

Abstract
INT131 is a potent non-thiazolidinedione (TZD)-selective peroxisome proliferator-activated receptor-γ modulator being developed for the treatment of type 2 diabetes. In preclinical studies and a phase II clinical trial, INT131 has been shown to lower glucose levels and ameliorate insulin resistance without typical TZD side effects. To determine whether the insulin-sensitizing action of INT131 is mediated by effects on insulin-mediated glucose homeostasis and insulin signaling, high-fat diet-induced obese (DIO) insulin-resistant mice treated with INT131 were studied. INT131's effects on bone density were also investigated. Treatment with INT131 enhanced systemic insulin sensitivity, as revealed by lower insulin levels in the fasted state and an increase in the area above the curve during an insulin tolerance test. These effects were independent of changes in adiposity. Insulin-stimulated PI3K activity in skeletal muscle and adipose tissue of DIO mice was significantly reduced ∼50–65%, but this was restored completely by INT131 therapy. The INT131 effects on PI3K activity are most likely due to increased IRS-1 tyrosine phosphorylation. Concurrently, insulin-mediated Akt phosphorylation also increased after INT131 treatment in DIO mice. Importantly, INT131 therapy caused a significant increase in bone mineral density without alteration in circulating osteocalcin in these mice. These data suggest that a newly developed insulin-sensitizing agent, INT131, normalizes obesity-related defects in insulin action on PI3K signaling in insulin target tissues by a mechanism involved in glycemic control. If these data are confirmed in humans, INT131 could be used for treating type 2 diabetes without loss in bone mass.
Keywords: thiazolidinedione, peroxisome proliferator-activated receptor-γ, insulin resistance, type 2 diabetes, obesity
insulin increases glucose transport and metabolism by activating a cascade of signaling events in the skeletal muscle and adipose tissue (10). Resistance to insulin's effects on glucose metabolism in these tissues is a major contributor to the pathogenesis of insulin-resistant states such as obesity and type 2 diabetes (18). Intense studies have focused on identifying the key steps in the insulin-signaling pathway that are responsible for stimulation of glucose transport, which is dysregulated in insulin-resistant states (36). It is clear that the activation of phosphatidylinositol 3-kinase (PI3K) is necessary for the regulation of insulin-induced glucose transport in insulin target tissues/cells (12, 28, 34). Impaired insulin-dependent PI3K activity has been observed in the skeletal muscle of type 2 diabetic subjects (6, 25) and insulin-resistant animals (11, 19).
Thiazolidinediones (TZDs) are a class of insulin-sensitizing agents used to treat type 2 diabetic patients (37). The molecular targets of these compounds are thought to include the nuclear receptor peroxisome proliferator activated receptor-γ (PPARγ), which regulates the expression of numerous genes that play a role in glucose and lipid metabolism (20). Evidence suggests that TZDs ameliorate insulin resistance in skeletal muscle of humans primarily by increasing insulin-stimulated glucose disposal. This effect is thought to be mediated via the PI3K signaling pathway in skeletal muscle (4, 17, 24). However, there have been discussions about the therapeutic effects of the currently available TZDs in the management of type 2 diabetes (21). The major concern of the TZD PPARγ agonists is that they have problems in fluid retention and weight gain and the increased risk of congestive heart failure (21, 33, 39). Additionally, TZD treatment has been shown to decrease bone mineral density in type 2 diabetic humans and animals (16, 21, 27, 40). Moreover, increased numbers of peripheral fractures were seen in patients with type 2 diabetes treated with pioglitazone or rosiglitazone (16, 39). Thus, it is critical to develop a new agent that minimizes unwanted side effects.
INT131 is a potent non-TZD-specific PPARγ modulator and is currently in clinical trials for treatment of type 2 diabetes (15, 26, 32). INT131 binds to PPARγ in the same binding pocket as other TZDs but occupies a unique area in the pocket and contacts the receptor at distinct points from the TZDs (32). Because of this unique property, INT131 may have distinct patterns of gene transcript induced by PPARγ, which ultimately leads to different biological effects on glucose metabolism and adipocyte function. Indeed, INT131 has little effect on lipid accumulation in 3T3-L1 adipocytes, whereas TZDs enhance this (32). This effect is thought to be due to modest induction of adipogenic molecules, which are target genes of PPARγ (32). This agent is also potent and highly efficacious in reducing glucose levels in animal models of type 2 diabetes but causes much less weight gain and volume expansion than rosiglitazone (14, 15). However, the molecular mechanisms underlying the insulin-sensitizing action of INT131 are largely unknown, although they are thought to result, at least in part, from increased circulating adiponectin levels (15).
In this study, we investigated whether the insulin-sensitizing effects of INT131 could involve the reversal of the demonstrated defect in insulin-stimulated signaling in an insulin resistance model made by high-fat feeding. Our results show that INT131 therapy ameliorates insulin sensitivity by improving insulin signaling in diet-induced obese (DIO) mice in the absence of adverse effects on bone density.
MATERIALS AND METHODS
Animals
Study of db/db mice.
Male leptin receptor-deficient (db/db; 6 wk of age) and lean littermates were obtained from The Jackson Laboratory and fed a standard chow diet (PMI Feeds, St. Louis, MO) ad libitum. After 2 wk of environmental adjustment, db/db mice at 9 wk of age were treated with a vehicle (1% methylcellulose), INT131 (0.1, 1, or 10 mg/kg body wt), or pioglitazone (10 mg/kg body wt) by oral gavage for 2 wk (n = 5–6 in each group).
