

Abstract
Increased dispersion of repolarization has been suggested to underlie increased arrhythmogenesis in human heart failure (HF). However, no detailed repolarization mapping data were available to support the presence of increased dispersion of repolarization in failing human heart. In the present study, we aimed to determine the existence of enhanced repolarization dispersion in the right ventricular (RV) endocardium from failing human heart and examine its association with arrhythmia inducibility. RV free wall preparations were dissected from five failing and five nonfailing human hearts, cannulated and coronary perfused. RV endocardium was optically mapped from an ∼6.3 × 6.3 cm2 field of view. Action potential duration (APD), dispersion of APD, and conduction velocity (CV) were quantified for basic cycle lengths (BCL) ranging from 2,000 ms to the functional refractory period. We found that RV APD was significantly prolonged within the failing group compared with the nonfailing group (560 ± 44 vs. 448 ± 39 ms, at BCL = 2,000 ms, P < 0.05). Dispersion of APD was increased in three failing hearts (161 ± 5 vs. 86 ± 19 ms, at BCL = 2,000 ms). APD alternans were induced by rapid pacing in these same three failing hearts. CV was significantly reduced in the failing group compared with the nonfailing group (81 ± 11 vs. 98 ± 8 cm/s, at BCL = 2,000 ms). Arrhythmias could be induced in two failing hearts exhibiting an abnormally steep CV restitution and increased dispersion of repolarization due to APD alternans. Dispersion of repolarization is enhanced across the RV endocardium in the failing human heart. This dispersion, together with APD alternans and abnormal CV restitution, could be responsible for the arrhythmia susceptibility in human HF.
Keywords: action potential duration, conduction abnormality, human heart failure, dispersion of repolarization, right ventricle
ventricular arrhythmia and sudden cardiac death is common in patients with heart failure (HF) (12, 29). However, the understanding of the origin and nature of arrhythmia in HF remains incomplete, particularly in humans. While clinical studies that demonstrate an increased QT dispersion and prolonged interval between T wave's peak and end (1, 13, 31) suggest a link between dispersion of repolarization and arrhythmia in HF patients, studies into the mechanism of repolarization dispersion in HF are limited and report inconsistent findings. In a tachypacing-induced HF dog model, increased dispersion of left ventricular (LV) repolarization was found to be present transmurally (between endocardium and epicardium) due to a disproportionate action potential duration (APD) prolongation of the M cells. However, this was not confirmed in the human heart with advanced HF. It was found that the dispersion of repolarization from endocardium to epicardium was decreased in failing human heart due to a greater APD prolongation of the subepicardium compared with either midmyocardium or subendocardium (18). This phenomenon was recapitulated in a pressure-overload HF mouse model (37) and in a different study using a canine tachypacing-induced HF model (24).
The aforementioned experimental data suggest that dispersion of repolarization might not be present transmurally but present across other regions of the failing myocardium. Several clinical electrophysiology (EP) studies have examined dispersion of repolarization in the right ventricle (RV) endocardium, due to technical accessibility of this region via catheter, and these studies have identified the RV endocardium as an important substrate for the generation of arrhythmias in various cardiac disorders (6, 16, 30). However, to our knowledge, dispersion of repolarization on RV endocardium has not been studied in patients with advanced HF. Also, despite the advantage that clinical EP studies are conducted in vivo, these studies are limited in the surface area that can be thoroughly mapped. With our unique experimental preparation and imaging modality, we have been able to examine dispersion in an area encompassing a major portion of the RV endocardium, enabling us to detect potential differences in repolarization dispersion. In this study, we aimed to test the hypothesis that dispersion of repolarization at the RV endocardium might be enhanced in the failing human heart and that this increased RV repolarization dispersion is correlated with increased arrhythmia vulnerability. We also aimed to assess remodeling of RV gene expression during HF to identify targets that were remodeled in the RV during HF. To test these hypotheses, we optically recorded endocardial action potentials (AP) on the isolated coronary-perfused RV preparation. Alternatively to monophasic action potential (MAP) electrode recordings, optical recordings of AP have several thousand recording sites. This permits the quantification of the dispersion of repolarization at high spatial resolution. We also conducted gene expression analysis on RV tissues with a panel of 96 gene targets known to be important for regulating normal cardiac electrophysiological properties.
