

Abstract
Cytoplasmic inclusion of RNA binding protein FUS/TLS in neurons and glial cells is a characteristic pathology of a subgroup of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Dysregulation of RNA metabolism caused by FUS cytoplasmic inclusion emerges to be a key event in FUS-associated ALS/FTD pathogenesis. Our recent discovery of a FUS autoregulatory mechanism and its dysregulation in ALS-FUS mutants demonstrated that dysregulated alternative splicing can directly exacerbate the pathological FUS accumulation. We show here that FUS targets RNA for pre-mRNA alternative splicing and for the processing of long intron-containing transcripts, and that these targets are enriched for genes in neurogenesis and gene expression regulation. We also identify that FUS RNA targets are enriched for genes in the DNA damage response pathway. Together, the data support a model in which dysregulated RNA metabolism and DNA damage repair together may render neurons more vulnerable and accelerate neurodegeneration in ALS and FTD.
Keywords: FUS/TLS, RNA metabolism, alternative splicing, DNA damage response, amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD)
Introduction
FUS/TLS (fused in sarcoma or translocated in liposarcoma) was originally identified as a translocated gene in human liposarcoma and leukemia.1 Recently FUS cytoplasmic inclusion in neurons and glial cells was identified as a characteristic pathology of a subgroup of amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig’s disease)2,3 and frontotemporal dementia (FTD).4 ALS and FTD are clinically, pathologically, and mechanistically linked, and now are considered as one continuum of a broad neurodegenerative disorder.4 In ALS, the degeneration of upper and lower motor neurons in the brain and spinal cord causes progressive muscle wasting and paralysis.5 ALS patients typically die from respiratory failure within 3–5 y of symptom onset.5 FUS is mutated in about 5% of familial ALS and rare cases of sporadic ALS.6 Over 40 FUS mutations have been identified in ALS since 2009.6 Most FUS mutations are dominant missense point mutations, with very few insertion, deletion or truncations observed.6 The mutations are clustered in two major regions, the extreme C-terminus containing a nuclear localization signal (NLS) and the first glycine-rich region located between the amino-transcriptional domain and the carboxy-RNA binding domains.6 FTD, neuropathologically, is known as frontotemporal lobar degeneration (FTLD), which is characterized by atrophy of the frontal and temporal brain lobes.4 In FTD, degeneration of frontal and temporal cortices affects cognitive functions such as speech, language, and personality.4 FTD is the second most common dementia after Alzheimer disease, particularly in patients with a disease onset before 65 y.4 FUS mutation in FTD is very rare. However, cytoplasmic inclusion of wild-type FUS protein occurs in about 9% of FTD, which is pathologically classified as FTLD-FUS.6
FUS is an RNA binding protein and is predominantly localized in the nucleus. FUS regulates several key steps of RNA metabolism in the nucleus, including transcription, alternative splicing, and nuclear-cytoplasmic mRNA transport,1 which are pivotal to various biological processes, including neurogenesis. FUS also functions in the cytoplasm, specifically in neurons, to facilitate mRNA transport to dendritic spines for site-specific translation, which affects dendritic spine morphology and synaptic formation.7 ALS-associated FUS mutants that are retained in the cytoplasm are recruited to stress granules, which may contribute to the formation of FUS cytoplasmic inclusions.8-10
In ALS and FTD, the FUS mutant or wild-type proteins trapped in the cytoplasmic inclusions are unable to perform the normal function of FUS in the cytoplasm, such as site-specific mRNA transport, which affects neuron morphology and function. Moreover, the cytoplasmic inclusion is usually accompanied by a concomitant decrease of FUS protein levels in the nucleus,11 which likely results in the dysregulation of FUS-dependent RNA metabolism in the nucleus, including transcription and alternative splicing. Emerging evidence suggests that dysregulated FUS-dependent RNA metabolism may be a central mechanism underlying FUS-associated ALS and FTD pathogenesis.11
