

Abstract
Omega-3 polyunsaturated fatty acids (n-3 PUFAs) enhance insulin sensitivity and glucose homeostasis in rodent models of insulin resistance. These beneficial effects have been linked with anti-inflammatory properties, but emerging data suggest that the mechanisms may also converge on mitochondria. We evaluated the influence of dietary n-3 PUFAs on mitochondrial physiology and muscle lipid metabolites in the context of high-fat diet (HFD) in mice. Mice were fed control diets (10% fat), HFD (60% fat), or HFD with fish oil (HFD+FO, 3.4% kcal from n-3 PUFAs) for 10 wk. Body mass and fat mass increased similarly in HFD and HFD+FO, but n-3 PUFAs attenuated the glucose intolerance that developed with HFD and increased expression of genes that regulate glucose metabolism in skeletal muscle. Despite similar muscle triglyceride levels in HFD and HFD+FO, long-chain acyl-CoAs and ceramides were lower in the presence of fish oil. Mitochondrial abundance and oxidative capacity were similarly increased in HFD and HFD+FO compared with controls. Hydrogen peroxide production was similarly elevated in HFD and HFD+FO in isolated mitochondria but not in permeabilized muscle fibers, likely due to increased activity and expression of catalase. These results support a hypothesis that n-3 PUFAs protect glucose tolerance, in part by preventing the accumulation of bioactive lipid mediators that interfere with insulin action. Furthermore, the respiratory function of skeletal muscle mitochondria does not appear to be a major factor in sphingolipid accumulation, glucose intolerance, or the protective effects of n-3 PUFAs.
Keywords: fish oil, omega-3 fatty acids, essential fatty acids, polyunsaturated fatty acids, insulin sensitivity, sphingolipid, ceramide, long-chain acyl-coenzyme A, high-fat diet, mitochondria
a growing body of evidence implicates ectopic lipid accumulation within insulin-sensitive tissues as a mechanism underlying insulin resistance (47). More specifically, ceramides, diacylglycerols, and long-chain acyl-CoAs (LCACoA) have emerged as the primary groups of bioactive lipid mediators that are linked with the development of insulin resistance (1, 13, 57, 63). Furthermore, people with type 2 diabetes exhibit lower abundance of mitochondria in skeletal muscle (30, 51), spawning a hypothesis that ectopic lipid accumulation is a consequence of decreased mitochondrial function and the subsequent blunting of fat oxidation (41, 46). Indeed, high fat intake has been reported to decrease mitochondrial content and function (8) and the expression of peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC-1α), a key transcriptional coactivator of genes involved in mitochondrial biogenesis (11, 52). However, other reports indicate that high-fat feeding increased activity of mitochondrial enzymes, stimulated mitochondrial biogenesis, increased expression of mitochondrial genes, and increased mitochondrial function (14, 19, 34, 36, 58). Although the precise role of mitochondria in the etiology of insulin resistance continues to be unclear, these organelles represent an attractive therapeutic target for prevention or treatment of insulin resistance, type 2 diabetes, and related metabolic derangements.
Diet and exercise are highly effective countermeasures for insulin resistance (18), but alternative therapeutic strategies are needed for individuals who cannot adopt such lifestyle changes. One such strategy may be dietary intake of omega-3 polyunsaturated fatty acids (n-3 PUFAs), which include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) from fish oil. Many rodent studies show that n-3 PUFAs prevent insulin resistance and improve glucose homeostasis (29, 37, 39, 53, 54), but the underlying mechanisms are only beginning to emerge. A leading hypothesis is that n-3 PUFAs reverse insulin resistance by suppressing inflammation (39, 50). Accumulating evidence also suggests that the mechanisms of the insulin-sensitizing effects of n-3 PUFAs may converge on mitochondria. Specifically, n-3 PUFAs prevented insulin resistance in wild-type mice but not in mice lacking the α2 catalytic subunit of AMP-activated protein kinase (AMPK) (27) or PPARα (37). Both AMPK and PPARα are known to regulate genes involved in mitochondrial biogenesis and fatty acid oxidation (15, 59). Data from these knockout models suggest that mitochondria may play a critical role in the protective effects of n-3 PUFAs on insulin sensitivity.
The present study was conducted to determine whether dietary n-3 PUFAs influence mitochondrial physiology and accumulation of lipid metabolites in the context of diet-induced insulin resistance. We fed mice high-fat diets with or without n-3 PUFAs for 10 wk. We hypothesized that the insulin-sensitizing effects of dietary n-3 PUFAs would be paralleled by attenuated accumulation of lipid metabolites subsequent to enhanced mitochondrial function in skeletal muscle.
METHODS AND PROCEDURES
Animals.
The Mayo Clinic Institutional Animal Care and Use Committee approved the protocol. Male C57BL6 mice were obtained from Jackson Laboratory (Bar Harbor, ME) at 10 mo of age. Mice were housed individually in a temperature-controlled facility with a 12:12-h light-dark cycle and ad libitum access to chow and water. Animals were provided with nestlets for minimal environmental enrichment. After 10 days of acclimation, body composition was measured by Echo-MRI (Echo Medical Systems, Houston, TX), and mice were randomly assigned to one of three groups: normal-fat diet (NFD; 20% kcal protein, 70% kcal carbohydrate, 10% kcal fat), high-fat diet (HFD; 20% kcal protein, 20% kcal carbohydrate, 60% kcal fat), or HFD + fish oil (HFD+FO; 20% kcal protein, 20% kcal carbohydrate, 60% kcal fat + 23% kcal from menhaden oil). The HFD+FO diet provided 3.4% kcal from EPA and DHA, stabilized with tert-butylhydroquinone (tBHQ, 200 ppm) as an antioxidant and stored at −80°C. The custom-designed diets were formulated by Research Diets (New Brunswick, NJ). Tables 1 and 2 give the macronutrient composition and lipid distribution, respectively, of each diet. Mice were given ad libitum access to chow and water, and fresh chow was given 2–3 times/wk. Body composition was measured again at 5 and 10 wk.
Table 1.
Composition of purified diets
| NFD |
HFD |
HFD+FO |
||||
|---|---|---|---|---|---|---|
| g | kcal % | g | kcal % | g | kcal % | |
| Protein | 19 | 20 | 26 | 20 | 26 | 20 |
| Carbohydrates | 67 | 70 | 26 | 20 | 26 | 20 |
| Fat | 4 | 10 | 35 | 60 | 35 | 60 |
| kcal/g | 3.8 | 5.2 | 5.2 | |||
| g | kcal | g | kcal | g | kcal | |
| Casein, 80 mesh | 200 | 800 | 200 | 800 | 200 | 800 |
| l-Cystine | 3 | 12 | 3 | 12 | 3 | 12 |
| Corn starch | 506.2 | 2025 | 0 | 0 | 0 | 0 |
| Maltodextrin 10 | 125 | 500 | 125 | 500 | 125 | 500 |
| Sucrose | 68.8 | 275 | 68.8 | 275 | 68.8 | 275 |
| Cellulose, BW200 | 50 | 0 | 50 | 0 | 50 | 0 |
| Corn oil | 15 | 135 | 15 | 135 | 15 | 135 |
| Lard | 30 | 270 | 255 | 2295 | 193 | 1737 |
| Menhaden oil | 0 | 0 | 0 | 0 | 62 | 558 |
| Mineral mix | 10 | 0 | 10 | 0 | 10 | 0 |
| Dicalcium phosphate | 13 | 0 | 13 | 0 | 13 | 0 |
| Calcium carbonate | 5.5 | 0 | 5.5 | 0 | 5.5 | 0 |
| Potassium citrate | 16.5 | 0 | 16.5 | 0 | 16.5 | 0 |
| Vitamin mix V10001 | 10 | 40 | 10 | 40 | 10 | 40 |
| Choline bitartrate | 2 | 0 | 2 | 0 | 2 | 0 |
NFD, normal-fat diet; HFD, high-fat diet; HFD+FO, HFD + fish oil.
Table 2.