METHODS
The protocol for this study was approved by the Washington University Institutional Review Board. Failing human hearts (HF, n = 5, Table 1) were obtained at the time of cardiac transplantation performed at the Barnes-Jewish Hospital, Washington University School of Medicine. Donor hearts with normal RV function were provided by Mid-America Transplant Service (St. Louis, MO) and used as controls representing the nonfailing group (n = 5, Table 1).
Table 1.
Patient information
| Group No. | Gender | Age, yr | Diagnosis |
|---|---|---|---|
| Failing 1 | Male | 53 | Nonischemic cardiomyopathy |
| Failing 2 | Male | 55 | Ischemic cardiomyopahty |
| Failing 3 | Female | 67 | Nonischemic cardiomyopathy |
| Failing 4 | Male | 59 | Nonischemic cardiomyopathy |
| Failing 5 | Female | 53 | Nonischemic cardiomyopathy |
| Nonfailing 1 | Female | 51 | Cerebrovascular/strock |
| Nonfailing 2 | Male | 31 | Gunshot wound to the head |
| Nonfailing 3 | Male | 40 | Head trauma, motor vehicle accident |
| Nonfailing 4 | Female | 59 | Cerebrovascular/stroke |
| Nonfailing 5 | Male | 76 | Cerebrovascular/stroke |
Optical mapping experiments.
Explanted hearts were cardioplegically arrested in the operating room (18, 26). The RV free wall was dissected out along the ventricular septum and right coronary artery. The RV outflow tract was cut open so that the preparation could be laid flat. The origin of the right coronary artery was cannulated by a 16-gauge cannula. The quality of perfusion was verified by methylene blue dye. The preparation was perfused by oxygenated Tyrode solution at 37°C. Major arterial leaks were ligated to maintain a constant perfusion pressure of 60–70 mmHg. The preparation was fully immersed in a chamber with Tyrode solution to assure appropriate superfusion. To remove the toxic metabolites, a 30-min washout with Tyrode solution was performed before the conduction of the experimental protocol. The width of the flattened RV free-wall preparation (from RV outflow tract to the septum) was 115 ± 10 mm in failing vs. 91 ± 6 mm in the nonfailing group (P < 0.01). Care was taken so that the tissues were not stretched when they were pinned down onto a silicon sheet. Tissue was immobilized with 10–20 μM blebbistatin to suppress the motion artifacts in optical recordings (11). Optical APs were recorded from the majority of endocardium of RV free wall (63 ± 7 by 63 ± 7 mm).
We commenced pacing at a cycle length (CL) of 2,000 ms and then gradually decreased the CL until reaching the functional refractory period or until an arrhythmia was induced. All of the preparations were stimulated at the bottom of the field of view (close to the apex) at a voltage twice the stimulation threshold. Figure 1 shows a representative RV preparation and AP recordings from a nonfailing human heart. The APD at 80% repolarization (APD80) was measured simultaneously from all recording sites. Assuming that the conduction wavefront was perpendicular to the endocardial surface, we measured the global conduction velocity (CV) across the field of view by dividing the distance along the fastest direction of conduction from the pacing site to the atrioventricular groove by the corresponding conduction time. Dispersion of APD was defined as the difference between 95th percentile and 5th percentile of APD80 from all the recorded sites. While local spatial gradient of repolarization is an indicator for the dispersion of repolarization, its quantification is not well suited for RV endocardium because of its trabeculated structure (Fig. 1A). Because of the trabeculation, RV endocardium is not a flat surface and therefore the distance between any two points can be underestimated by our two-dimensional mapping. This is especially true for the gradient over the edges of the trabeculae carneae (Fig. 1A), where the measured gradient was an extreme overestimation of the actual value due to an underestimation of the actual distance.
Fig. 1.