Here we report that our FUS CLIP-seq data from HeLa cells suggest FUS regulates alternative splicing of pre-mRNAs and processing of long-intron containing transcripts. We identified FUS RNA targets are associated with neurogenesis and gene expression regulation, supporting that the dysregulation of FUS-dependent RNA metabolism may lead to neurodegeneration. As a proof of principle, we demonstrated that FUS autoregulates its own protein homeostasis by alternative splicing, and that dysregulation of FUS autoregulation in ALS-FUS mutants likely exacerbates the pathological FUS accumulation in ALS.12
While dysregulation of FUS-dependent RNA metabolism may initiate or drive neurodegeneration, deficiency of FUS-modulated DNA damage repair may render neurons more vulnerable to stress and accelerate neurodegeneration. It is well known that mutations of genes related to DNA damage response and repair are genetic predispositions to both cancer and neurological defects. For instance, ataxia telangiectasia (AT) with ATM mutations, AT-like disorders (ATLD) with MRE11 mutations and Nijmegen breakage syndrome (NBS) with NBS1 mutations are all neurological disorders with increased risk of lymphoid malignancy.13 We and others have demonstrated that FUS is required for DNA damage repair and the maintenance of genomic stability.14,15 Emerging evidence suggests that ALS-FUS mutants are defective in DNA damage repair and RNA splicing of genes that regulate neuronal function during DNA damage response.16,17 Here we analyzed our and others’ CLIP-seq data and show that FUS binds RNA encoding proteins important for DNA damage response and repair pathways. These data suggest that dysregulation in RNA metabolism may be a mechanism underlying FUS-dependent DNA damage response and repair.
FUS Regulates the Alternative Splicing of Genes Crucial for Neurogenesis and Gene Expression Regulation
Recently, the application of UV crosslinking RNA immunoprecipitation and deep sequencing (CLIP-seq) together with RNA-seq has provided tremendous information about FUS RNA targets, and more importantly, deep insight into the function of FUS in RNA metabolism.12,18-22 We and others identified that introns of pre-mRNA account for 60–80% of all FUS binding sites,12,18-22 suggesting the primary function of FUS is to regulate pre-mRNA processing such as alternative splicing. Our CLIP-seq analysis revealed that the most abundant FUS-associated alternative splicing event is cassette exon.12 Overall there are 652 FUS-associated alternative splicing events, and cassette exon events account for 32%.12 The number of cassette exons events associated with FUS-CLIP clusters is significantly higher (Z-score = 26.55) than that associated with random controls.12 FUS is associated with cassette exons of 180 genes in HeLa cells. Among these 180 genes, 21 genes were identified as FUS RNA targets in all five previously reported FUS CLIP-seq assays of different tissues and cell lines; and 151 genes were identified in at least two previous FUS-CLIP assays.18-22 These data suggest that cassette exon is a primary alternative splicing event regulated by FUS.
We and others’ CLIP-seq data show that FUS binding is enriched in introns flanking cassette exons, particularly proximal to splice sites flanking the cassette exons.12,19,20,22 FUS binds intronic regions to either activate or repress the splicing of these cassette exons.12,19,20,22 FUS deficiency results in the expression changes of over 600 RNAs and the splicing changes of over 370 RNAs in the mouse brain.20 FUS alternative splice targets in mouse brain, such as Mapt (Tau), Ntng1, Ndrg2, Camk2a, Nrxn1 and Nlgn1, play crucial roles in axonogenesis, cytoskeleton organization, cell adhesion, and synaptic formation and function.20 The dysregulation of these RNA targets may cause synaptic dysfunction, axon withdrawal and denervation, which leads to neurodegeneration in ALS and FTD.
