG, editors. International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules. Second. West Sussex, United Kingdom: John Wiley & Sons, Ltd; 2012. pp. 282–295. [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Read RJ. Pushing the Boundaries of Molecular Replacement with Maximum Likelihood. Acta Crystallogr. D Biol. Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a Comprehensive Python-based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic Ligand Builder and Optimization Workbench (eLBOW): A Tool for Ligand Coordinate and Restraint Generation. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell JA, Cao Y, Gunn JR, Day T, Gallicchio E, Zhou Z, Levy R, Farid R. PrimeX and the Schrödinger Computational Chemistry Suite of Programs. In: Arnold E, Himmel DM, Rossmann MG, editors. International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules. Second. John Wiley & Sons, Ltd, West Sussex, United Kingdom; 2012. pp. 534–538. [Google Scholar]
- 46.Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-Resolution Structures of HIV-1 Reverse Transcriptase/TMC278 Complexes: Strategic Flexibility Explains Potency Against Resistance Mutations. Proc. Natl. Acad. Sci. U S A. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLano Scientific. In: The PyMOL Molecular Graphics System. DeLano LW, editor. 2002. [Google Scholar]
- 48.Kleywegt GJ, Brünger AT. Checking Your Imagination: Applications of the Free R Value. Structure. 1996;4:897–904. doi: 10.1016/s0969-2126(96)00097-4. [DOI] [PubMed] [Google Scholar]
- 49.Kleywegt GJ, Bergfors T, Senn H, LeMotte P, Gsell B, Shudo K, Jones TA. Crystal Structures of Cellular Retinoic Acid Binding Proteins I and II in Complex with All-trans-retinoic Acid and a Synthetic Retinoid. Structure. 1994;2:1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- 50.Luzzati V. Traitement Statistique des Erreurs dans la Determination des Structure Cristallines. Acta. Cryst. 1952;5:802–810. [Google Scholar]
- 51.Engh RA, Huber R. Accurate Bond and Angle Parameters for X-ray Protein Structure Refinement. Acta Cryst. 1991;A47:392–400. [Google Scholar]
- 52.Jacobo-Molina A, Ding J, Nanni RG, Clark AD, Ju X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Crystal Structure of Human Immunodeficiency Virus Type 1 Reverse Transcriptase Complexed with Double-stranded DNA at 3.0 Å Resolution Shows Bent DNA. Proc. Natl. Acad. Sci. USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal Engineering of HIV-1 Reverse Transcriptase for Structure-based Drug Design. Nucleic Acids Res. 2008;36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuroda DG, Bauman JD, Challa JR, Patel D, Troxler T, Das K, Arnold E, Hochstrasser RM. Snapshot of the Equilibrium Dynamics of a Drug Bound to HIV-1 Reverse Transcriptase. Nat. Chem. 2013;5:174–181. doi: 10.1038/nchem.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Himmel DM, Das K, Clark AD, Jr, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. Crystal Structures for HIV-1 Reverse Transcriptase in Complexes with Three Pyridinone Derivatives: A New Class of Non-nucleoside Inhibitors Effective Against a Broad Range of Drug-Resistant Strains. J. Med. Chem. 2005;48:7582–7581. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.