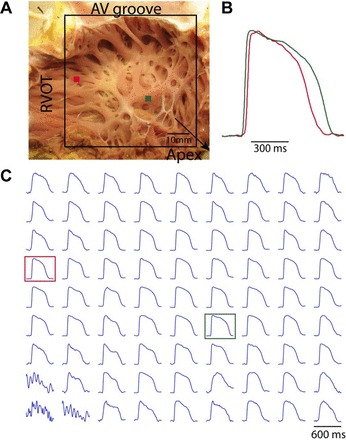
Representative right ventricular (RV) free wall preparation and optical action potentials (AP). A: RV free wall preparation and mapping field of view (FOV). B: close-up view of two AP recordings. C: representative optical AP from an evenly spaced array of locations spanning the whole FOV. Red and green dots in A and squares in C correspond to recordings in B. RVOT, right ventricular outflow tract.
Statistical comparisons were made between the failing group and nonfailing group. For functional, optical imaging results, levels of significance were determined by the two-way ANOVA. Bonferroni adjustment was used to account for multiple comparisons. P < 0.05 was considered statistically significant. Values were given as means ± SD.
Gene expression assay and false discovery rate analysis.
A TaqMan low-density array was used to quantify the expression patterns of the genes encoding ion channels, membrane transporters, calcium-handling proteins, and regulatory proteins, as described previously (14, 15, 28). Briefly, transmural tissue segments for RNA extraction were harvested near the RV apex, immediately caudal to the functional tissue preparation. Total RNA was extracted using the RNEasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA), and RNA yield was measured using a Nanodrop 1000 (Thermo Scientific, Rockford, IL). Custom low-density TaqMan gene arrays were used in a two-step RT-PCR process: 1) total RNA was transcribed into cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), and 2) the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) was used for subsequent reactions. We selected 96 genes to analyze, with GAPDH as a reference gene for normalization.
Gene expression changes were quantified using the relative quantification method with Applied Biosystems SDS 2.1 software. The Grubb's test was used to exclude sample outliers with GenEx software (MultiD, Weihenstephan, Germany). Multiple comparisons statistical testing was conducted using the false discovery rate (FDR) method, which was implemented using the Significance Analysis of Microarrays (SAM) software package (Stanford University, Stanford, California). The significance threshold was selected to be as small as possible while still enabling identification of potentially remodeled targets.
RESULTS
AP in failing human RV.
APD prolongation is considered a hallmark change in the failing myocardium (34). However, prolongation of APD was not observed in endocardium of LV from failing human heart (18). To determine whether APD is prolonged in the RV endocardium of failing human heart, we averaged all APDs for each heart at various pacing CLs. Figure 2 shows APDs at multiple CLs for each individual heart (left) and for each group (right). It can be seen that APDs in the failing group are significantly longer than the nonfailing group. This prolongation is further demonstrated in Fig. 3, which shows representative AP recordings from each individual heart paced at a CL of 2,000 ms. Three different columns of recordings are APs from regions with maximum APD, mean APD, and minimum APD from each heart.
Fig. 2.
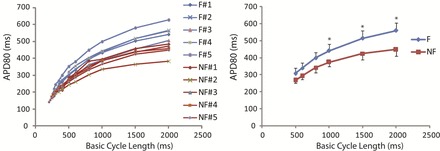
Individual action potential duration (APD) restitution and summary APD restitution. Left, APD at 80% repolarization (APD80) at various basic cycle lengths (BCL) from failing (blue lines, F) and nonfailing (red lines, NF) human hearts. Right, averaged APD80 of failing and nonfailing hearts. Because not every heart was paced faster than BCL of 500 ms, only results for cycle lengths from 500 to 2,000 ms are shown. *P < 0.05 for the comparison between failing and nonfailing groups.
Fig. 3.
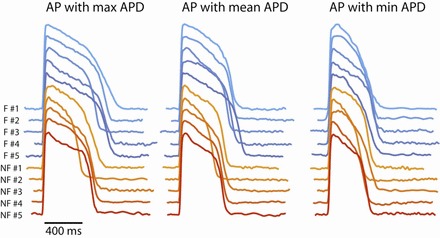
Representative AP recordings from individual failing (blue traces) and nonfailing (red traces) human hearts at the pacing cycle length (CL) of 2,000 ms. Left, AP recordings with the maximum APD in the FOV. Middle, the AP recordings with the mean APD in the FOV. Right, the AP recordings with the minimum APD in the FOV.