Our CLIP-seq data in HeLa cells revealed that FUS-associated cassette exons are significantly enriched for genes regulating transcription (P ≤ 0.05, FDR ≤ 0.05, Fig. 1A). Our analysis of the most evolutionarily conserved FUS RNA targets in HeLa cells showed that FUS targets RNA of genes regulating RNA processing (P ≤ 0.05, FDR ≤ 0.05, Fig. 1B). Genes that regulate transcription and RNA processing play critical roles in gene expression and impact all cellular processes. FUS RNA targets in HeLa cells such as GLI2, STAT3, hnRNPK, SFPQ are known to regulate neuronal development,23 suggesting FUS may regulate neurogenesis through modulating the alternative splicing of critical transcription or splicing regulators. Most of the FUS-associated transcription or splicing regulators that we identified in HeLa cells were identified in at least three previously reported FUS CLIP-seq assays, suggesting they comprise cell-type independent conserved FUS RNA targets (Table 1). This notion is consistent with two previous reports showing that FUS binds to conserved introns of RNA binding proteins in induced pluripotent stem cell-derived mouse neurons21 and that FUS is required for the proper splicing of genes regulating transcription in Xenopus embryos,24 respectively. Taken together, our data suggest that one of the important functions of FUS is to modulate the alternative splicing of genes that themselves are critical players in gene expression to control important cellular processes including neurogenesis.

Figure 1. FUS targets RNAs that regulate transcription and RNA splicing. (A) Analysis of Gene Ontology (GO) biological process terms for genes encoding FUS-associated cassette exons shows a significant enrichment of transcription regulation (false discovery rate (FDR) ≤ 0.05). (B) Analysis of GO biological process terms for evolutionarily conserved FUS RNA targets shows a significant enrichment of RNA splicing (FDR ≤ 0.05). Conservation is determined by PhastCons vert46 score. FUS RNA targets in which 70% of the nucleotides are evolutionarily conserved were used for GO analysis. GO analysis were performed using the DAVID Bioinformatics Resources 6.7. For both (A) and (B), blue bars represent fold enrichment as compared with all RNAs expressed in HeLa cells.30 The red line represents FDR values that are adjusted p values using the Benjamini-Hochberg procedure.
Table 1. List of FUS RNA targets* that regulate transcription and RNA splicing.
| Gene Symbol | Zhou et al.12 | Hoell et al.18 | Lagier-Tourenne et al.20 | Ishigaki et al.19 | Rogelj et al.22 | Nakaya et al.21 |
|---|---|---|---|---|---|---|
| ARHGEF10L | √ | √ | √ | √ | √ | □ |
| GLI2 | √ | √ | √ | √ | √ | □ |
| IKZF2 | √ | √ | √ | √ | √ | □ |
| MAML3 | √ | √ | √ | √ | √ | □ |
| MAPK14 | √ | √ | √ | √ | √ | □ |
| MORF4L2 | √ | √ | □ | □ | □ | □ |
| PBX1 | √ | √ | √ | √ | √ | √ |
| RUNX1 | √ | √ | √ | √ | √ | □ |
| RUNX2 | √ | √ | √ | √ | □ | □ |
| SMAD2 | √ | √ | √ | √ | √ | □ |
| STAT3 | √ | √ | √ | √ | □ | □ |
| TBL1X | √ | √ | □ | □ | □ | □ |
| TCF7L2 | √ | √ | √ | √ | √ | □ |
| ZNF148 | √ | √ | √ | √ | √ | □ |
| TBL1XR1 | √ | √ | √ | √ | √ | □ |
| FUS | √ | √ | √ | √ | √ | □ |
| HNRNPK | √ | √ | √ | √ | □ | □ |
| HNRPDL | √ | √ | √ | √ | □ | □ |