Lipid content of purified diets (in g)
| NFD | HFD | HFD+FO | |
|---|---|---|---|
| Corn oil | 15 | 15 | 15 |
| Lard | 30 | 255 | 193 |
| Menhaden oil | 0 | 0 | 62 |
| Total | 45 | 270 | 270 |
| C12, Lauric | 0 | 0 | 0.1 |
| C14, Myristic | 0.3 | 2.3 | 6 |
| C14:1, Myristoleic | 0.2 | 1.3 | 1 |
| C15, Pentadecanoic | 0 | 0 | 0.3 |
| C16, Palmitic | 8.5 | 60 | 55 |
| C16:1, Palmitoleic | 1.1 | 9.7 | 13.4 |
| C16:2, Hexadecanoic | 0 | 0 | 1 |
| C16:3, Hexadecatriaenoic | 0 | 0 | 0.9 |
| C16:4, Hexadecatetraenoic | 0 | 0 | 0.9 |
| C17, Heptadecanoic | 0 | 0 | 0.3 |
| C18, Stearic | 4.3 | 34.2 | 27.5 |
| C18:1, Oleic | 16.1 | 108.5 | 89 |
| C18:2, Linoleic | 11.6 | 30.9 | 26.8 |
| C18:3, Linolenic | 0.5 | 2.8 | 3.1 |
| C18:4, Stearidonic | 0 | 0 | 1.9 |
| C20, Arachidic | 0 | 0 | 0.1 |
| C20:1, Gadoleic | 0 | 0 | 0.9 |
| C20:2, Auricolic | 0 | 0 | 0.1 |
| C20:3, Bishomopinolenic | 0 | 0 | 0.2 |
| C20:4, Arachidonic | 0.5 | 4.3 | 4.6 |
| C20:5, EPA | 0 | 0 | 8.8 |
| C21:5, Heneicosapentaenoic | 0 | 0 | 0.5 |
| C22, Behenic | 0 | 0 | 0.1 |
| C22:1, Erucic | 0 | 0 | 0.2 |
| C22:4, Adrenic | 0 | 0 | 0.1 |
| C22:5, Docosapentaenoic | 0 | 0 | 1.7 |
| C22:6, DHA | 0 | 0 | 7.6 |
| C24, Lignoceric | 0 | 0 | 0.4 |
| C24:1, Selacholeic | 0 | 0 | 0.1 |
| Total | 43 | 254.1 | 252.7 |
| Saturated | 13.1 | 96.5 | 89.8 |
| Monounsaturated | 17.4 | 119.5 | 104.7 |
| Polyunsaturated | 12.6 | 38 | 59.6 |
| Saturated, % | 30.3 | 38 | 35.5 |
| Monounsaturated, % | 40.3 | 47 | 41.4 |
| Polyunsaturated, % | 29.3 | 15 | 23.6 |
Oral glucose tolerance tests.
Oral glucose tolerance tests (OGTT) were performed following a 6-h fast as described previously (20). Fasting blood glucose was measured from the tail vein (AlphaTrak; Abbott Laboratories, Abbott Park, IL), and plasma was collected and frozen for measuring fasting insulin concentrations. Blood glucose was measured in duplicate at 5, 10, 15, 30, 60, and 120 min following oral glucose administration (2 g/kg body wt). Plasma was collected at 15 min for insulin measurements. Plasma insulin was measured in triplicate using an enzyme-linked immunosorbent assay (Crystal Chem, Downers Grove, IL). OGTTs were performed before randomization and again after 5 and 10 wk of the dietary intervention.
Whole body metabolic outcomes.
An 18-chamber open-circuit cage calorimeter system (CLAMS, Columbus Instruments) was used to measure oxygen consumption (V̇o2) and carbon dioxide production (V̇co2) of individual mice, as previously described (26). Indirect calorimetry was performed in a constant light- (12-h light and dark phases) and temperature- (∼23°C) controlled room. Oxygen and carbon dioxide analyzers were calibrated with certified primary standard calibration gas (20.49% oxygen, 5,031 ppm carbon dioxide, and nitrogen balanced). Mice were then acclimated to the chambers for ∼12 h followed by the start of the 48-h experimental period at 6 AM. Measurements were conducted for 24 h of fed and 24 h of fasted conditions. The respiratory exchange ratio (RER) was calculated from V̇o2 and V̇co2 values. Energy expenditure was calculated using the equation [(3.815 + 1.232 × RER) × V̇o2]. Food intake was measured as part of the cage calorimetry experiments using an integrated balance accounting for spillage of food. Physical activity levels were measured by photocells.
Mitochondrial energetics.
Approximately 1 wk following the 10-wk OGTT, mice were euthanized by overdose of pentobarbital sodium. One whole quadriceps muscle group was immediately removed and placed in ice-cold BIOPS buffer (2.77 mM CaK2EGTA, 7.23 mM K2EGTA, 5.77 mM Na2ATP, 6.56 mM MgCl2 6H2O, 20 mM taurine, 15 mM Na2phosphocreatine, 20 mM imidazole, 0.5 mM DTT, and 50 mM MES). The muscle was transferred to a chilled glass dish containing homogenizing buffer (100 mM KCl, 50 mM Tris, 5 mM MgCl2, 1.8 mM ATP, and 1 mM EDTA, pH 7.2). Tissue was minced, treated with protease for 2 min, and gently homogenized on ice for 10 min using a motor-driven Potter-Elvehjem tissue grinder, as described previously (32). Mitochondria were separated by differential centrifugation (32) and resuspended in a preservation buffer (225 mM sucrose, 44 mM KH2PO4, 12.5 mM Mg acetate, and 6 mM EDTA) based on starting wet weight of the muscle tissue (1:4 wt/vol). High-resolution respirometry was used to evaluate mitochondrial bioenergetics using an Oroboros Oxygraph 2K (Oroboros Instruments, Innsbruk, AT). A 50-μl aliquot of the mitochondrial suspension was added to each 2-ml oxygraph chamber containing respiration buffer (MiRO5, 110 mM sucrose, 60 mM potassium lactobionate, 0.5 mM EGTA, 1 g/l BSA essentially fat free, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, pH 7.1). A stepwise titration protocol was used to assess states 1, 2, 3, and 4 with electron flow through various respiratory chain complexes (16). Respiration was measured in isolated mitochondria devoid of substrates (state 1), in the presence of 10 mM glutamate and 2 mM malate (state 2), followed by 2.5 mM ADP to stimulate state 3 respiration with electron flow through complex I. Cytochrome c was added at 10 μM to assess integrity of the outer mitochondrial membrane. Next, 10 mM succinate was added to provide convergent electron flow through complex I and II, followed by 0.5 μM rotenone to inhibit complex I. State 4 respiration was measured in the presence of 2 μg/ml oligomycin to inhibit ATP synthase activity. Last, 2.5 μM antimycin A was added to inhibit mitochondrial oxygen consumption to confirm that oxygen consumption was of mitochondrial origin. A parallel experiment was conducted to measure mitochondrial capacity for lipid oxidation. Mitochondria were respired in the presence of palmitoyl-l-carnitine (0.005 mM) and malate (2 mM) without adenylates (state 2), in the presence of 2.5 mM ADP (state 3), and 2 μg/ml oligomycin (state 4). Oxygen flux rates were calculated for each step and corrected for background oxygen kinetics using Datlab software (Oroboros Instruments, Innsbruck, Austria). Protein content of the mitochondrial suspension was measured using a colorimetric assay (Pierce 660-nm Protein Assay). Oxygen flux rates were expressed per tissue wet weight and per milligram of mitochondrial protein.
Mitochondrial coupling efficiency was evaluated from the respiratory control ratio (RCR) as the quotient of state 3 and state 4 respiration rates. Mitochondrial phosphorylation efficiency was evaluated from the ADP:O ratio, determined by quantifying the amount of oxygen consumed in response to a nonsaturating (15 μM) pulse of ADP. The actual concentration of ADP was measured in triplicate using the EnzyLight ADP Assay Kit (BioAssay Systems, Hayward, CA). To determine the time point where oxygen consumption increased in response to the ADP pulse and returned to baseline after phosphorylating all of the ADP, oxygen concentration was plotted for each experiment, and a segmented, bilinear regression model was used to fit the data (PROC NLIN in SAS; Statistical Analysis Software Institute, Cary, NC), according to the following function (60):
where Y is the predicted [O2], a0 is the intercept of the first line, a1 is the slope of the first line, b1 is the slope of the second line, and ip is the inflection point. For all cases where time t < ip, id was assigned a value of 1, indicating that the data point belonged to the first line. When t ≥ ip, id was assigned a value of 0, indicating that the data point belonged to the second line. The NLIN procedure was used to simultaneously estimate a0, a1, b1, and ip, based on a two-stage iterative fitting procedure that minimized the error sums of squares for the above model. The difference in oxygen concentration across the two inflection points was used to compute ADP:O. All measurements in isolated mitochondria were performed in duplicate and averaged.
Mitochondrial abundance.