Study of diet-induced obese mice.
Male C57BL/6 mice (6 wk of age; The Jackson Laboratory) were fed a high-fat diet for 15 wk, with 58% kcal from fat (D12331; Research Diets). At 21 wk of age, mice were treated with a vehicle (1% methylcellulose), INT131 (10 mg/kg of body wt), or pioglitazone (10 mg/kg) by oral gavage for 7 wk (n = 8–12 in each group). Mice fed a normal chow diet were also treated with a vehicle and used as a normal control group.
General protocols.
All of the mice were allowed to access their experimental or control diets ad libitum and were housed under controlled temperature at 24°C and a 12:12-h light-dark cycle, with light from 6:30 AM to 6:30 PM. All aspects of animal care and experimentation were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Metabolic and Other Measurements
Blood was collected from randomly fed or overnight-fasted mice via tail bleed between 8 and 10 AM. Blood glucose was measured using a OneTouch Ultra glucose meter (LifeScan, Milpitas, CA). Serum insulin (Crystal Chem, Chicago, IL) and osteocalcin (Biomedical Technologies) were measured by enzyme-linked immunosorbent assay. Bone mineral density (BMD) was measured in the whole body except for the skull and tail, which were measured with dual X-ray absorptiometry (Lunar PIXIMUS, Madison, WI). For the glucose tolerance test (GTT), male mice were fasted overnight, and blood glucose was measured immediately before and 15, 30, 45, 60, 90, and 120 min after an intraperitoneal injection of glucose (1.0 g/kg body wt). For the insulin-tolerance test (ITT), food was removed for 5 h in the morning, and blood glucose was measured immediately before and 15, 30, 60, 90, and 120 min after an intraperitoneal injection of human insulin (1.5 U/kg body wt of db/db mice or 0.75 U/kg body wt of DIO mice; Humulin R; Lilly). For ITT, the area above the curve is the appropriate analysis because insulin promotes glucose disposal, and the glucose values go down from baseline after insulin injection. The area is calculated on the basis of trapezoidal areas defined by each data point as a function of time. Thus, the greater the area above the curve, the more insulin is promoting glucose disposal, reflecting increased insulin sensitivity. By contrast, with a GTT curve, glucose rises above the baseline as a function of time, and there it is appropriate to calculate the area under the curve.
Acute Insulin Stimulation
For injection experiments, DIO mice treated with INT131 or pioglitazone were fasted overnight. On the day of the experiment, a bolus injection of insulin (10 U/kg body wt; Humulin R) was administered through the tail vein. Six minutes later, the gastrocnemius muscle, epididymal adipose tissue, interscapular brown adipose tissue, and heart were rapidly removed, weighed, frozen in liquid nitrogen, and stored at −80°C until analysis.
Preparation of Muscle Lysates
Fifty milligrams of tissue was homogenized using a polytron at half-maximum speed for 1 min on ice in 500 ml of buffer A (20 mM Tris, pH 7.5, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 2 mM Na3VO4) containing 1% Nonidet P-40, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Tissue lysates were solubilized by continuous stirring for 1 h at 4°C and centrifuged for 10 min at 14,000 g. The supernatants were stored at −80°C until analysis.
Determination of PI3K activity
Tissue lysates (500 μg of protein) were subjected to immunoprecipitation for 4 h at 4°C with 5 μl of a polyclonal insulin receptor substrate-1 (IRS-1) antibody (1:100 dilution; gift from Dr. Morris White, Children's Hospital, Boston, MA) coupled to protein A-sepharose (Sigma, St. Louis, MO). The immune complex was washed, and PI3K activity was determined as described previously (22).
Immunoblotting Analysis
Tissue lysates (20–50 μg of protein) were resolved by SDS-PAGE and transferred to nitrocellulose membranes, as described previously (29). The membranes were incubated with polyclonal antibodies against phospho-Tyr1162 IR (Invitrogen), phospho-Tyr612 IRS-1 (Invitrogen), phospho-Ser473 Akt (Cell Signaling Technology), phospho-Thr308 Akt (Cell Signaling Technology), phospho-Thr389 S6 kinase (Cell Signaling Technology), phospho-Ser240/244 S6 ribosomal protein (Cell Signaling Technology), IR (Santa Cruz Biotechnology), p110β catalytic subunit of PI3K (Millipore), Akt (Santa Cruz Biotechnology), S6 kinase (Cell Signaling Technology), S6 ribosomal protein (Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology). The bands were visualized with enhanced chemiluminescence and quantified by densitometry (8). All phosphorylation data were normalized by total protein levels.
Statistical Analysis
Data are expressed as means ± SE. Differences between two groups were assessed using unpaired two-tailed t-tests and among more than two groups by ANOVA. Data involving more than two repeated measurements were assessed by repeated-measures ANOVA. When a significant difference was found in ANOVA, post hoc analyses were performed with Fisher's protected least significant difference test. Differences were considered significant at P < 0.05. Analyses were performed using StatView software (BrainPower).