| SFRS5 | √ | □ | □ | □ | □ | □ |
| SFPQ | √ | √ | √ | √ | √ | □ |
| HNRNPA2B1 | √ | √ | √ | √ | □ | √ |
| HNRNPD | √ | √ | √ | √ | □ | □ |
| SYNCRIP | √ | √ | √ | √ | √ | □ |
| SKIV2L2 | √ | √ | √ | □ | □ | □ |
| RBM39 | √ | √ | √ | √ | □ | □ |
| SFSR12 | √ | □ | □ | √ | □ | □ |
| TRIT1 | √ | √ | √ | □ | √ | □ |
While thousands of FUS RNA targets have been identified, only a few targets have been characterized in detail to elucidate how dysregulated alternative splicing of specific RNA targets can contribute to ALS pathogenesis. For example, exon 10 of the microtubule-associated protein Mapt/Tau is a known target of FUS. Knockdown of FUS in rat hippocampal neurons promotes Mapt/Tau exon 10 inclusion (Tau 4R isoform), and consequently causes cytoskeleton disorganization, shortened axons and enlarged growth cones that may contribute to neurodegeneration.25 FUS regulates the splicing of brain-derived neurotrophic factor (BDNF).16 ALS-associated FUS-R521C mutant neurons showed increased binding to 5′ splice junction of exon 2, 4 and 6 of BDNF, which resulted in the reduction of both BDNF mRNA and protein.16 The reduction of BDNF partially contributes to the defects of dendritic growth and synaptic structure observed in FUS-R521C motor neurons.16 We recently identified FUS represses the splicing of a cassette exon (exon 7) of its own pre-mRNA.12 Through modulating the alternative splicing of exon 7, FUS controls its own protein homeostasis. ALS-FUS mutants that harbor mutations in the nuclear localization signals (NLS) show dysregulated FUS autoregulation, which likely forms a feed-forward mechanism exacerbating FUS cytoplasmic accumulation. FUS cytoplasmic accumulation and aggregation have been inversely correlated with the age of ALS onset. Taking advantage of this FUS autoregulatory mechanism, we developed an antisense oligonucleotide that can potentially downregulate pathological FUS accumulation in both ALS and FTD to slow down disease progression.
In summary, identification of FUS RNA targets suggests that FUS regulates the splicing of genes associated with neurogenesis and gene expression; and that dysregulated splicing of these RNA targets may lead to neurodegeneration. Functional validation of these FUS RNA targets may reveal many potential strategies for new therapeutic intervention in ALS and FTD.
FUS Regulates the Processing of Long Intron-Containing Transcripts in Neurogenesis
It has been reported that FUS binds to long-intron (> 100 kb) containing transcripts that are preferentially expressed in the brain.20,22 Interestingly, we identified that FUS also preferentially binds to long introns in HeLa cells in our CLIP-seq assay. FUS CLIP clusters mapped to 47.5% of transcripts that are expressed in HeLa cells and longer than 100 kb (Fig. 2A). To ensure that this enrichment of FUS CLIP tags in long introns is not simply due to the length of introns (larger target region) or the expression level of the transcripts, an adjusted value normalized to the transcript expression level and representative of the number of FUS CLIP clusters per hundred kilobase (kb) intron was calculated (Fig. 2A, see legend). Indeed, more FUS CLIP clusters per 100 kb were detected in long introns (0.62 