Dispersion of APD.
Figure 3 not only shows the prolongation of APD in the failing group but also suggests the presence of increased span of APDs in some failing hearts, as evident from the bigger difference between maximum and minimum APDs. The global dispersion of APD is further quantified by calculating the difference between 95th and 5th percentile of all the APDs for each heart. The results are shown in Fig. 4, which shows the dispersion of APD for each heart (left) and each group (right). It can be seen that three failing hearts (nos. 1, 2, and 5) had increased dispersion of APD at CL >800 ms, whereas the other two failing hearts showed similar dispersion of APD to the nonfailing group.
Fig. 4.
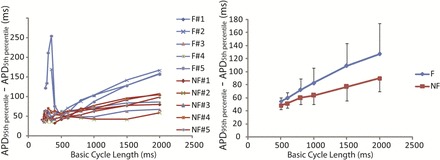
Dispersion of repolarization quantified by APD80 at the 95th percentile (APD95th percentile) minus APD80 at the 5th percentile (APD5th percentile). Left, dispersion of repolarization at various BCL from failing (blue lines) and nonfailing (red lines) human hearts. Right, averaged dispersion of repolarization within failing and nonfailing groups for CL from 500 to 2,000 ms. It can be seen from the left that three failing hearts (nos. 1, 2, and 5) stand out from the other hearts by showing significant enhancement of dispersion of repolarization at slow heart rate and that the two failing hearts (nos. 2 and 5) had pronounced dispersion at fast heart rate as well.
One example with increased dispersion of APD at the CL of 2,000 ms from the failing group is shown in Fig. 5, top, together with an example of nonfailing heart shown in Fig. 5, bottom. Initiated at the pacing site at the apex (bottom of the view), activation uniformly propagated toward the base in both groups (Fig. 5, left). On the other hand, the APDs are more spatially heterogeneous (from 420 to 640 ms) in the failing example than the nonfailing one (from 400 to 530 ms; Fig. 5, right), therefore indicating an increased dispersion of repolarization in the failing heart.
Fig. 5.
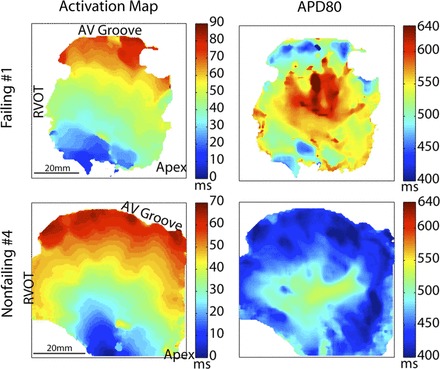
Representative examples of dispersion of repolarization at the endocardium of RV free wall from a failing heart (row on top) and a nonfailing heart (row on bottom) at the pacing CL of 2,000 ms. Left, maps of activation (from blue to red) after a stimulus at the bottom of the FOV. Middle, maps of APD80. Right, FOV with inverted color for better visualization of the structure.
Dynamics of CV.
Abnormal activation delay at rapid activation rate could increase dispersion of conduction and repolarization (22). To determine the conduction abnormality in the failing RV, we quantified the global CV restitution for all hearts as shown in Fig. 6. CV was significantly smaller in the failing group than the nonfailing group (Fig. 6, right). In all of the nonfailing hearts and three of the failing hearts, as we gradually decreased the pacing CL, the CV remained constant until the basic CL approached 500 ms. In contrast, the decrease of CV at decreasing CL started at a much longer basic CL (1,000 ms) in two failing human hearts (failing hearts 2 and 5; Fig. 6, left). This abnormal CV restitution has been previously reported to associate with the origin of arrhythmia (10). Interestingly, these were the only two hearts in our study in which arrhythmias could be induced. In addition, these two hearts were among the previously identified three hearts that had enhanced dispersion of APD.
Fig. 6.