RESULTS
Effect of INT131 on Glucose Level and Insulin Sensitivity in Mice with Obesity and Type 2 Diabetes
It is clear that TDZs have a glucose-lowering and antidiabetic effect by enhancing insulin's ability to increase glucose use in rodents and humans (15, 33). To determine whether INT131 treatment, a potent non-TZD-selective PPARγ modulator (SPPARM), alters glucose metabolism in vivo, obese diabetic db/db mice were treated with 0.1–10 mg/kg for 2 wk. As an experimental control, pioglitazone (10 mg/kg) was used for all studies. After 2 wk of treatment, glucose levels in the fed state of db/db mice were not significantly different at doses of 0.1–1.0 mg INT131 treatment. However, at a dose of 10 mg, INT131 caused a significant decrease in glucose levels in db/db mice compared with vehicle-treated db/db mice (Fig. 1, A and B). As expected, fed glucose levels were greatly reduced by ∼60% in pioglitazone-treated db/db mice (Fig. 1, A and B). The magnitude of reduction of glucose levels in INT131-treated mice is similar to that of pioglitazone-treated db/db mice. The levels of glucose in the fasting state were also notably decreased by INT131 treatment in db/db mice (not shown).
Fig. 1.
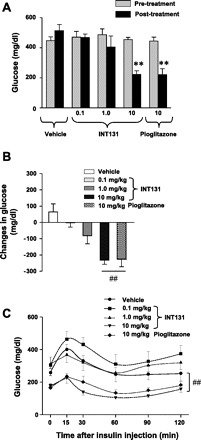
Effects of INT131 treatment on glucose homeostasis in db/db mice. Male db/db mice were treated with a vehicle, INT131 (0.1, 1, or 10 mg/kg), or pioglitazone (10 mg/kg) by oral gavage for 2 wk. Fed glucose levels (A) and changes in glucose level during the treatments (B) were measured. C: an insulin tolerance test (ITT) was performed 5 h after food removal. Mice were injected intraperitoneally with insulin at 1.5 U/kg body wt. Blood glucose was measured from tail bleeds at the indicated time. Data are means ± SE for 5–6 mice. **P < 0.01 vs. pretreatment db/db mice; ##P < 0.01 vs. vehicle-treated mice.
To determine the effects of INT131 on whole body insulin sensitivity, we performed ITTs on db/db mice. During the course of ITT, glucose levels at all time points examined were significantly decreased in db/db mice treated with 10 mg of INT131 compared with db/db mice treated with a vehicle. Similar results were observed in pioglitazone-treated db/db mice (Fig. 1C). Compared with vehicle-treated mice, insulin-stimulated percent decreases in glucose levels during the ITT (15–60 min) were significantly enhanced by 113% for INT131-treated mice and 77% for pioglitazone-treated mice. These data were calculated by normalization to baseline glucose levels (0 time point) and expressed as insulin-stimulated percent changes in glucose levels. However, lower doses (0.1 or 1 mg) of INT131 had no effect on glucose levels in db/db animals (Fig. 1C). Collectively, these results suggest that INT131, with an effective antidiabetic dose of 10 mg/kg of body wt, had a blood glucose-lowering effect and insulin-sensitizing effect in mice with obesity and type 2 diabetes. These effects are very similar to those of pioglitazone, which is widely used for treatment of type 2 diabetic patients.
Effect of INT131 on Metabolic Parameters in DIO Mice
To further determine whether INT131 treatment improves glucose homeostasis in a physiological insulin-resistant state, mice were fed a high-fat diet for 15 wk, followed by 7 wk of INT131 treatment. At the end of high-fat feeding, body weight was notably increased in high-fat-fed mice compared with chow diet-fed mice. However, both INT131 and pioglitazone had no effect on weight gain in high-fat feeding (Fig. 2A). Interestingly, when normalizing body weight, epididymal fat mass was decreased significantly by pioglitazone treatment compared with the value of the vehicle treatment (Fig. 2B). INT131 therapy tended to decrease epididymal fat amount, but the difference was not statistically significant (Fig. 2B). No differences were observed in interscapular brown adipose tissue or liver weight among the groups (data not shown). A high-fat diet caused a significant decrease in heart weight in mice. Interestingly, the mass of the heart by INT131 treatment was reduced compared with vehicle treatment, but pioglitazone treatment had no effects (Fig. 2C).
Fig. 2.
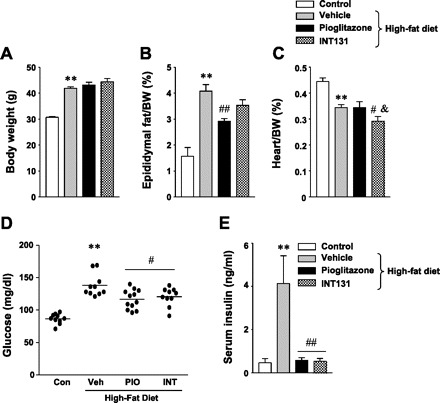
Effect of INT131 (INT) on metabolic parameters in diet-induced obese mice. Male mice were fed a high-fat diet for 15 wk and were treated with a vehicle (Veh), INT (10 mg/kg), or pioglitazone (PIO; 10 mg/kg) by oral gavage for 7 wk. Body weight (A), epididymal fat mass (B), heart weight (C), fasting glucose levels (D), and serum insulin levels (E) were measured in diet-induced mice at 21 wk of age. Data are means ± SE for 8–12 mice. **P < 0.01 vs. control mice; #P < 0.05, ##P < 0.01 vs. Veh-treated mice; &P < 0.05 vs. PIO-treated mice. Con, control.