for introns ≥ 100 kb) than were detected in shorter introns (0.22 for introns 10 - 100 kb and 0.12 for introns ≤ 10 kb) (Fig. 2A), suggesting that FUS preferentially binds to long introns. We also showed FUS binds to long introns in a typical sawtooth-like pattern that shows substantially more CLIP tags on the 5′ end and a gradual decrease toward the 3′ end of introns (Fig. 2B, C), as was previously observed in mouse brains.20,22 The sawtooth-like binding pattern suggests that FUS may regulate the transcription elongation of these long-intron-containing transcripts.20,22 It is also possible that FUS may facilitate the proper splicing or regulate the stability of long-intron-containing transcripts.20,22
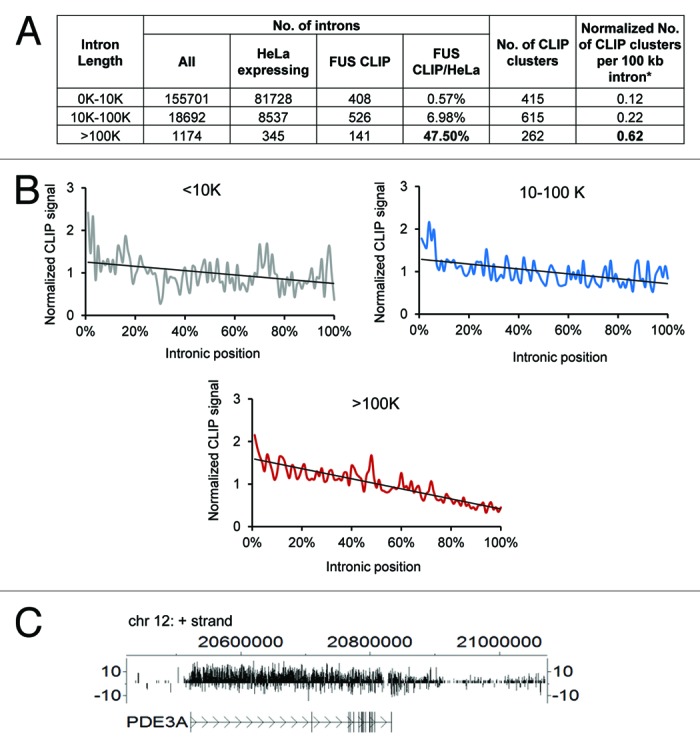
Figure 2. FUS binds to long intron-containing transcripts. (A) The ratio of FUS-targeted introns over the total number of introns in each intron size group. To minimize the bias caused by intron length and transcript expression levels, a value that is normalized to the transcript expression level and represents the number of CLIP clusters per 100 kb intron was calculated for each size group. This value equals (the number of clusters/sum ((intron length (kb) × intron expression level (FPKM))) × 100. All introns in each size group expressed in HeLa cells30 were summarized. (B) Distribution of FUS CLIP sequence tags across introns of different sizes. The graph was generated by plotting the normalized CLIP signals (Y axis) against the relative positions of the CLIP sequence tags within the introns (X axis) as previously described.20 The relative positions (X axis) were computed by setting intron 5′ end as 0% and 3′ end as 100%. To adjust the differences in the number of CLIP tags between different transcripts, each CLIP tag was counted as a fraction of the total number of tags within a transcript. The fractions at the same relative positions (from 0% to 100%) of different introns were summed, representing the normalized CLIP signals (Y axis). (C) CisGenome V2 browser view (www.biostat.jhsph.edu/~hji/cisgenome/) showing the binding pattern of FUS CLIP tags across PDE3A, a representative gene containing an intron longer than 100 kb.
FUS-associated long introns in HeLa cells are enriched for two groups of genes (Fig. S1). One group of genes regulates neuronal development, including PRKCA, ALCAM, IGF1R, CDH13, PTPRM, NTNG1, and GLI2. The other group of genes regulates transcription, including HDAC4, CDH13, EBF1, PBX1, MAML3, GLI2, TBL1X, MYST4, NFIB, and BMP6. Interestingly, all these transcripts except IGF1R, are common FUS RNA targets identified in at least four CLIP-seq assays, including both neuronal and non-neuronal cells.18-22 These data together suggest that the mechanism underlying FUS binding long-intron-containing transcripts is conserved in different cell types and may determine fundamental cellular processes required for neurogenesis. The dysregulation of FUS-dependent long-intron-containing transcripts would affect neuronal cells more than other types of cells since these transcripts are preferentially expressed in neuronal cells.20 Genes that are known to regulate axon growth such as ALCAM, CDH13, and NTNG1 are worth further investigation.