Skeletal muscle mitochondrial abundance was determined using electron microscopy and real-time PCR quantitation of mitochondrial DNA abundance. A portion of the quadriceps muscle was immediately placed in Trumps fixative (30% formaldehyde, 10% glutaraldehyde) followed by staining with uranyl acetate. Tissues were embedded in epoxy resin and sectioned for light microscopy to confirm orientation longitudinally along muscle fibers. Microtome thin sections stained with lead citrate were examined by a transmission electron microscope (JEOL ExII, Peabody, MA) at ×25,000 magnification by a blinded investigator. Ten digital images were acquired for each sample, which were analyzed using NIH Image J software for measurement of mitochondrial area and perimeter. Mitochondrial density by area was computed as the total area (μm2) of mitochondria divided by the area of the field of view, calibrated to a digital scale bar for each micrograph. For mtDNA abundance, DNA was extracted from frozen muscle using a QIAamp DNA minikit (Qiagen, Valencia, CA). Relative mtDNA copy numbers were determined by real-time PCR (Applied Biosystems 7900HT Sequence Detection System) using primer/probe sets targeted to mtDNA-encoded NADH dehydrogenase subunits 1 (ND1) and 4 (ND4). The primer and probe sequences for ND1 were as follows: (GenBank acc. no. NC_005089) forward primer AAGGAGAATCAGAATTAGTATCAGGGTT, reverse primer TAGTACTCTGCTATAAAGAATAACGCGAAT, and probe ACGTAGAATACGCAGCCGGCC. The primer and probe sequences for ND4 were as follows: (GenBank acc. no. NC_005089) forward primer TCCAACTACGAACGGATCCA, reverse primer AAGTGGGAAGACCATTTGAAGTC, and probe AGCCGTACTATAATCATGGCCCGA. Samples were run in duplicate and normalized for the nuclear housekeeping gene 28S ribosomal DNA.
Gene expression.
Total RNA was isolated from ∼20 mg of skeletal muscle tissue using the Qiagen RNeasy Mini Kit according to the manufacturer's instructions. cDNA was prepared using the Taqman reverse transcription kit (Life Technologies) per the manufacturer's instructions. Real-time PCR was performed on a Viia7 Real Time PCR System (Life Technologies) using the Taqman gene expression assays. The following mouse gene expression assays (Life Technologies) were used for quantitative real-time PCR: PPARγ (Mm01184322_m1), PPARα (Mm00440939_m1), PGC1α (Mm01208835_m1), GLUT4 (Mm01245502_m1), glycogen synthase-1 (GYS1, Mm000472712_m1), insulin receptor substrate 1 (IRS1, Mm01278327_m1), nuclear respiratory factor 1 (NRF1, Mm01135606), transcription factor A, mitochondria (TFAM, Mm00447485_m1), superoxide dismutase-1 (SOD1, Mm01344233_g1), superoxide dismutase-2 (SOD2, Mm01313000_m1), catalase (CAT, Mm00437992_m1), and glutathione peroxidase-1 (GPX1 Mm00656767_g1). Reactions were performed in a 384-well assay format with a relative standard curve. All samples were plated in duplicate, and each plate contained one experimental gene and an endogenous control, mouse β-actin.
H2O2 production in isolated mitochondria and permeabilized muscle fibers.
The reactive oxygen species (ROS) emitting potential of isolated mitochondria was evaluated under state 2 conditions. A Fluorolog 3 (Horiba Jobin Yvon) spectrofluorometer with temperature control and continuous stirring was used to monitor Amplex Red (Invitrogen) oxidation in a 50-μl aliquot of the freshly isolated mitochondrial suspension. Isolated mitochondria were placed in a quartz cuvette with 2 ml 0f buffer z containing (in mM) 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO4, 3 MgCl2-6H2O, and 5 mg/ml BSA (pH 7.4, 295 mOsm) (2). Amplex Red oxidation was measured in the presence of glutamate (10 mM), malate (2 mM), and succinate (10 mM). The fluorescent signal was corrected for background autooxidation and calibrated to a standard curve. H2O2 production rates were expressed per tissue wet weight and per protein concentration of the mitochondrial suspension.
H2O2 emission was also measured in permeabilized muscle fibers as described previously (3, 32a). Duplicate sets of fiber bundles (∼5 mg) were mechanically separated using sharp forceps and incubated on ice in saponin (50 μg/ml) for 30 min. Permeabilized fibers were then washed in buffer containing (in mM) 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO4, 3 MgCl2-6H2O, 0.05 pyruvate, and 0.02 malate and 5 mg/ml BSA (pH 7.4, 295 mOsm). Fibers were preincubated in 10 mM pyrophosphate to deplete endogenous adenylates. Fiber bundles were placed in a quartz cuvette with 2 ml of buffer z containing 2 μg/ml oligomycin followed by a stepwise addition of glutamate (5 μM) and malate (2 μM), and succinate (0.25–24 mM) to stimulate H2O2 production under state 4 conditions. H2O2 production rates were expressed per tissue wet weight.
Antioxidant enzyme activities.
Muscle SOD and catalase activities were measured in cell lysates spectrophotometrically. Catalase activity was determined using a direct kinetic assay by following the action of catalase on H2O2 (32a). Total SOD activity was determined from the consumption of xanthine oxidase-generated superoxide radical by endogenous SOD (Cayman Chemical, Ann Arbor, MI).
DNA oxidation.
The DNA adduct biomarker of oxidative stress, 8-oxo-2′-deoxyguanosine (8-oxo-dG), was measured by LC/MS/MS as described previously (32a). Following extraction of DNA (DNA Extractor TIS Kit; Wako Richmond, VA), an internal standard solution was added before hydrolysis as instructed in the 8-OHdG Assay Preparation Reagent Set from Wako Chemicals. A Waters C18 BEH 2.1 × 50-mm column at 0.4 ml/min flow rate via Waters Acquity UPLC system (Milford, MA) was used to separate dG and 8-OHdG. All ions were run in negative electrospray ionization using selected-reaction monitoring (SRM) for transitions on a TSQ Quantum Ultra from Thermo Scientific (Waltham, MA).
Triglyceride content.
Muscle tryclycerides were quantified enzymatically as described previously (17). Approximately 20 mg of frozen muscle was thawed on ice and dissected under a stereomicroscope (×10–40; Motic Microscopes, Hong Kong, China) to remove connective tissue, adipocytes, and blood vessels. The dissected tissue was homogenized on ice in Folch solvent (chloroform-methanol, 2:1). The homogenate was filtered and centrifuged at 2,000 rpm to separate intracellular free glycerol and glucose (upper phase) and total lipids (lower phase). The separated triglycerides were hydrolyzed in 2.5% methanolic H2SO4 at 80°C and quantified enzymatically (24).
Muscle lipid metabolites.
Skeletal muscle LCACoA was measured using methods described previously (6). Briefly, LCACoAs were extracted using ACN-2-propanol-methanol (3:1:1, vol/vol/vol). A known amount of heptadecanoyl-CoA was added as an internal standard. The molecules (C14:0-CoA, C16:0-CoA, C16:1-CoA, C18:2-CoA, C18:1-CoA, C18:0-CoA, C20:0-CoA) were separated on reverse-phase UPLC column using a binary gradient with ammonium hydroxide (NH4OH) in water and NH4OH in ACN. The LCACoA was quantified using SRM on a triple quadrupole mass spectrometer in positive electrospray ionization (ESI) mode. myristoyl coenzyme A (C14-CoA), palmitoyl coenzyme A (C16-CoA), palmitoleoyl coenzyme A (C16:1-CoA), stearoyl coenzyme A (C18-CoA), oleoyl coenzyme A (C18:1-CoA), linoleoyl coenzyme A (C18:2-CoA), arachidonoyl coenzyme A (C20-CoA), as well as internal standard heptadecanoyl coenzyme A (C17-CoA) were purchased from Avanti Polar Lipids (Alabaster, Al). The HPLC grade acetonitrile and HPLC grade water were obtained from Fisher Chemical (Pittsburg, PA). NH4OH was obtained from Sigma (St. Louis, MO).
Sphingolipids were measured as described previously (7). Sphingolipids were extracted using isopropanol-water-ethyl acetate (35:5:60, vol/vol/vol). Quantitative measurement of sphingolipids (Sph, dhSph, S1P, C14:0-Cer, C16:0-Cer, C18:1-Cer, C18:0-Cer, C20:0-Cer, C24:1-Cer, C24:0-Cer) were made using a Thermo TSQ Quantum Ultra mass spectrometer using positive ion ESI source with SRM. The chromatographic separation was performed using a Waters ACQUITY Ultra Performance Liquid Chromatography (UPLC). The analytic column was a reverse-phase Acquity C8 UPLC BEH column 2.1 × 150 mm, 1.7 μm (Waters, Milford, MA). Chromatographic separation was conducted in binary gradient using 2 mM ammonium formate, 0.15% formic acid in methanol as solvent A, and 1.5 mM ammonium formate, 0.1% formic acid in water as solvent B at the flow rate of 0.4 ml/min. C17Sph, C17S1P, and C17:0-ceramide were used as an internal standards. Standards utilizing 18C-sphingoid bases: sphingosine d18:1 (Sph), sphinganine d18:0 (dhSph), sphingosine-1-phosphate d18:1 (S1P), d18:1/14:0-Cer - ceramides containing myristic acid (C14:0-Cer), d18:1/16:0-Cer - ceramides containing palmitic acid (C16:0-Cer), d18:1/17:0-Cer - ceramides containing margaric acid (C17:0-Cer) - internal standard for ceramides, d18:1/18:0-Cer - ceramides containing stearic acid (C18:0-Cer), d18:1/18:1-Cer - ceramides containing oleic acid (C18:1-Cer), d18:1/20:0-Cer - ceramides containing arachidic acid (C20:0-Cer), d18:1/24:0-Cer - ceramides containing lignoceric acid (C24:0-Cer), d18:1/24:1-Cer - ceramides containing nervonic acid (C24:1-Cer), as well as internal standards utilizing 17C-sphingoid bases: sphingosine (d17:1-Sph) - internal standard for sphingosine and sphinganine, sphingosine 1-phosphate (d17:1-S1P) - internal standard for sphingosine 1-phosphate, were purchased from Avanti Polar Lipids. HPLC-grade methanol was purchased from EMD Chemicals (Gibbstown, NJ) and HPLC-grade water from Fisher Chemical (Pittsburg, PA). Formic acid, ammonium formate, and ethanol were obtained from Sigma-Aldrich.