After 7 wk of treatment, fasting blood glucose levels were decreased significantly in INT131-treated mice compared with vehicle-treated mice (Fig. 2D). INT131 showed a glucose-lowering effect similar to pioglitazone (Fig. 2D). In addition, treatment of mice fed a high-fat diet with INT131 caused a significant decrease in insulin levels compared with vehicle-treated mice (Fig. 2E). Similar effects were seen in pioglitazone-treated mice (Fig. 2E). These data suggest that INT131 may exert beneficial effects on glycemic control in insulin-resistant mice with obesity.
INT131 Increases Insulin Sensitivity in DIO Mice
To determine the effects of INT131 on glucose tolerance and insulin sensitivity, we performed GTTs and ITTs in DIO mice. As expected, glucose tolerance was impaired in high-fat-fed mice compared with control mice. However, neither INT131 nor pioglitazone treatment had an effect on glucose tolerance in DIO mice, as indicated by similar areas under the glucose curve during the GTT (Fig. 3, A and B). DIO mice were insulin resistant, as evidenced by the lack of a decrease in blood glucose levels after insulin injection (0.75 U/kg body wt), whereas in mice treated with INT131 or pioglitazone, glucose levels decreased significantly after insulin injection (Fig. 3C). The area above the glucose curve was decreased by 70% in DIO mice (P < 0.001) compared with control mice (Fig. 3D). However, this decrease was improved ∼40% by INT131 or pioglitazone treatment (Fig. 3D), suggesting the enhancement of insulin sensitivity.
Fig. 3.
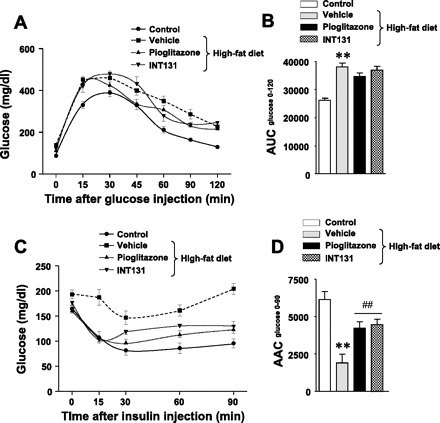
Insulin-sensitizing effect of INT131 in diet-induced obese mice. Male mice were fed a high-fat diet for 15 wk, and mice were treated further with a vehicle, INT131 (10 mg/kg), or pioglitazone (10 mg/kg) by oral gavage for 7 wk. A: a glucose tolerance test (GTT) was performed on overnight-fasted mice with intraperitoneal injections of glucose at 1.0 g/kg body wt. Blood glucose was determined from tail bleeds at the indicated time. B: the area under the glucose curve was calculated during the GTT. C: an ITT was performed 5 h after food removal. Mice were injected intraperitoneally with insulin at 1.0 U/kg body wt. Blood glucose was measured from tail bleeds at the indicated time. D: the area above the glucose curve was calculated during the ITT. Data are means ± SE for 8–12 mice. **P < 0.01 vs. control mice; ##P < 0.01 vs. vehicle-treated mice.
INT131 Ameliorates Insulin Signaling in Skeletal Muscle of DIO Mice
To explore the mechanism by which INT131 improves insulin sensitivity in DIO mice, we measured the ability of insulin to activate PI3K and multiple distal pathways in skeletal muscle. Because a low dose of INT131 had no effect on insulin sensitivity (not shown), we studied only a high dose of animals that were treated with 10 mg/kg of INT131 or pioglitazone. Insulin-stimulated PI3K activity associated with IRS-1 in skeletal muscle of DIO mice was significantly reduced by ∼50% compared with normal diet-fed mice. However, this was completely restored by INT131 therapy (Fig. 4). The INT131 effects on PI3K are most likely due to increased IRS-1 tyrosine phosphorylation (Fig. 5). Similarly, INT131 treatment was associated with normalization of impaired Akt activation in the skeletal muscle of DIO mice (Fig. 5). Interestingly, S6 phosphorylation induced by insulin was decreased dramatically after INT131 treatment despite normal S6K phosphorylation (Fig. 5). The levels of IRS-1 protein in skeletal muscle were notably decreased by a high-fat diet, but these effects were ameliorated by INT131 or pioglitazone treatment (Fig. 5). Protein expressions of other signaling molecules, including p110β, Akt, S6K, and S6, were unaltered after INT131 and pioglitazone treatment (Fig. 5).
Fig. 4.