FUS RNA Targets Encode Proteins in the DNA Damage Response and Repair Pathways
Emerging evidence suggests that the defects of FUS-regulated DNA damage repair may render neurons more vulnerable to stress and promote neurodegeneration.16,17 We and others have demonstrated that FUS is required for DNA damage repair.14,15,17 FUS is involved in DNA double strand break repair including both homologous recombination (HR) and non-homologous end joining repair (NHEJ) pathways.17 Deficiency of FUS in mice results in hypersensitivity to ionizing irradiation and widespread genomic instability on the chromosomal level.14,15 FUS null mice also show defects in B cell development and spermatogenesis in which physiological DNA damage repair occurs frequently.14,15 Recent studies suggest that defects of FUS-regulated DNA damage repair may underlie neurodegeneration in ALS.16,17 ALS-FUS mutants R244C, R514S, R521C are deficient in DNA double-strand repair including both HR and NHEJ.17 Spinal cord motor neurons and cortical neurons of ALS-FUS patients harboring R521C, P525L mutations showed marked increase of DNA damage when compared with the controls.17 Consistent with the observation in patients’ tissues, a mouse study showed that the brain and the spinal cord of FUS-R521C mice exhibited increased DNA damage at selective DNA loci, such as the BDNF gene.16 BDNF plays an important role in promoting dendritic growth and synaptic formation.26 Neurons expressing FUS-R521C showed retraction in dendritic growth, similar to BDNF-deficient neurons.16 These data suggest that defects of FUS-regulated DNA damage repair at the loci of genes important for neuronal development and function may promote neurodegeneration.
The function of FUS in DNA damage repair on the molecular level is not yet clear. In response to DNA damage, FUS is rapidly recruited to DNA damage sites prior to H2AX phosphorylation, and interacts with HDAC1.17 FUS may be recruited to modulate chromatin conformation changes, stabilize DNA damage repair complexes, or regulate transcription and alternative splicing. Our recent analysis of CLIP-seq data suggests that FUS may regulate the alternative splicing of genes crucial in DNA damage response and repair pathways. We searched for FUS RNA targets that fall into the GO functional category of cellular response to DNA damage stimulus (GO: 0006974) in ours and five other previously published FUS CLIP-seq data.12,18-22 We identified 382 genes that were detected in at least two studies (data not shown) and applied Ingenuity Pathway Analysis (IPA) to these genes. The top 20 statistically significant (P ≤ 0.05) pathways are shown in Figure 3A. BRCA1 breast cancer signaling, ATM signaling (Fig. 3B), DNA double-strand break repair and cell cycle checkpoints are among the most enriched pathways. Remarkably, about 60% of the genes in the DNA double-strand break repair HR pathway (Fig. S2A) and NHEJ pathway (Fig. S2B) are FUS RNA targets. These data suggest that FUS may regulate the alternative splicing of these DNA damage response or repair genes in response to DNA double strand breaks. It was reported that EWS, a family member closely related to FUS, modulates the alternative splicing of ABL1, CHEK2, and MAP4K2 in response to UV-induced single strand DNA damage and genotoxic stress signaling.27 A similar mechanism likely exists for FUS-dependent DNA damage response. Our analysis here suggests for the first time that RNA metabolism may represent a novel mechanism underlying FUS-dependent DNA damage response and repair. Given that FUS is phosphorylated by ATM in response to DNA double-strand breaks,28 it is tempting to speculate that FUS may link the ATM signaling pathway to RNA metabolism. FUS may modulate alternative splicing to coordinate crucial DNA damage response and repair pathways. Genetic lesions in DNA damage repair and DNA damage response signaling genes are well known to cause cell death and neurodegeneration.13 Defective DNA damage repair caused by ALS-associated FUS mutations may render neurons more vulnerable to stress and promote neurodegeneration. In line with this notion, ALS with FUS mutations, in general, show an earlier disease onset when compared with other ALS mutations.29

Figure 3. FUS targets RNA of genes in DNA damage response and repair pathways. (A) Ingenuity Pathways Analysis (IPA) of FUS RNA targets that fall into the GO functional category of “cellular response to DNA damage stimulus” (GO: 0006974). FUS RNA targets from ours12 and five others’ CLIP-seq assays were analyzed.18-22 The RNA targets that fall into the GO: 006974 category and were also identified by at least two research groups are used for IPA. The blue bars represent P values. The pink line represents the ratio of FUS RNA targets over the total number of genes in each pathway. (B) FUS RNA targets that map to the ATM signaling pathway by IPA analysis. FUS RNA targets are highlighted in purple.