Statistical analyses.
Data are presented as means ± SE. Outcome variables were analyzed by ANOVA with group (NFD, HFD, HFD+FO) as the main effect. When significant (P < 0.05) main effects (1-way ANOVA) or interactions (2-way ANOVA) were found, Tukey's HSD procedure was used for pairwise comparisons across groups and time points while maintaining 5% type I error rate. Analysis of covariance (ANCOVA) was used to analyze energy expenditure and food intake using body weight as a covariate. All statistical analyses were performed using JMP Software (SAS Institute, Cary, NC). The inflection point analyses for ADP:O measurements were performed using the PROC NLIN procedure using SAS Software.
RESULTS
Food intake and body composition.
Both groups receiving HFD gained weight, but at no point did HFD and HFD+FO differ in body weight (Fig. 1A). Food intake during the light and dark cycles were similar in all three groups, using body weight as a covariate (Fig. 1E). After 5 and 10 wk of diet, fat mass was similarly elevated in HFD and HFD+FO groups (Fig. 1B). At 10 wk, lean mass was significantly increased in HFD and HFD+FO compared with NFD (Fig. 1C). Percent body fat was higher in HFD and HFD+FO than in NFD at both 5 and 10 wk (Fig. 1D).
Fig. 1.
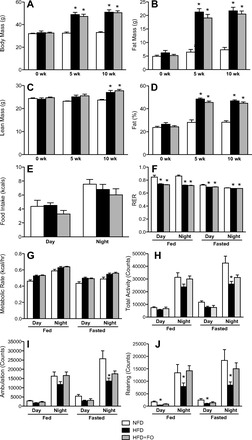
Ten weeks of high-fat diet (HFD) increases body mass and fat mass in mice with or without fish oil. Body mass (A), fat mass (B), lean mass (C), and percent body fat (D) were measured by EchoMRI at baseline and after 5 and 10 wk of dietary intervention. Respiratory exchange ratio (RER; F) was derived from V̇o2 and V̇co2 measured by indirect calorimetry. Metabolic rate (G) was calculated from V̇o2 and RER. Food intake (E) was measured during calorimetry experiments. Total cage activity counts (H), ambulatory activity (I), and rearing activity (J) were measured by photocell beam breaks during calorimetry measurements. Bars represent means ± SE for normal-fat diet (NFD), HFD, and HFD with fish oil (HFD+FO). *Significant statistical differences from NFD (P < 0.05, Tukey's HSD).
Whole body metabolic parameters and physical activity.
RER was lower in HFD and HFD+FO than in NFD under all conditions (Fig. 1F). Notably, RER was higher during fed conditions compared with fasting conditions in NFD, reflecting a shift in substrate use with nutrient availability, but this shift was not evident in HFD or HFD+FO (Fig. 1F). Metabolic rate did not differ between groups under any condition using body weight as a covariate (Fig. 1G). The total activity counts (Fig. 1H), ambulation counts (Fig. 1I), and rearing counts (Fig. 1J) show a pattern where activity was lower in HFD than in NFD but maintained in HFD+FO. This pattern was statistically significant at night under fasting conditions for total activity (Fig. 1H) and ambulation (Fig. 1I), and under all conditions for rearing (Fig. 1J).
OGTT.
At baseline, all groups exhibited similar blood glucose (Fig. 2A) and plasma insulin (Fig. 2C) responses to an oral glucose load (2 g/kg). The glucose area above baseline (AAB) during the 2 h following glucose administration was similar in all three groups (Fig. 2B). After 5 wk of respective diets, glucose tolerance deteriorated in HFD mice, reflected by increased glucose AAB (Fig. 2, D and E). At 10 wk, glucose AAB normalized somewhat in HFD but remained significantly elevated from controls (Fig. 2, G and H). The improvement of glucose tolerance between 5 and 10 wk coincided with increased plasma insulin levels at the 15-min time point at 10 wk (Fig. 2I). Fish oil attenuated the impairment in glucose intolerance with HFD. At 5 and 10 wk, glucose AAB was not different from NFD controls (Fig. 2, E and H). At 10 wk, plasma insulin at 15 min was significantly higher in HFD+FO and HFD compared with NFD (Fig. 2I), consistent with the development of insulin resistance. Notably, although HFD and HFD+FO exhibited similarly elevated insulin during the 10-wk OGTT, unlike HFD, HFD+FO maintained glucose AAB similar to controls, suggesting greater insulin sensitivity in HFD+FO compared with HFD. After mice were euthanized at 10 wk, GLUT4 mRNA expression was higher in HFD+FO than in HFD. mRNA expression of IRS1 and GYS1 were all significantly elevated in HFD+FO compared with NFD and HFD (Fig. 2J).
Fig. 2.
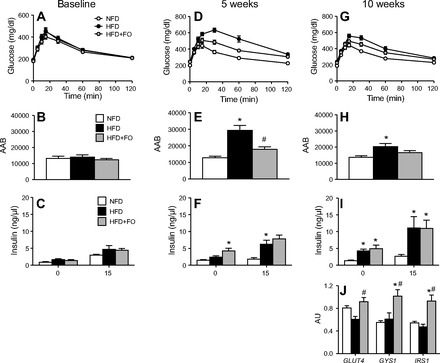
Fish oil prevents decline in glucose tolerance with Hfd. Blood glucose measured over 2 h following an oral glucose bolus at baseline (A), 5 wk of diet (D), and 10 wk of diet (G). Glucose responses were quantified from the area above baseline (AAB) at baseline (B), 5 wk (E), and 10 wk of diet (H). Plasma insulin was measured in the fasted state and 15 min following glucose bolus at baseline (C), 5 wk (F), and 10 wk (I). Gene transcript levels (J) of glucose transporter type 4 (GLUT4), glycogen synthase-1 (GYS1), and insulin receptor substrate 1 (IRS1) were measured in muscle tissue at 10 wk. Bars represent means ± SE for NFD, HFD, and HFD+FO. *Significant statistical differences from NFD; #significantly different from HFD (P < 0.05, Tukey's HSD).
Intracellular lipids.
Total muscle triglycerides were significantly elevated after 10 wk of HFD (0.104 ± 0.011 mg/g tissue) compared with NFD (0.050 ± 0.005 mg/g tissue). This lipid accumulation with high-fat feeding was not prevented by fish oil (0.096 ± 0.013 mg/g tissue). We next profiled LCACoA and ceramides in different cellular fractions in skeletal muscle. Muscle LCACoA content was significantly elevated in HFD compared with NFD in the muscle homogenate, sarcoplasmic, and mitochondrial fractions (Table 3). Although LCACoA levels were also elevated in HFD+FO compared with NFD, the levels were significantly lower than observed in HFD in all fractions (Table 3). Total muscle ceramide content was significantly greater in HFD than NFD in total homogenate, sarcoplasmic, and mitochondrial fractions (Table 4), with dominating signals coming from the greater abundance C16 and C18 species. Other species such as the C14 and C24 species showed no change or decrease with HFD. Fish oil had a similar effect on ceramides as it did on LCACoA but varied considerably depending on the individual species. Notably, fish oil attenuated the HFD-induced increase in the most abundant C18 ceramide levels in all fractions (Table 4) but increased the abundance of C14 and C24:1 species.
Table 3.