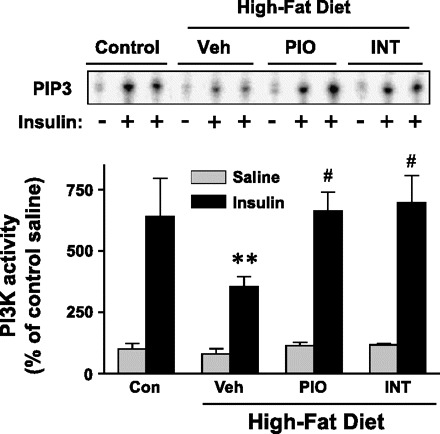
INT131 treatment ameliorates insulin-stimulated phosphatidylinositol 3-kinase (PI3K) activity in the skeletal muscle of diet-induced mice. Male mice were fed a high-fat diet for 15 wk, and mice were treated further with a vehicle, INT131 (10 mg/kg), or pioglitazone (10 mg/kg) by oral gavage for 7 wk. After an overnight fast, mice were injected intraperitoneally with saline (gray bars) or 10 U/kg insulin (black bars). Ten minutes later, muscle was removed. Muscle lysates were subjected to immunoprecipitation with an insulin receptor substrate-1 antibody. PI3K activity was measured and quantitated using a phosphorimager. Data are means ± SE for 8–12 mice. **P < 0.01 vs. control mice; #P < 0.05 vs. vehicle-treated mice.
Fig. 5.
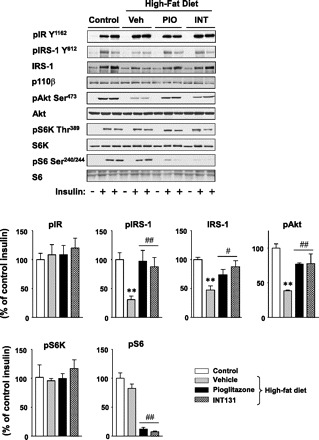
INT treatment improves insulin signaling in skeletal muscle of diet-induced mice. Muscle lysates were subjected to SDS-PAGE and immunoblot with antibodies as indicated. The bands were quantitated using densitometry and normalized by total protein levels. Data are means ± SE for 8–12 mice. **P < 0.01 vs. control mice; #P < 0.05; ##P < 0.01 vs. vehicle-treated mice. Note: the insulin receptor substrate-1 (IRS-1) antibody reacts more strongly with phosphorylated IRS-1 protein. PIO, pioglitazone.
INT131 Improves Insulin Signaling in Adipose Tissue of DIO Mice
To further investigate the mechanism of INT131-mediated insulin-sensitizing effects, we measured insulin signaling in adipose tissue of DIO mice treated with INT131. Similar to muscle, in adipose tissue of DIO mice, IRS-1-associated PI3K activity induced by insulin was also greatly reduced, by ∼62%, compared with control mice. Importantly, INT131 therapy but not pioglitazone therapy normalized this defect of PI3K activity to the normal level (Fig. 6). These effects arose mainly from both increased IRS-1 tyrosine phosphorylation and increased p110β catalytic subunit expression of PI3K in adipose tissue (Fig. 7). IRS-1 protein levels were unaltered by INT131 therapy. Interestingly, insulin-stimulated Akt phosphorylation in DIO mice was increased by high-fat feeding and further increased with INT131 treatment (Fig. 7). Treatment of DIO mice with INT131 also restored impaired IR tyrosine phosphorylation without change in IR protein levels (Fig. 7). INT131 treatment increased pS6K phosphorylation, whereas it decreased S6 phosphorylation compared with vehicle-treated DIO mice (Fig. 7).
Fig. 6.
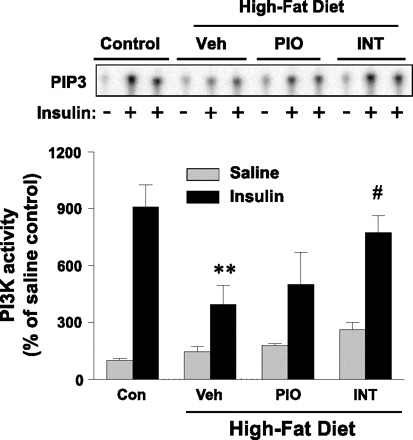
INT treatment improves insulin-stimulated PI3K activity in adipose tissue of diet-induced mice. Male mice were fed a high-fat diet for 15 wk, and mice were treated further with a Veh, INT (10 mg/kg), or PIO (10 mg/kg) by oral gavage for 7 wk. After an overnight fast, mice were injected intraperitoneally with saline (gray bar) or 10 U/kg insulin (black bars). Six minutes later, muscle was removed. Adipose lysates were subjected to immunoprecipitation with an IRS-1 antibody. PI3K activity was measured and quantitated using a phosphorimager. **P < 0.01 vs. control mice; #P < 0.05 vs. Veh-treated mice.
Fig. 7.
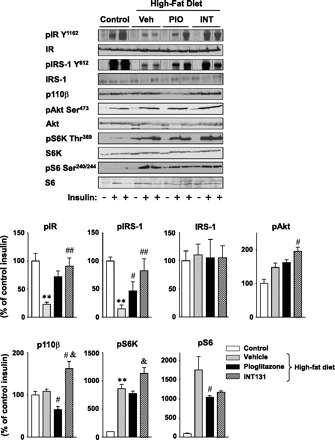
Effect of INT131 treatment on insulin signaling in adipose tissue of diet-induced mice. Adipose lysates were subjected to SDS-PAGE and immunoblot with antibodies as indicated. The bands were quantitated using densitometry and normalized by total protein levels. Note: the blots for phospho (p)-IR, p-IRS-1, and IRS-1 are rearranged within the same blots, and the lines in the blots indicate the rearrangement of the blots. Data are means ± SE for 8–12 mice. **P < 0.01 vs. control mice; #P < 0.05 (P = 0.05 in the case of p-Akt); ##P < 0.01 vs. Veh-treated mice; &P < 0.05 vs. PIO-treated mice.