Taken together, these data suggest that DNA damage repair is defective in neurons expressing ALS-FUS mutants16,17 and RNA metabolism may represent a novel mechanism underlying FUS-dependent DNA damage repair.
Conclusion
Here we propose a model that dysregulated RNA metabolism and DNA damage repair collectively drive and accelerate neurodegeneration in ALS and broadly in FTD and other neurodegenerative disease (Fig. 4). FUS regulates the alternative splicing of pre-mRNAs and the processing of long intron containing transcripts that play crucial roles in neuronal development, neuronal function and gene expression regulation. FUS protein that is retained in the cytoplasmic inclusions in ALS and FTD is deficient in the processing of these pre-mRNAs, which may cause synaptic dysfunction, axon withdrawal, and denervation, and drive neurodegeneration. RNA metabolism may also represent a novel mechanism underlying FUS-dependent DNA damage response. ALS-FUS mutants are deficient in alternative splicing and DNA damage repair, and have limited stress-coping capacity, which may render neurons more vulnerable and accelerate neurodegeneration.
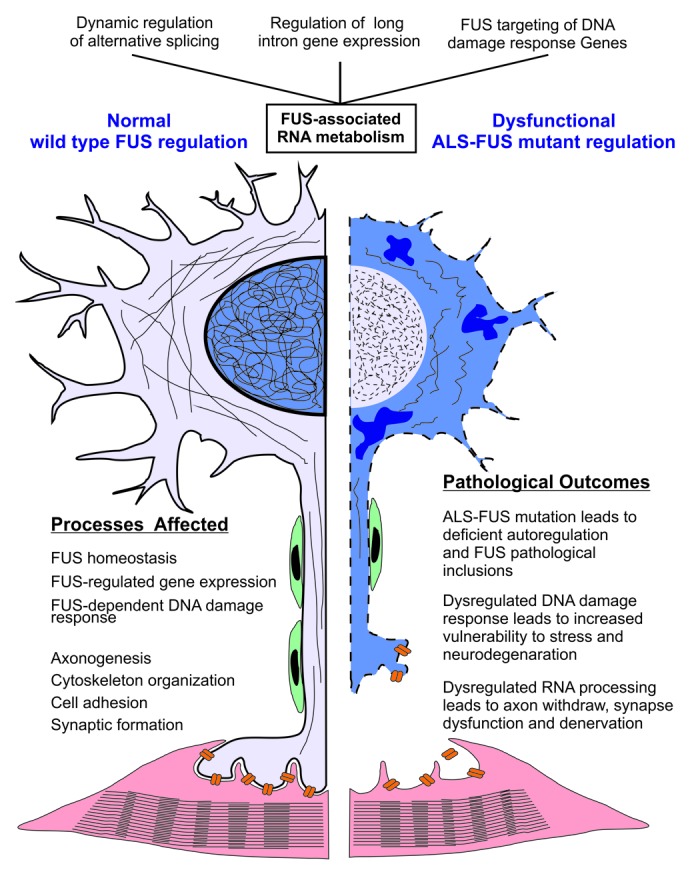
Figure 4. Dysregulation of FUS-associated RNA metabolism and DNA damage response collectively drive and accelerate neurodegeneration in ALS-FUS. Three major areas of FUS-associated RNA metabolism highlighted in this paper are listed at the top of the figure. Molecular and cellular processes regulated by wildtype FUS-dependent RNA metabolism in a normal, healthy, motor neuron are highlighted in the left panel of the figure. The pathological outcomes resulting from mutated ALS-FUS-dependent dysregulation are highlighted in the right panel. ALS-FUS mutations result in a triple-hit on motor neurons of ALS patients. i) The ALS-FUS mutation itself results in the direct accumulation of cytoplasmic inclusions, ii) FUS-dependent RNA metabolism of target genes required for the healthy maintenance of neuronal processes is dysregulated, and iii) DNA damage response required for the maintenance and survival of long-lived post-mitotic neuronal cells is dysregulated. The later is a new etiological dimension of ALS-FUS, and may explain the relative severity of ALS-FUS as a familial form of ALS.