Long-chain acyl-CoA species in whole-muscle homogenates and subcellular fractions after 10 wk of NFD, HFD, and HFD+FO
| Fraction | Species | NFD | HFD | HFD+FO |
|---|---|---|---|---|
| C14 | 1.59 ± 0.09a | 3.03 ± 0.17b | 2.34 ± 0.17c | |
| C16 | 5.80 ± 0.19a | 9.51 ± 0.37b | 8.52 ± 0.24b | |
| C16:1 | 1.84 ± 0.08a | 3.99 ± 0.15b | 3.09 ± 0.18c | |
| Homogenate | C18 | 3.31 ± 0.10a | 6.28 ± 0.25b | 5.35 ± 0.25c |
| C18:1 | 31.21 ± 1.99a | 61.45 ± 2.85b | 44.55 ± 3.03c | |
| C18:2 | 4.66 ± 0.34a | 7.22 ± 0.27b | 5.72 ± 0.42a | |
| C20 | 3.89 ± 0.67a | 7.35 ± 0.32b | 6.76 ± 0.26b | |
| Total | 52.31 ± 2.09a | 98.83 ± 3.23b | 76.32 ± 3.55c | |
| C14 | 0.38 ± 0.01a | 3.03 ± 0.17b | 2.34 ± 0.17c | |
| C16 | 1.88 ± 0.07a | 9.51 ± 0.37b | 8.52 ± 0.24a | |
| C16:1 | 0.71 ± 0.03a | 3.99 ± 0.15b | 3.09 ± 0.18b | |
| Sarcoplasmic | C18 | 3.28 ± 0.16a | 6.28 ± 0.25b | 5.35 ± 0.25b |
| Fraction | C18:1 | 6.01 ± 0.37a | 61.45 ± 2.85b | 44.55 ± 3.03c |
| C18:2 | 2.01 ± 0.13a | 7.22 ± 0.27b | 5.72 ± 0.42ab | |
| C20 | 3.99 ± 0.27a | 7.35 ± 0.32a | 6.76 ± 0.26a | |
| Total | 18.26 ± 0.56a | 98.83 ± 3.23b | 76.32 ± 3.55c | |
| C14 | 0.022 ± 0.001a | 0.024 ± 0.0003b | 0.024 ± 0.0002ab | |
| C16 | 0.226 ± 0.006a | 0.265 ± 0.007b | 0.247 ± 0.015ab | |
| C16:1 | ND | ND | ND | |
| Mitochondrial | C18 | ND | ND | ND |
| Fraction | C18:1 | 0.351 ± 0.013a | 0.624 ± 0.010b | 0.539 ± 0.025c |
| C18:2 | 0.151 ± 0.001a | 0.152 ± 0.001a | 0.153 ± 0.003a | |
| C20 | ND | ND | ND | |
| Total | 0.750 ± 0.015a | 1.065 ± 0.014b | 0.963 ± 0.032c |
Data are presented as means ± SE as ng/mg tissue. Groups with different superscript letters are significantly different (P < 0.05, Tukey's HSD).
Table 4.
Ceramide species in whole-muscle homogenates and subcellular fractions after 10 wk of NFD, HFD, and HFD+FO/
| Fraction | Species | NFD | HFD | HFD+FO |
|---|---|---|---|---|
| C14 | 0.021 ± 0.001a | 0.023 ± 0.002a | 0.022 ± 0.002a | |
| C16 | 0.503 ± 0.035a | 0.660 ± 0.024b | 0.616 ± 0.031b | |
| C18 | 7.380 ± 0.306a | 9.405 ± 0.273b | 8.077 ± 0.171a | |
| Homogenate | C18:1 | 1.585 ± 0.058a | 2.331 ± 0.090b | 2.155 ± 0.080b |
| C20 | 0.264 ± 0.024a | 0.260 ± 0.016a | 0.179 ± 0.020b | |
| C24 | 2.737 ± 0.056a | 2.454 ± 0.061a | 2.756 ± 0.043a | |
| C24:1 | 0.557 ± 0.014a | 0.554 ± 0.026b | 0.571 ± 0.019a | |
| Total | 13.047 ± 0.428a | 15.689 ± 0.409b | 14.377 ± 0.164ab | |
| C14 | 0.019 ± 0.002a | 0.017 ± 0.001a | 0.019 ± 0.002a | |
| C16 | 0.328 ± 0.023a | 0.414 ± 0.033a | 0.396 ± 0.031a | |
| C18 | 4.137 ± 0.160a | 5.661 ± 0.132b | 4.620 ± 0.255a | |
| Sarcoplasmic | C18:1 | 1.065 ± 0.042a | 1.721 ± 0.081b | 1.477 ± 0.06ca |
| Fraction | C20 | 0.129 ± 0.011a | 0.134 ± 0.016a | 0.067 ± 0.011b |
| C24 | 1.923 ± 0.047a | 1.890 ± 0.028a | 2.026 ± 0.046a | |
| C24:1 | 0.250 ± 0.017a | 0.273 ± 0.016a | 0.270 ± 0.024a | |
| Total | 7.852 ± 0.204a | 10.110 ± 0.253b | 8.876 ± 0.310c | |
| C14 | 0.003 ± 0.0002a | 0.003 ± 0.0003a | 0.005 ± 0.001b | |
| C16 | 0.066 ± 0.004a | 0.098 ± 0.008b | 0.092 ± 0.007b | |
| C18 | 1.352 ± 0.097a | 1.798 ± 0.075b | 1.591 ± 0.087ab | |
| Mitochondrial | C18:1 | 0.207 ± 0.011a | 0.343 ± 0.018b | 0.349 ± 0.021b |
| Fraction | C20 | 0.048 ± 0.004ab | 0.052 ± 0.004a | 0.035 ± 0.004b |
| C24 | 0.400 ± 0.016a | 0.401 ± 0.008a | 0.447 ± 0.015a | |
| C24:1 | 0.092 ± 0.006a | 0.108 ± 0.007a | 0.102 ± 0.005a | |
| Total | 2.168 ± 0.126a | 2.804 ± 0.108b | 2.620 ± 0.115b |
Data are presented as means ± SE as ng/mg tissue. Groups with different superscript letters are significantly different (P < 0.05, Tukey's HSD).
Mitochondrial content.
Representative electron micrographs of skeletal muscle mitochondria are shown for NFD (Fig. 3A), HFD (Fig. 3B), and HFD+FO (Fig. 3C). Analysis of micrographs revealed no morphological differences across groups (Fig. 3D). Mitochondrial density was increased significantly in muscle from HFD+FO compared with NFD, with a nonsignificant trend for HFD (Fig. 3E). Mitochondrial DNA abundance, measured by RT-PCR for subunits 1 and 4 of the mitochondrial-encoded NADH dehydrogenase genes, were not different across the three groups (Fig. 3F). mRNA expression of PPARα and PPARγ were significantly increased in HFD+FO (Fig. 3G). PGC1α expression was significantly lower in HFD compared with NFD but maintained similar to controls in HFD+FO (Fig. 3G). NRF1, a downstream transcription factor regulated by PGC1α, was significantly elevated in HFD+FO, with a similar but nonsignificant trend for TFAM (Fig. 3G).
Fig. 3.
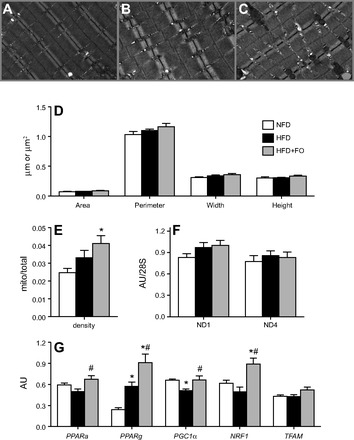
Mitochondrial abundance is increased with fish oil by electron microscopy with no change in mitochondrial DNA copy number. A, B, C: representative transmission electron micrographs (×25,000 magnification) of skeletal muscle from NFD (A), HFD (B), and HFD+FO (C) mice. D: average perimeter, width, height, and area of mitochondria were measured from digitized image. E: mitochondrial density by area was determined from digitized electron micrographs. F: mitochondrial DNA abundance was measured by RT-PCR using mitochondrial-encoded NADH dehydrogenase subunits 1 (ND1) and 4 (ND4) expressed relative to 28S as a nuclear housekeeping gene. G: gene transcript levels of peroxisome proliferator-activated receptor-α (PPARα), PPARγ (PPARγ), PPARγ coactivator 1α (PGC1α), nuclear respiratory factor 1 (NRF1), and transcription factor A, mitochondria (TFAM) were measured in muscle tissue at 10 wk. Bars represent means ± SE for NFD, HFD, and HFD+FO. *Significant statistical differences from NFD; #significantly different from HFD (P < 0.05, Tukey's HSD).
Mitochondrial function.
Skeletal muscle mitochondrial respiratory capacity was evaluated in the presence of carbohydrate (CHO)-based substrates (Fig. 4, A and C) and lipid-based substrates (Fig. 4, B and D). When oxygen flux was normalized to tissue wet weight, oxidative capacity was similarly elevated in HFD and HFD+FO compared with NFD for both substrates (Fig. 4, A and B). When oxygen flux was normalized to mitochondrial protein, oxidative capacity was similar across all groups for CHO-based substrates (Fig. 4C). In contrast, HFD and HFD+FO showed significantly higher state 3 respiration with lipid-based substrates even after normalization to mitochondrial protein (Fig. 4D). Mitochondrial efficiency was evaluated from RCRs as an index of mitochondrial proton leak (Fig. 4E) and from ADP:O ratios as an index of phosphorylation efficiency (Fig. 4F). RCR was similar across groups for CHO-based substrates but was greater in HFD and HFD+FO for lipid-based substrates (Fig. 4E). Similar but nonsignificant trends were observed for ADP:O (Fig. 4F).