Effect of INT131 on BMD and Serum Osteocalcin Level
To test the possibility that INT131 could play a role in regulating bone metabolism, we measured BMD and serum osteocalcin levels in DIO mice that were treated with INT131. Circulating osteocalcin level, a biochemical marker of bone formation, was reduced in DIO mice compared with control mice (Fig. 8B). After 7 wk of treatment with INT131 or pioglitazone, osteocalcin level tended to increase, but differences were not statistically significant (P < 0.10; Fig. 8B). Interestingly, BMD was increased significantly in INT131-treated mice compared with DIO mice treated with a vehicle or pioglitazone. However, pioglitazone treatment had no effect on BMD in DIO mice (Fig. 8A).
Fig. 8.
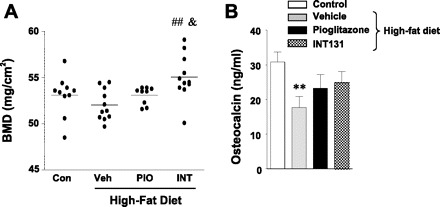
Effects of INT131 treatment on bone mass and osteocalcin levels in diet-induced mice. Male mice were fed a high-fat diet for 15 wk, and mice were treated further with a Veh, INT (10 mg/kg), or PIO (10 mg/kg) by oral gavage for 7 wk. A: bone mineral density (BMD) was measured using a dual X-ray absorptiometry. B: serum osteocalcin levels were measured in diet-induced mice at 21 wk of age. Data are means ± SE for 8–12 mice. ##P < 0.01 vs. Veh-treated mice; &P < 0.05 vs. PIO-treated mice.
DISCUSSION
Normal insulin signaling is essential in maintaining glucose homeostasis (36). In the insulin-signaling pathway, IRS-1 and PI3K are thought to be key steps necessary for the regulation of insulin-dependent glucose transport and metabolism in insulin-sensitive tissues (11, 19, 36). Impairment in insulin-stimulated PI3K activity associated with IRS-1 is an important contributor to the pathogenesis of insulin-resistant states such as obesity and type 2 diabetes (18, 41). The present study was designed to determine whether the defects in PI3K signaling observed in insulin-resistant animals could be restored by treatment with the insulin-sensitizing agent INT131, a new non-TZD SPPARM. Here, we show that INT131 treatment ameliorates the impairment of insulin-stimulated PI3K activity and enhances insulin-stimulated Akt activation in skeletal muscle and adipose tissue of insulin-resistant obese mice. INT131 therapy also improves BMD in these mice. These data suggest that the insulin-sensitizing action of INT131 on whole body glucose metabolism in insulin-resistant obese mice could be due to enhanced insulin signaling in skeletal muscle and adipose tissue. These effects concordantly occurred with enhancement of bone metabolism.
In past years, insulin-sensitizing TZDs agents have been widely used for patients with type 2 diabetes (38). However, there is accumulating evidence that undesirable side effects, including body weight gain, anemia, fluid retention, and congestive heart failure, were seen in type 2 diabetic patients or animals treated with these compounds (13, 16, 27). Recent studies have developed a number of selective peroxisome proliferator-activated receptor modulators, including T2384, FK614, and PAR-1622, all of which are partial PPARγ agonists (23, 30, 31). The beneficiary effect of these insulin-sensitizing compounds is that antihyperglycemic activity occurred independently of typical side effects in diabetic animals (14). Specifically, PAR-1622 or INT131 therapy does not increase plasma volume in ICR mice, whereas rosiglitazone therapy increases this (23, 26). Collectively, these data raise the possibility that insulin sensitizers of selective PPARγ modulators, including INT131, are likely to be promising antidiabetic drugs that have excellent antihyperglycemic activity with a broad safety profile for fluid retention.
In this study, INT131 therapy was efficacious in decreasing plasma glucose levels and increasing insulin-stimulated glucose disposal in obese diabetic mice and DIO mice, suggesting improved insulin sensitivity. The antihyperglycemic action of this agent is aroused mainly from increased insulin stimulation of IRS-1/PI3K/Akt signaling in skeletal muscle, which is predominantly responsible for insulin-mediated glucose disposal. These data are supported further by the previous findings from human studies that troglitazone treatment restored insulin-stimulated PI3K and Akt activity in skeletal muscle of obese type 2 diabetic subjects (24). Thus, our data clearly suggest that the molecular mechanism by which INT131 exerts its effects on glycemic control is mediated through the key molecules of the insulin-signaling pathway, including PI3K signaling in skeletal muscle.