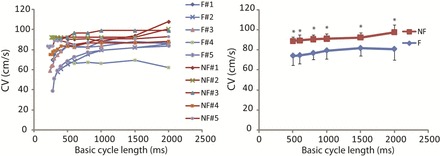
Conduction velocity (CV) restitution. Left, individual CV restitution from failing (blue lines) and nonfailing (red lines) human hearts. Right, averaged CV restitution. *P < 0.05 between failing and nonfailing group. Note that the CV was significantly decreased in the failing hearts. Also note that CV restitutions in failing hearts 2 and 5 have a nonflat slope over a much wider range of BCL.
APD alternans.
APD alternans were induced by rapid pacing in the three failing hearts with large APD dispersion (failing hearts 1, 2, and 5) as well as in one nonfailing heart (no. 5). The CL at the onset of APD alternans was 367 ± 29 ms (vs. 220 ms in the nonfailing heart). APD alternans are evident from the AP recordings during pacing shown in Fig. 7A. Moreover, the pattern of APD alternans is heterogeneous. While AP recording in one location (site a in Fig. 7A) does not show APD alternans, other sites (sites b, c, d, and e in Fig. 7A) demonstrate alternans. While site b shows a 2:2 alternans pattern, sites c, d, and e show a 3:3 alternans pattern. These spatially discordant APD alternans significantly increased the dispersion of repolarization at fast heart rates (CL <400 ms) in two hearts (failing hearts 2 and 5 in Fig. 4) and could provide the substrate for reentrant arrhythmia. Indeed, arrhythmia was induced in these two hearts, which also had abnormal CV restitution (Fig. 6).
Fig. 7.
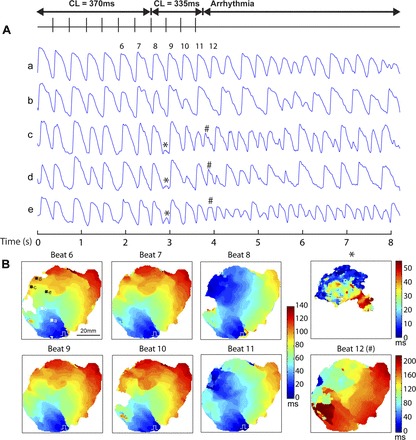
Arrhythmia induced by rapid pacing in failing heart 2. A: AP from five sites (a, b, c, d, and e) during a recording where pacing CL was switched from 370 to 335 ms, which induced arrhythmia. Alternans of APD is evident from recording sites b, c, d, and e. *Low-amplitude activation. #Apparent ectopic focal beat (beat 12) that triggered the arrhythmia. B: sequential activation maps of beat 6 to beat 12.
Induction of arrhythmia by rapid pacing.
The initiation of arrhythmia by rapid pacing in one failing human heart (no. 2) is illustrated in Fig. 7. Interestingly, before the initiation of arrhythmia, apparent ectopic focal beats occurred every three paced beats (at beats 2, 5, 8, and 11), which echoes the 3:3 pattern of APD alternans. These apparent ectopic focal beats started simultaneously with the paced beat, and the initiated waves would propagate and collide with the paced beat (see beats 8 and 11 in Fig. 7B). Following beat 8, there is a low-amplitude ectopic “beat” (Fig. 7) that did not trigger arrhythmia. Beat 12 (Fig. 7) is an ectopic beat that also occurred before the next pacing stimulus that initiated the arrhythmia.
RV gene expression analysis.
To investigate the molecular correlates of AP prolongation and dispersion of repolarization in the failing human RV, we performed TaqMan gene array analysis of 96 targets that are involved in regulating cardiac electrophysiology. These genes include many ion channels and membrane transporters, as well as their regulatory proteins, signaling molecules, and transcription factors. We compared gene expression between nonfailing and failing human RV tissues (n = 4 for each group) from most of the same hearts used for our functional mapping study (failing nos. 1, 2, 4, and 5; nonfailing nos. 1, 3, 4, and 5). Using FDR analysis, we found 17 genes (Table 2) that are potentially downregulated in the failing RV compared with the nonfailing RV (FDR = 10.89%). We did not identify any significantly upregulated genes in the failing human heart. Figure 8 shows average relative quantification gene expression values for the potentially altered genes in HF. Genes are ordered based on SAM d score, and error bars represent SDs.