Fig. 4.
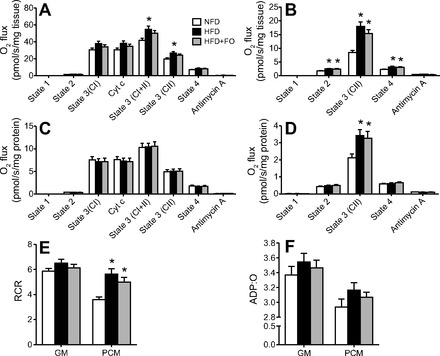
HFD increases mitochondrial respiratory capacity with no effect of fish oil. Respiration rates were measured in isolated mitochondria from skeletal muscle with substrates targeting complex I (CI), complex I+II (CI+II), and complex II (CII) (A) as well as lipid-based substrates (B). Nonmitochondrial V̇o2 was measured in the presence of antimycin A. Respiration rates were expressed per tissue wet (A and B) and mitochondrial protein content (C and D). Respiratory control ratio (RCR; state 3/state 4; E) and phosphorylation efficiency (ADP:O; F) were measured in isolated mitochondria. Bars represent means ± SE for NFD, HFD, and HFD+FO. *Significant statistical differences from NFD; #significantly different from HFD (P < 0.05, Tukey's HSD).
H2O2 emission.
H2O2 production from isolated mitochondria was significantly increased in HFD and HFD+FO compared with NFD when expressed per milligram muscle wet weight (Fig. 5A) but was similar across groups when normalized to mitochondrial protein (Fig. 5B). All groups exhibited similar H2O2 emission rates from permeabilized muscle fibers in response to increasing succinate concentrations (Fig. 5C). SOD1 mRNA expression was lower in both HFD groups compared with NFD, significantly so for HFD+FO with a trend in HFD (Fig. 5D). In contrast, CAT mRNA expression was significantly higher in HFD compared with NFD. Catalase expression was highest in HFD+FO and was significantly elevated over NFD and HFD (Fig. 5D). SOD2 and GPX1 expression did not differ by group (Fig. 5D). Similar to mRNA expression, catalase activity was significantly increased in muscle from HFD and HFD+FO mice, with no effect of fish oil (Fig. 5E). Total SOD activity did not differ across groups (Fig. 5F). The DNA adduct biomarker of oxidative stress, 8-oxo-dG, was not different across the three groups of mice (Fig. 5G).
Fig. 5.
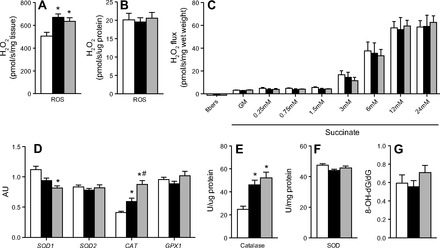
Hfd increases mitochondrial H2O2 production and catalase activity with no effect of fish oil. H2O2 production was measured in isolated mitochondria under state 4 conditions by spectrofluorometer, expressed relative to tissue wet weight (A) and relative to mitochondrial protein content (B). H2O2 emission was measured in permeabilized muscle fibers during a stepwise increase in succinate under state 4 conditions. Gene transcript levels (D) of superoxide dismutase-1 (SOD1), superoxide dismutase-2 (SOD2), catalase (CAT), and glutathione peroxidase (GPX1) were measured in muscle tissue at 10 wk. Activity of catalase (E) and total SOD (F) were measured spectrophotometrically. Skeletal muscle oxidative damage (G) was determined from 8-oxo-2′-deoxyguanosine (8-oxo-dG), a major product of DNA oxidation measured by mass spectrometry. Bars represent means ± SE for NFD, HFD, and HFD+FO. *Significant statistical differences from NFD; #significantly different from HFD (P < 0.05, Tukey's HSD).
DISCUSSION
The experiments described herein were designed to evaluate the hypothesis that dietary n-3 PUFAs attenuate diet-induced glucose intolerance by decreasing ectopic lipid accumulation subsequent to increased mitochondrial function in skeletal muscle. We found that 10 wk of HFD increased lipid metabolite levels and decreased insulin sensitivity, but this was partially prevented by substituting 3.4% kcal of saturated fat with n-3 PUFAs. Furthermore, we found that mitochondrial oxidative capacity was not impaired by HFD. The only feature of “mitochondrial dysfunction” that we observed with HFD was an increase in mitochondrial H2O2 production, but this was effectively buffered by an increase in the expression and activity of catalase, likely explaining why there was no change in net H2O2 production from permeabilized muscle fibers. Although mitochondrial abundance and respiratory capacity were significantly increased in mice fed n-3 PUFAs compared with controls, the increase was similar to the mice fed HFD without fish oil. These data indicate that the respiratory function and oxidant emission of skeletal muscle mitochondria are not major factors in ectopic lipid accumulation, diet-induced insulin resistance, or the protective effects of n-3 PUFAs on insulin sensitivity.
Ad libitum access to a HFD caused profound impairment in glucose tolerance following an oral glucose load. The glucose intolerance with HFD was most evident at 5 wk. Although glucose tolerance normalized somewhat at 10 wk in HFD, plasma insulin levels at the 15-min time point were significantly elevated, suggesting pancreatic β-cell compensation in these mice. Glucose tolerance in HFD+FO remained similar to NFD controls throughout the 10-wk dietary intervention, indicating that fish oil prevents the deterioration of glucose tolerance with high-fat feeding. It is important to point out that, despite preserved glucose tolerance, HFD+FO exhibited evidence of insulin resistance. At 10 wk, both HFD and HFD+FO showed significantly higher plasma insulin compared with controls at the 15-min time point of the OGTT. Depite similarly elevated plasma insulin in HFD and HFD+FO, the prevailing blood glucose level was lower in HFD+FO, indicating that fish oil partially preserves insulin sensitivity despite HFD. Consistent with this notion, the transcript levels of GLUT4, GYS1, and IRS1 were all significantly elevated in the presence of fish oil. These observations are in agreement with several previous studies that showed that dietary n-3 PUFAs prevented the reduction in glucose tolerance and insulin sensitivity in rodents (29, 37, 39, 53, 54).
The mechanisms by which n-3 PUFAs preserve insulin sensitivity are not completely understood, but modulation of inflammation is a leading mechanistic explanation. Omega-3 fatty acids appear to have anti-inflammatory effects (50), and a recent paper demonstrates that n-3 PUFAs activate a G protein-coupled receptor, which suppresses the expression and pathways of inflammatory cytokines (39). Here we show that n-3 PUFAs also attenuated the accumulation of ceramides and their immediate precursors, LCACoA, in response to high-fat feeding. Studies in rodents and humans show that LCACoA levels are closely associated with insulin action (4, 13). Ceramide, a downstream synthesis product of sphingoid base and LCACoA, is also suspected as a bioactive lipid mediator that interferes with insulin signaling (43, 48, 55, 56). Our data suggest that n-3 PUFAs may also protect insulin sensitivity by attenuating the accumulation of lipid metabolites that interfere with insulin signaling. Consistent with this finding, others reported that n-3 PUFAs combined with rosiglitazone blunts the increase in skeletal muscle ceramide content with high-fat feeding (31). The effects of n-3 PUFAs on inflammation and sphingolipid levels may not be coincidental. The inflammatory cytokine TNF-α stimulates de novo ceramide synthesis (62). Ceramide can be metabolized to the biologically active sphingosine 1-phosphate (S1P), which plays a role in inflammation and immune cell function (10, 33). Furthermore, inflammatory cytokines activate sphingosine kinase-1, an enzyme necessary for conversion of ceramide to S1P (5). The role of sphingosine kinase and S1P in inflammation was demonstrated in a mouse asthma model where inflammation was prevented by inhibiting S1P or sphingosine kinase receptors. (25, 38). These data suggest an attractive link between lipid metabolites in insulin-sensitive tissues and inflammation.