With FUS RNA targets having been identified, further research focusing on the molecular mechanism underlying the dysregulation of FUS-dependent RNA metabolism in ALS and FTD will shed light on the pathogenic mechanism of ALS and FTD and, more broadly, other neurodegenerative disease. Moreover, research on FUS-dependent RNA metabolism in response to DNA damage may provide novel insights into the disease mechanism for both neurodegeneration and cancer.
Supplementary Material
Acknowledgment
We thank Mohammad Golam Sabbir for the artwork of Figure 4. This work was supported by grants from Canada Research Chairs (CIHR) and CancerCare Manitoba to GGH. YZ was supported by a studentship from the Manitoba Health Research Council. Funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Glossary
Abbreviations:
- FUS/TLS
fused in sarcoma/translocated in liposarcoma
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- NLS
nuclear localization signal
- CLIP-seq
UV crosslinking RNA immunoprecipitation and deep sequencing
- HR
homologous recombination
- NHEJ
non-homologous end joining repair
References
- 1.Law WJ, Cann KL, Hicks GG. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic. 2006;5:8–14. doi: 10.1093/bfgp/ell015. [DOI] [PubMed] [Google Scholar]
- 2.Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 3.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Langenhove T, van der Zee J, Van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med. 2012;44:817–28. doi: 10.3109/07853890.2012.665471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:639–49. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- 6.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci. 2005;118:5755–65. doi: 10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- 8.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr., Sapp P, McKenna-Yasek D, Brown RH, Jr., Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobágyi T, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22:2676–88. doi: 10.1093/hmg/ddt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, Liu S, Liu G, Oztürk A, Hicks GG. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9:e1003895. doi: 10.1371/journal.pgen.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 14.Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24:175–9. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- 15.Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000;19:453–62. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124:981–99. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16:1383–91. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18:1428–31. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. 2012;2:529. doi: 10.1038/srep00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–97. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakaya T, Alexiou P, Maragkakis M, Chang A, Mourelatos Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA. 2013;19:498–509. doi: 10.1261/rna.037804.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dillon AK, Fujita SC, Matise MP, Jarjour AA, Kennedy TE, Kollmus H, Arnold HH, Weiner JA, Sanes JR, Kaprielian Z. Molecular control of spinal accessory motor neuron/axon development in the mouse spinal cord. J Neurosci. 2005;25:10119–30. doi: 10.1523/JNEUROSCI.3455-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dichmann DS, Harland RM. fus/TLS orchestrates splicing of developmental regulators during gastrulation. Genes Dev. 2012;26:1351–63. doi: 10.1101/gad.187278.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orozco D, Tahirovic S, Rentzsch K, Schwenk BM, Haass C, Edbauer D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012;13:759–64. doi: 10.1038/embor.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–87. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paronetto MP, Miñana B, Valcárcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43:353–68. doi: 10.1016/j.molcel.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 28.Gardiner M, Toth R, Vandermoere F, Morrice NA, Rouse J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem J. 2008;415:297–307. doi: 10.1042/BJ20081135. [DOI] [PubMed] [Google Scholar]
- 29.Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, Brown RH, Jr., Shapiro BE, Lomen-Hoerth C. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010;20:1069–76. doi: 10.1111/j.1750-3639.2010.00413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.