Table 2.
Significantly remodeled genes in the right ventricle of failing human heart: TaqMan gene expression analysis
| Gene | Significant Genes (FDR 10.89%) |
||
|---|---|---|---|
| Encoded protein | Score (d) | Fold change | |
| SCN1A | Neural/muscle Na+ channel | −2.80 | 0.17 |
| KCNK3 | TASK two-pore K+ channel | −2.53 | 0.12 |
| CNN1 | Calponin 1 | −2.24 | 0.20 |
| KCNJ12 | Kir2.2 (potential IK1 contributor) | −2.24 | 0.43 |
| HCN4 | Hyperpolarization-activated K+ channel | −2.22 | 0.33 |
| TBX2 | T-box transcription factor | −1.96 | 0.58 |
| ADRB3 | β3-Adrenoreceptor | −1.83 | 0.09 |
| ATP1B1 | Na+-K+-ATPase β1-subunit | −1.82 | 0.58 |
| KCNK6 | TWIK2 two-pore K+ channel | −1.79 | 0.39 |
| PANX1 | Pannexin 1 | −1.70 | 0.42 |
| GJC1 | Cx45 | −1.63 | 0.48 |
| ATP2B4 | Sarcolemmal Ca2+-ATPase | −1.62 | 0.69 |
| ITPR3 | Inositol 1,4,5-triphosphate receptor 3 | −1.57 | 0.41 |
| TBX3 | T-box transcription factor | −1.55 | 0.38 |
| KCNH2 | Kv11.1 (IKr) | −1.52 | 0.54 |
| ADRB2 | β2-Adrenoreceptor | −1.49 | 0.58 |
| PPP3CA | Protein phosphatase 3 | −1.49 | 0.54 |
The protein encoded by the identified genes are indicated, along with the Significant Analysis Microarrays d score and fold expression change for each target.
FDR, false discovery rate; Kir2.2, inward rectifying potassium channel; IK1, inward rectifying background potassium current; Cx, connexin; IKr, rapidly activating delayed-rectifier potassium current..
Fig. 8.
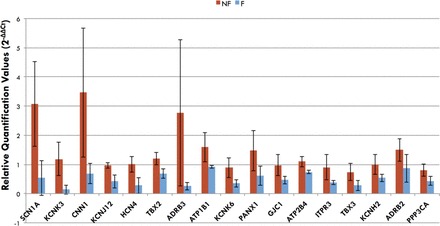
Average gene expression for significantly changed genes in failing compared (n = 4) with nonfailing (n = 4) RV. Relative quantification values are plotted, and error bars represent SD. Several repolarizing potassium channel genes and genes involved in conduction were identified (see Table 2).
While gene expression changes may not be directly related to functional ionic current alterations, we find it interesting that we identified several less-investigated targets that are potentially downregulated in the failing human RV. These results suggest that we may need to look more broadly to find the molecular correlates for repolarization and conduction changes in human HF.
DISCUSSION
In this study, we examined the remodeling of repolarization and conduction in the RV endocardium of failing human hearts. We observed consistent prolongation of repolarization and slowing of conduction in the failing group. Enhanced APD dispersion, APD alternans, and CV abnormality were not present in all of the failing hearts, but these measures appear to be closely associated with the arrhythmia inducibility.
Prolongation of APD in human HF.
APD prolongation has been consistently observed in various animal models of HF in both isolated cells and tissues and has been reviewed previously (35). Compared with animal models of HF, human studies of APD remodeling in HF are rather limited and mostly in the isolated cells (4, 5, 23, 25) or isolated small-sized RV trabeculae (19, 36), where spatial heterogeneity could not be assessed. We have recently explored transmural heterogeneity in the human LV wedge preparations and found that prolongation of APD was only observed at the subepicardium but not at the subendocardium and midmyocardium (18). In contrast to the absence of APD prolongation in the LV endocardium (18), prolongation of APD was a consistent phenomenon in the RV endocardium in this study. We also identified several potassium channel genes, including those for background two-pore domain channels, the inwardly rectifying background potassium current and the rapidly activating delayed-rectifier potassium current, that are downregulated in the failing RV. Decreased expression of these potassium channel genes could promote AP prolongation in the RV during HF.