Long-chain fatty acyl-CoAs are utilized for ceramide biosynthesis, but can also be oxidized as fuel in mitochondria. We hypothesized that n-3 PUFAs would decrease the accumulation of LCACoAs and ceramides, in part by diverting more LCACoA into β-oxidation. This hypothesis was based on preceding literature suggesting a potential role for mitochondria in the insulin-sensitizing effects of n-3 PUFAs. One study found that n-3 PUFAs prevented insulin resistance in wild-type mice but not in mice lacking the α2 catalytic subunit of AMPK (27). Similarly, others found that n-3 PUFAs prevented insulin resistance in wild-type mice but not in mice lacking PPARα (37). Both AMPK and PPARα are known to upregulate genes involved in mitochondrial biogenesis and fatty acid oxidation (15, 59). Furthermore, n-3 PUFAs are strong PPAR ligands (12), and EPA incubation increased PGC-1α, Tfam, cytochrome c oxidase expression, mitochondrial membrane potential, and ATP levels in glioma cells (28). As PPAR, AMPK, and PGC-1α agonists, n-3 PUFAs could stimulate mitochondrial biogenesis, increase mitochondrial oxidative capacity, increase lipid oxidation, and decrease the abundance of intracellular lipid metabolites that would otherwise interfere with insulin signaling. Our data are not consistent with this hypothesis. Consistent with previous reports (12, 28), we also found that fish oil increased gene transcripts for PPARα and PPARγ as well as genes known to regulate mitochondrial biogenesis (PGC1α, NRF1, TFAM). These data suggest that fish oil supplementation may increase the abundance of mitochondria in skeletal muscle. Although mitochondrial abundance measured by electron microscopy was significantly higher in skeletal muscle from HFD+FO compared with NFD, the increase was not different from that observed in mice fed HFD without fish oil. Furthermore, there was only a slight trend for increased mtDNA abundance, which was similar in HFD and HFD+FO. Mitochondrial oxidative capacity and coupling efficiency were significantly increased compared with NFD mice, but similar increases were also observed in HFD without fish oil. These data do not exclude the possibility that n-3 PUFAs stimulate mitochondrial biogenesis but indicate that in the context of high-fat feeding there is no enhancement in mitochondrial content or function that can account for the significant differences in lipid metabolite levels or insulin sensitivity between HFD and HFD+FO. Of interest, we show that, despite attenuated LCACoA and ceramide levels with fish oil administration, there were no differences in muscle triglycerides between HFD and HFD+FO. This observation that fish oil did not attenuate ectopic triglyceride accumulation is consistent with the absence of any enhancement in fat oxidation measured in vitro or in vivo at the whole body level. Lower sphingolipid levels in HFD+FO despite similar triglyceride levels may be explained by decreased in acyl-CoA synthase activity, which is elevated with high-fat feeding (61).
Insulin resistance induced by HFD is also linked with increased cellular redox state and elevated mitochondrial ROS production (3). We show that high-fat feeding increases H2O2 production from isolated mitochondria, with no protective effect of fish oil. The fact that the stimulatory effect of HFD on mitochondrial H2O2 production is observed relative to tissue weight but not when normalized to mitochondrial protein content, suggests that the increase in H2O2 production is related to the increase in mitochondrial abundance in skeletal muscle with HFD rather than an intrinsic feature of the organelle. Despite the increased H2O2 production in isolated mitochondria, we did not observe any increase in H2O2 emission in permeabilized muscle fibers from HFD or HFD+FO. Unlike isolated mitochondria, permeabilized muscle fibers retain the endogenous antioxidants that scavenge ROS. Indeed, n-3 PUFAs demonstrate antioxidant properties in human aortic endothelial cells (44), highlighting the possibility that n-3 PUFAs may not alter ROS production but may attenuate oxidative stress through oxidant scavenging. In this study, we found that catalase transcript level and activity were markedly higher in both HFD and HFD+FO compared with controls, but no effect of fish oil. It is conceivable that the effect of HFD on increasing catalase activity may explain why H2O2 production is elevated in isolated mitochondria but not permeabilized muscle fibers. Together, these data indicate that, although fish oil increased the transcript levels of catalase, fish oil did not influence mitochondrial ROS production or activity of endogenous antioxidants and cannot explain why fish oil protects glucose tolerance and insulin sensitivity.
Our observations that muscle mitochondrial function is increased by high-fat feeding despite insulin resistance is not without precedent (19, 22, 58) but conflicts with what is commonly believed to be a typical phenotype of insulin resistance. Derangements in skeletal muscle mitochondrial bioenergetics have been observed in people with insulin resistance or type 2 diabetes (30, 35, 40, 42, 49). These observations, combined with reports of decreased mitochondrial function in lean offspring of patients with type 2 diabetes (41), highlight a potential role for mitochondria in the development of insulin resistance (41, 46). A possible explanation is that lipid metabolites accumulate because of decreased oxidation by mitochondria. Alternatively, lipid oversupply may negatively influence mitochondrial biology. Some insight into this issue comes from studies where lipid supply is acutely elevated by lipid infusion (9, 21, 45) or short-term HFD (52). These studies found that lipid oversupply decreased mitochondrial gene expression (21, 45, 52) and insulin-stimulated mitochondrial ATP flux (9). Conflicting data come from rodent studies where mitochondrial content and function are increased by high-fat feeding (19, 22, 58). The current study is consistent with the latter, showing that 10 wk of HFD significantly increased oxidative capacity of skeletal muscle mitochondria despite insulin resistance. The markedly increased levels of ceramide, LCACoA, and triglycerides in skeletal muscle cannot be explained on the basis of decreased mitochondrial lipid oxidation. Rather, HFD animals appear to be well adapted to lipid oxidation as evidenced from not only increased mitochondrial lipid oxidative capacity but consistently lower RER values during whole body calorimetry measurements. The current data add to an accumulating body of evidence arguing against the hypothesis that mitochondrial oxidative capacity is a mediator of insulin resistance (23).
In conclusion, we have shown that dietary n-3 PUFAs protect against diet-induced insulin resistance. To this point, others have identified suppression of inflammation as the primary mechanism underlying the insulin-sensitizing effects of n-3 PUFAs. Here, we identify another potential mechanism to explain the protective effects of n-3 PUFAs, specifically the attenuation of bioactive lipid mediators that interfere with insulin action. A potential link between tissue sphingolipid levels and inflammatory state awaits further investigation. Although the precise mechanisms by which n-3 PUFAs attenuate sphingolipid accumulation remain unresolved, our data indicate that n-3 PUFAs do not enhance mitochondrial function beyond what is observed with high-fat feeding, nor do they alter the redox state of skeletal muscle. These data indicate that the respiratory function of skeletal muscle mitochondria is not a major factor in ectopic lipid accumulation, diet-induced insulin resistance, or the protective effects of n-3 PUFAs on insulin sensitivity. An intriguing area for future investigation is the influence of n-3 PUFAs on spontaneous physical activity. The current study clearly shows that fish oil increases rearing, ambulation, and total cage activity compared with mice fed a high-fat diet alone. Similarly, cachectic patients with pancreatic cancer exhibited increased physical activity when supplemented with EPA (36a), and others found a strong correlation between DHA levels in red blood cells and physical activity in women (33a). It is difficult to ignore the possibility that some beneficial effects of omega-3 supplementation may result from increased spontaneous physical activity.
GRANTS
This publication was supported by CTSA Grant No. KL2 TR-000136 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: I.R.L. and K.S.N. conception and design of research; I.R.L., A.B.-Z., M.L.J., J.M.C.-S., D.R.J., N.K.L., and P.Z. performed experiments; I.R.L., A.B.-Z., M.L.J., J.M.C.-S., D.R.J., N.K.L., M.D.J., and P.Z. analyzed data; I.R.L., A.B.-Z., M.L.J., N.K.L., M.D.J., K.S.N., and P.Z. interpreted results of experiments; I.R.L. and N.K.L. prepared figures; I.R.L. and P.Z. drafted manuscript; I.R.L., A.B.-Z., M.L.J., J.M.C.-S., N.K.L., M.D.J., K.S.N., and P.Z. edited and revised manuscript; I.R.L., A.B.-Z., M.L.J., J.M.S., D.R.J., N.K.L., M.D.J., K.S.N., and P.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Kate Klaus, Dawn Morse, Mai Persson, Charles Ford, Eileena Li, and Melissa Aakre for technical expertise.