Given that INT131 can only partially activate classical PPARγ function (32), it is hypothesized that this property may lead to different outcomes of the adipogenic phenotype when insulin-sensitive cells/tissues are exposed to TZDs and/or INT131. Consistent with this notion, we found that epididymal fat mass in DIO mice was unaltered by INT131 but significantly decreased by pioglitazone despite similar body weight gain. This could be explained at least in part by the different redistribution of fat mass in these mice, as indicated by the evidence that the TZD-mediated weight gain may be due to an increase in subcutaneous adipose tissue and a decrease in visceral fat (38). Although our study does not provide the precise mechanism of INT131's action on adipose tissue redistribution from visceral to peripheral fat depots, it is interesting that there is a different property on fat accumulation between a classic TZD and a selective modulator PPARγ INT131. Further data supporting this aspect include the experimental findings that INT131 treatment, but not pioglitazone treatment, greatly enhances insulin-mediated PI3K activity in adipose tissue, although improvements in glucose and insulin concentrations were similar with the two compounds. The improvement of PI3K induced by INT131 treatment may be due, at least in part, to increased expression of the p110β catalytic subunit of PI3K in adipose tissue but not in muscle. Therefore, it is likely that INT131 acts differentially on gene expression of p110β in a tissue-specific manner by unidentified mechanisms. However, what degree of enhancement in insulin activation of PI3K signaling in adipose tissue after INT131 treatment contributes to increased whole body glucose homeostasis remains unclear at this time.
A number of studies have demonstrated that PPARγ activity plays an important role in the maintenance of bone homeostasis (1, 2, 9, 13, 35). Inhibition of PPARγ activation leads to increased bone mass, which is likely due to increased osteoblastogenesis from bone marrow progenitor cells (1, 40). However, activation of PPARγ induced by rosiglitazone, a full agonist of PPARγ and antidiabetic agent, causes significant reductions in bone mineral density and bone volume and changes in bone microarchitecture (2, 27). Indeed, in type 2 diabetic patients treated with TZDs, bone loss is found (3, 5), causing an increase in the risk of a fracture and unnecessary stress on the bone skeleton. These unwanted side effects have been of concern in treating type 2 diabetic patients with TZDs. In this regard, the present study suggest that INT131 may be a desirable drug that does not affect bone mass for humans with type 2 diabetes, as evidenced by the finding that INT131 therapy, but not pioglitazone, significantly improves bone mineral density in insulin-resistant obese mice. X-ray crystallography studies revealed that INT131 binds to PPARγ in the same binding pocket as TZDs but does not bind to PPARγ in exactly the same way as other full PPARγ agonists (32). This feature could induce different conformational changes in PPARγ affecting the status of PPARγ activity differently. These factors may explain the different outcome on bone mass in INT131-treated mice compared with pioglitazone-treated mice. Other potential mechanisms to explain INT131's effects on increased bone mass could be an increased number of osteoblasts or a decreased number of adipocytes in the skeleton. Future studies are required for a full investigation of bone metabolism and formation in response to INT131 in conjunction with clinical implications.
Recent evidence demonstrated that the cyclin-dependent kinase 5-mediated phosphorylation of PPARγ leads to the dysregulated expression of genes, including adipsin and adiponectin, both of which are inappropriately regulated in obesity, without altering its adipogenic capacity (7), suggesting a critical role for PPARγ phosphorylation in insulin resistance. This leads to the attractive possibility that the separation between PPARγ transcriptional activity and PPARγ phosphorylation may enable the development of selective antidiabetic modulators that will be of benefit for both glucose metabolism and undesirable side effects.
In conclusion, our results suggest that INT131 as a new selective PPARγ modulator is a favorable antidiabetic agent that has a significant glucose-lowering and insulin-sensitizing effect in insulin-resistant obese mice. The mechanism responsible for this is involved in enhanced insulin's ability to increase key components of signaling in skeletal muscle and adipose tissue. Specifically, INT131 has a safety profile for bone mass compared with other TZD PPARγ agonists.
GRANTS
This work was supported by a grant from InteKrin Therapeutic.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.H.L., H.H., and K.C. performed the experiments; D.H.L. and Y.-B.K. analyzed the data; D.H.L. prepared the figures; C.S.M. and Y.-B.K. interpreted the results of the experiments; C.S.M. edited and revised the manuscript; Y.-B.K. did the conception and design of the research; Y.-B.K. drafted the manuscript; Y.-B.K. approved the final version of the manuscript.