Prolonged duration (or reduced repolarization reserve) increases the lability of AP and might increase the susceptibility to secondary depolarization, such as early afterdepolarization (EAD). It has been shown in simulation studies (20, 21) that EADs could occur at rapid rates if the repolarization reserve is sufficiently reduced. It has also been shown in tissue slices that rate acceleration could also promote EADs (7) in the settings of reduced repolarization reserve. Therefore, EADs might underlie the apparent ectopic focal beats during rapid pacing in the failing human heart in Fig. 7. On the other hand, optical mapping recorded AP mostly from the tissue surface and therefore cannot clearly distinguish “actual” focal beat from the “apparent” surface focal beat resulting from three-dimensional intramural reentry (2, 3). It remains to be determined which mechanism is more responsible for the arrhythmia inductions in failing human heart.
Dispersion of repolarization in human HF.
Increased QT dispersion in HF patients (1, 13, 31) suggested the existence of exaggerated dispersion of repolarization, which may predispose to ventricular arrhythmia. While increased transmural repolarization heterogeneity was not observed in the LV of failing human heart (18, 26), we demonstrated in this study prolongation of APD was heterogeneous at the RV endocardium in three failing hearts and thus increased the dispersion of repolarization at slow heart rates (Figs. 4 and 5). In two out of those three failing hearts, APD alternans was sufficiently pronounced to produce increased dispersion of APD at fast heart rates (Fig. 4). The mechanism why heterogeneous prolongation is associated with enhanced APD alternans susceptibility remains to be determined. One of the likely mechanisms might be decreased cellular coupling, because it could decrease the electrotonic effects and increase the heterogeneity of repolarization, as well as promote the incidence of APD alternans (9). We have recently shown that gap junctional coupling is indeed decreased in the human failing LV (17).
Remodeling of CV and arrhythmia.
We observed significantly decreased CV in the RV endocardium of the failing human heart. This is consistent with our previous findings in the LV of failing human heart (17, 18). The decreased CV could be due to decreased expression of gap junctional proteins, such as connexin (Cx) 43 (17) and Cx45. Our RV gene expression study suggested that Cx45 expression might be downregulated. Previous work by Yamada et al. (38) has found that Cx45 mRNA was not altered and Cx45 protein expression was upregulated in LV tissue from failing human hearts and has suggested that this upregulation in conjunction with Cx43 downregulation could result in conduction abnormality. While our gene expression results suggest the opposite, it remains to be determined whether Cx43 protein expression is generally downregulated in the failing myocardium. Also, more investigations, in model systems, will need to be conducted to demonstrate mechanistic associations between Cx45 expression and CV.
Abnormal CV restitution has been shown to contribute to the susceptibility to arrhythmia (8, 10, 32, 33). Our data strongly support the correlation between arrhythmia and abnormal CV restitution, where delayed activation occurred at a much slower heart rate (failing hearts 2 and 5 in Fig. 6A). This abnormal CV restitution also corresponds to a wider range of CL at which the restitution curve is steep. According to a computer simulation study (32), to produce spatially discordant alternans and subsequent induction of arrhythmia, pacing rate needs to be fast enough to engage the steep portion of CV restitution. Therefore, the wide range of steep portion of CV restitution could decrease the pacing rate threshold for the induction of discordant alternans. This could explain why abnormal CV dynamics and discordant APD alternans were observed in the same hearts in our study. This abnormal CV restitution has been previously reported in the failing human hearts by Kawara et al. (22) and was found to be closely associated with stringy and patchy fibrosis (22).
Variability in human HF.