REFERENCES
- 1. Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53: 25–31, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beha A, Juretschke HP, Kuhlmann J, Neumann-Haefelin C, Belz U, Gerl M, Kramer W, Roden M, Herling AW. Muscle type-specific fatty acid metabolism in insulin resistance: an integrated in vivo study in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab 290: E989–E997, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell Signal 17: 1203–1217, 2005 [DOI] [PubMed] [Google Scholar]
- 6. Blachnio-Zabielska AU, Koutsari C, Jensen MD. Measuring long-chain acyl-coenzyme A concentrations and enrichment using liquid chromatography/tandem mass spectrometry with selected reaction monitoring. Rapid Commun Mass Spectrom 25: 2223–2230, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blachnio-Zabielska AU, Persson XM, Koutsari C, Zabielski P, Jensen MD. A liquid chromatography/tandem mass spectrometry method for measuring the in vivo incorporation of plasma free fatty acids into intramyocellular ceramides in humans. Rapid Commun Mass Spectrom 26: 1134–1140, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 118: 789–800, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhausl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes 55: 136–140, 2006 [PubMed] [Google Scholar]
- 10. Chiba K, Matsuyuki H, Maeda Y, Sugahara K. Role of sphingosine 1-phosphate receptor type 1 in lymphocyte egress from secondary lymphoid tissues and thymus. Cell Mol Immunol 3: 11–19, 2006 [PubMed] [Google Scholar]
- 11. Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem 282: 15439–15450, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 20: 649–688, 1999 [DOI] [PubMed] [Google Scholar]
- 13. Ellis BA, Poynten A, Lowy AJ, Furler SM, Chisholm DJ, Kraegen EW, Cooney GJ. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol Endocrinol Metab 279: E554–E560, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Roves P, Huss JM, Han DH, Hancock CR, Iglesias-Gutierrez E, Chen M, Holloszy JO. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci USA 104: 10709–10713, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem 283: 35724–35734, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 41: 1837–1845, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Guo Z, Zhou L, Jensen MD. Acute hyperinsulinemia inhibits intramyocellular triglyceride synthesis in high-fat-fed obese rats. J Lipid Res 47: 2640–2646, 2006 [DOI] [PubMed] [Google Scholar]
- 18. Hamdy O, Goodyear LJ, Horton ES. Diet and exercise in type 2 diabetes mellitus. Endocrinol Metab Clin North Am 30: 883–907, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA 105: 7815–7820, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heikkinen S, Argmann CA, Champy MF, Auwerx J. Evaluation of glucose homeostasis. Curr Protoc Mol Biol Chapter 29: Unit 29B 23, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Hoeks J, Hesselink MK, Russell AP, Mensink M, Saris WH, Mensink RP, Schrauwen P. Peroxisome proliferator-activated receptor-gamma coactivator-1 and insulin resistance: acute effect of fatty acids. Diabetologia 49: 2419–2426, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Hoeks J, Wilde J, Hulshof MF, Berg SA, Schaart G, Dijk KW, Smit E, Mariman EC. High fat diet-induced changes in mouse muscle mitochondrial phospholipids do not impair mitochondrial respiration despite insulin resistance. PLoS One 6: e27274, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holloszy JO. Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am J Clin Nutr 89: 463S–466S, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Humphreys SM, Fisher RM, Frayn KN. Micro-method for measurement of sub-nanomole amounts of triacylglycerol. Ann Clin Biochem 27: 597–598, 1990 [DOI] [PubMed] [Google Scholar]
- 25. Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T, Willart MA, Hijdra D, Hoogsteden HC, Lambrecht BN. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest 116: 2935–2944, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7: 159–172, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jelenik T, Rossmeisl M, Kuda O, Jilkova ZM, Medrikova D, Kus V, Hensler M, Janovska P, Miksik I, Baranowski M, Gorski J, Hebrard S, Jensen TE, Flachs P, Hawley S, Viollet B, Kopecky J. AMP-activated protein kinase alpha2 subunit is required for the preservation of hepatic insulin sensitivity by n-3 polyunsaturated fatty acids. Diabetes 59: 2737–2746, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jeng JY, Lee WH, Tsai YH, Chen CY, Chao SY, Hsieh RH. Functional modulation of mitochondria by eicosapentaenoic acid provides protection against ceramide toxicity to C6 glioma cells. J Agric Food Chem 57: 11455–11462, 2009 [DOI] [PubMed] [Google Scholar]
- 29. Jucker BM, Cline GW, Barucci N, Shulman GI. Differential effects of safflower oil versus fish oil feeding on insulin-stimulated glycogen synthesis, glycolysis, and pyruvate dehydrogenase flux in skeletal muscle: a 13C nuclear magnetic resonance study. Diabetes 48: 134–140, 1999 [DOI] [PubMed] [Google Scholar]
- 30. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002 [DOI] [PubMed] [Google Scholar]
- 31. Kuda O, Jelenik T, Jilkova Z, Flachs P, Rossmeisl M, Hensler M, Kazdova L, Ogston N, Baranowski M, Gorski J, Janovska P, Kus V, Polak J, Mohamed-Ali V, Burcelin R, Cinti S, Bryhn M, Kopecky J. n-3 Fatty acids and rosiglitazone improve insulin sensitivity through additive stimulatory effects on muscle glycogen synthesis in mice fed a high-fat diet. Diabetologia 52: 941–951, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol 457: 349–372, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a. Lanza IR, Zabielski P, Klaus K, Morse DM, Heppelmann CJ, Bergen HR, Dasari S, Walrand S, Short KR, Johnson ML, Robinson M, Schimke J, Jakeitis DR, Asmann YW, Sun Z, Nair KS. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab 16: 11, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427: 355–360, 2004 [DOI] [PubMed] [Google Scholar]
- 33a. Magnusardottir AR, Steingrimsdottir L, Thorgeirsdottir H, Gunnlaugsson G, Skuladottir GV. Docosahexaenoic acid in red blood cells of women of reproductive age is positively associated with oral contraceptive use and physical activity. Prostaglandins Leukot Essent Fatty Acids 80: 27–32, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Miller WC, Bryce GR, Conlee RK. Adaptations to a high-fat diet that increase exercise endurance in male rats. J Appl Physiol 56: 78–83, 1984 [DOI] [PubMed] [Google Scholar]
- 35. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273, 2003 [DOI] [PubMed] [Google Scholar]
- 36. Nemeth PM, Rosser BW, Choksi RM, Norris BJ, Baker KM. Metabolic response to a high-fat diet in neonatal and adult rat muscle. Am J Physiol Cell Physiol 262: C282–C286, 1992 [DOI] [PubMed] [Google Scholar]
- 36a. Moses AW, Slater C, Preston T, Barber MD, Fearon KC. Reduced total energy expenditure and physical activity in cachectic patients with pancreatic cancer can be modulated by an energy and protein dense oral supplement enriched with n-3 fatty acids. Br J Cancer 90: 996–1002, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neschen S, Morino K, Dong J, Wang-Fischer Y, Cline GW, Romanelli AJ, Rossbacher JC, Moore IK, Regittnig W, Munoz DS, Kim JH, Shulman GI. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 56: 1034–1041, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Nishiuma T, Nishimura Y, Okada T, Kuramoto E, Kotani Y, Jahangeer S, Nakamura S. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol 294: L1085–L1093, 2008 [DOI] [PubMed] [Google Scholar]
- 39. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phielix E, Schrauwen-Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, Kooi ME, Moonen-Kornips E, Sels JP, Hesselink MK, Schrauwen P. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 57: 2943–2949, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J 382: 619–629, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Richard D, Kefi K, Barbe U, Bausero P, Visioli F. Polyunsaturated fatty acids as antioxidants. Pharmacol Res Off J Ital Pharmacol Soc 57: 451–455, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP, Mandarino LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 280: 10290–10297, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Roden M. Muscle triglycerides and mitochondrial function: possible mechanisms for the development of type 2 diabetes. Int J Obes (Lond) 29, Suppl 2: S111–S115, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274: 24202–24210, 1999 [DOI] [PubMed] [Google Scholar]
- 49. Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, Jeneson JA, Backes WH, van Echteld CJ, van Engelshoven JM, Mensink M, Schrauwen P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 50: 113–120, 2007 [DOI] [PubMed] [Google Scholar]
- 50. Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol 153, Suppl 1: S200–S215, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol 83: 166–171, 1997 [DOI] [PubMed] [Google Scholar]
- 52. Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 54: 1926–1933, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Storlien LH, Jenkins AB, Chisholm DJ, Pascoe WS, Khouri S, Kraegen EW. Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 40: 280–289, 1991 [DOI] [PubMed] [Google Scholar]
- 54. Storlien LH, Kraegen EW, Chisholm DJ, Ford GL, Bruce DG, Pascoe WS. Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 237: 885–888, 1987 [DOI] [PubMed] [Google Scholar]
- 55. Straczkowski M, Kowalska I, Baranowski M, Nikolajuk A, Otziomek E, Zabielski P, Adamska A, Blachnio A, Gorski J, Gorska M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 50: 2366–2373, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 53: 1215–1221, 2004 [DOI] [PubMed] [Google Scholar]
- 57. Turinsky J, O'Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem 265: 16880–16885, 1990 [PubMed] [Google Scholar]
- 58. Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56: 2085–2092, 2007 [DOI] [PubMed] [Google Scholar]
- 59. Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab 8: 249–256, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60. Wigmore DM, Damon BM, Pober DM, Kent-Braun JA. MRI measures of perfusion-related changes in human skeletal muscle during progressive contractions. J Appl Physiol 97: 2385–2394, 2004 [DOI] [PubMed] [Google Scholar]
- 61. Wu M, Liu H, Chen W, Fujimoto Y, Liu J. Hepatic expression of long-chain acyl-CoA synthetase 3 is upregulated in hyperlipidemic hamsters. Lipids 44: 989–998, 2009 [DOI] [PubMed] [Google Scholar]
- 62. Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LM, Choi DW, Hsu CY. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J Biol Chem 273: 16521–16526, 1998 [DOI] [PubMed] [Google Scholar]
- 63. Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277: 50230–50236, 2002 [DOI] [PubMed] [Google Scholar]