REFERENCES
- 1. Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest 113: 846–855, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology 146: 1226–1235, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Aubert RE, Herrera V, Chen W, Haffner SM, Pendergrass M. Rosiglitazone and pioglitazone increase fracture risk in women and men with type 2 diabetes. Diabetes Obes Metab 12: 716–721, 2010 [DOI] [PubMed] [Google Scholar]
- 4. Beeson M, Sajan MP, Dizon M, Grebenev D, Gomez-Daspet J, Miura A, Kanoh Y, Powe J, Bandyopadhyay G, Standaert ML, Farese RV. Activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes 52: 1926–1934, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Berberoglu Z, Yazici AC, Demirag NG. Effects of rosiglitazone on bone mineral density and remodelling parameters in Postmenopausal diabetic women: a 2-year follow-up study. Clin Endocrinol (Oxf) 73: 305–312, 2010 [DOI] [PubMed] [Google Scholar]
- 6. Björnholm M, Kawano Y, Lehtihet M, Zierath JR. Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes 46: 524–527, 1997 [DOI] [PubMed] [Google Scholar]
- 7. Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466: 451–456, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chun KH, Choi KD, Lee DH, Jung Y, Henry RR, Ciaraldi TP, Kim YB. In vivo activation of ROCK1 by insulin is impaired in skeletal muscle of humans with type 2 diabetes. Am J Physiol Endocrinol Metab 300: E536–E542, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cock TA, Back J, Elefteriou F, Karsenty G, Kastner P, Chan S, Auwerx J. Enhanced bone formation in lipodystrophic PPARgamma(hyp/hyp) mice relocates haematopoiesis to the spleen. EMBO Rep 5: 1007–1012, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeFronzo RA. Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes. Diabetes Rev 5: 177–269, 1997 [Google Scholar]
- 11. Folli F, Saad MJ, Backer JM, Kahn CR. Regulation of phosphatidylinositol 3-kinase activity in liver and muscle of animal models of insulin-resistant and insulin-deficient diabetes mellitus. J Clin Invest 92: 1787–1794, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frevert EU, Kahn BB. Differential effects of constitutively active phosphatidylinositol 3-kinase on glucose transport, glycogen synthase activity, and DNA synthesis in 3T3-L1 adipocytes. Mol Cell Biol 17: 190–198, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grey A. Skeletal consequences of thiazolidinedione therapy. Osteoporos Int 19: 129–137, 2008 [DOI] [PubMed] [Google Scholar]
- 14. Higgins LS, Depaoli AM. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am J Clin Nutr 91: 267S–272S, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Higgins LS, Mantzoros CS. The Development of INT131 as a Selective PPARgamma Modulator: Approach to a Safer Insulin Sensitizer. PPAR Res 2008: 936906, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet 373: 2125–2135, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 338: 867–872, 1998 [DOI] [PubMed] [Google Scholar]
- 18. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 106: 473–481, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kerouz NJ, Hörsch D, Pons S, Kahn CR. Differential regulation of insulin receptor substrates-1 and -2 (IRS-1 and IRS-2) and phosphatidylinositol 3-kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J Clin Invest 100: 3164–3172, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 405: 421–424, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Khanderia U, Pop-Busui R, Eagle KA. Thiazolidinediones in type 2 diabetes: a cardiology perspective. Ann Pharmacother 42: 1466–1474, 2008 [DOI] [PubMed] [Google Scholar]
- 22. Kienesberger PC, Lee D, Pulinilkunnil T, Brenner DS, Cai L, Magnes C, Koefeler HC, Streith IE, Rechberger GN, Haemmerle G, Flier JS, Zechner R, Kim YB, Kershaw EE. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J Biol Chem 284: 30218–30229, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim MK, Chae YN, Kim HS, Choi SH, Son MH, Kim SH, Kim JK, Moon HS, Park SK, Shin YA, Kim JG, Lee CH, Lim JI, Shin CY. PAR-1622 is a selective peroxisome proliferator-activated receptor gamma partial activator with preserved antidiabetic efficacy and broader safety profile for fluid retention. Arch Pharm Res 32: 721–727, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Kim YB, Ciaraldi TP, Kong A, Kim D, Chu N, Mohideen P, Mudaliar S, Henry RR, Kahn BB. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 51: 443–448, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Kim YB, Nikoulina SE, Ciaraldi TP, Henry RR, Kahn BB. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest 104: 733–741, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kintscher U, Goebel M. INT-131, a PPARgamma agonist for the treatment of type 2 diabetes. Curr Opin Investig Drugs 10: 381–387, 2009 [PubMed] [Google Scholar]
- 27. Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology 148: 2669–2680, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le Marchand-Brustel Y, Gautier N, Cormont M, Van Obberghen E. Wortmannin inhibits the action of insulin but not that of okadaic acid in skeletal muscle: comparison with fat cells. Endocrinology 136: 3564–3570, 1995 [DOI] [PubMed] [Google Scholar]
- 29. Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, White MF, Wei L, Kim YB. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem 284: 11776–11780, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Wang Z, Furukawa N, Escaron P, Weiszmann J, Lee G, Lindstrom M, Liu J, Liu X, Xu H, Plotnikova O, Prasad V, Walker N, Learned RM, Chen JL. T2384, a novel antidiabetic agent with unique peroxisome proliferator-activated receptor gamma binding properties. J Biol Chem 283: 9168–9176, 2008 [DOI] [PubMed] [Google Scholar]
- 31. Minoura H, Takeshita S, Kimura C, Hirosumi J, Takakura S, Kawamura I, Seki J, Manda T, Mutoh S. Mechanism by which a novel non-thiazolidinedione peroxisome proliferator-activated receptor gamma agonist, FK614, ameliorates insulin resistance in Zucker fatty rats. Diabetes Obes Metab 9: 369–378, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Motani A, Wang Z, Weiszmann J, McGee LR, Lee G, Liu Q, Staunton J, Fang Z, Fuentes H, Lindstrom M, Liu J, Biermann DH, Jaen J, Walker NP, Learned RM, Chen JL, Li Y. INT131: a selective modulator of PPAR gamma. J Mol Biol 386: 1301–1311, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356: 2457–2471, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 269: 3568–3573, 1994 [PubMed] [Google Scholar]
- 35. Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology 145: 401–406, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799–806, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes 45: 1661–1669, 1996 [DOI] [PubMed] [Google Scholar]
- 38. Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest 116: 581–589, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tzoulaki I, Molokhia M, Curcin V, Little MP, Millett CJ, Ng A, Hughes RI, Khunti K, Wilkins MR, Majeed A, Elliott P. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: retrospective cohort study using UK general practice research database. BMJ 339: b4731, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med 13: 1496–1503, 2007 [DOI] [PubMed] [Google Scholar]
- 41. White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283: E413–E422, 2002 [DOI] [PubMed] [Google Scholar]