In this study, while the RV endocardium from failing hearts shared similar functional remodeling (e.g., prolongation of APD), variability is present. We did not observe increased dispersion of APD, alternans, and abnormal CV in two out of five failing hearts. Moreover, the consistently observed prolongation of APD might have different mechanisms in different hearts (Fig. 2) because the prolongation was due to a slow repolarization phase (phase 3) in one failing heart (no. 1), whereas it was due to a longer plateau (phase 2) in other failing hearts. This variability suggests significant differences and heterogeneity of remodeling in each individual HF patient.
Limitations.
There are several limitations in this study. First, the etiology of HF, its duration, and medical treatment are varied and uncontrolled, and we did not have access to clinical data due to the deidentified tissue protocol. Second, other mechanisms might also contribute to the dispersion of repolarization. For example, heterogeneity of innervation might affect the dispersion of repolarization (27). Third, the isolated tissue preparations were mechanically unloaded due to the surgical opening of the tissue and the application of the excitation-contraction uncoupler blebbistatin. It remains to be determined how the mechanical loading affects the electrical properties in the human heart. Finally, the failing human hearts in this study were all in the end stage of HF. These results might not apply to the failing human heart in earlier stages of HF.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: Q.L., K.M.H., I.-W.W., and I.R.E. conception and design of research; Q.L., D.L.J., K.M.H., D.L., B.O., C.M.A., V.V.F., and I.-W.W. performed experiments; Q.L., D.L.J., K.M.H., D.L., and C.M.A. analyzed data; Q.L., K.M.H., and I.R.E. interpreted results of experiments; Q.L., K.M.H., and I.R.E. prepared figures; Q.L., K.M.H., and I.R.E. drafted manuscript; Q.L., K.M.H., B.O., and I.R.E. edited and revised manuscript; Q.L., K.M.H., and I.R.E. approved final version of manuscript.
REFERENCES
- 1. Barr CS, Naas A, Freeman M, Lang CC, Struthers AD. QT dispersion and sudden unexpected death in chronic heart failure. Lancet 343: 327–329, 1994 [DOI] [PubMed] [Google Scholar]
- 2. Baxter WT, Mironov SF, Zaitsev AV, Jalife J, Pertsov AM. Visualizing excitation waves inside cardiac muscle using transillumination. Biophys J 80: 516–530, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berenfeld O, Pertsov AM. Dynamics of intramural scroll waves in three-dimensional continuous myocardium with rotational anisotropy. J Theor Biol 199: 383–394, 1999 [DOI] [PubMed] [Google Scholar]
- 4. Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73: 379–385, 1993 [DOI] [PubMed] [Google Scholar]
- 5. Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85: 1046–1055, 1992 [DOI] [PubMed] [Google Scholar]
- 6. Bonatti V, Rolli A, Botti G. Recording of monophasic action potentials of the right ventricle in long QT syndromes complicated by severe ventricular arrhythmias. Eur Heart J 4: 168–179, 1983 [DOI] [PubMed] [Google Scholar]
- 7. Burashnikov A, Antzelevitch C. Acceleration-induced action potential prolongation and early afterdepolarizations. J Cardiovasc Electrophysiol 9: 934–948, 1998 [DOI] [PubMed] [Google Scholar]
- 8. Cao JM, Qu Z, Kim YH, Wu TJ, Garfinkel A, Weiss JN, Karagueuzian HS, Chen PS. Spatiotemporal heterogeneity in the induction of ventricular fibrillation by rapid pacing: importance of cardiac restitution properties. Circ Res 84: 1318–1331, 1999 [DOI] [PubMed] [Google Scholar]
- 9. Cherry EM, Fenton FH. Suppression of alternans and conduction blocks despite steep APD restitution: electrotonic, memory, and conduction velocity restitution effects. Am J Physiol Heart Circ Physiol 286: H2332–H2341, 2004 [DOI] [PubMed] [Google Scholar]
- 10. Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, Bhuiyan Z, Bezzina CR, Veldkamp MW, Linnenbank AC, van der Wal AC, Tan HL, Brugada P, Wilde AA, de Bakker JM. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112: 2769–2777, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm 4: 619–626, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Francis GS. Development of arrhythmias in the patient with congestive heart failure: pathophysiology, prevalence and prognosis. Am J Cardiol 57: 3B–7B, 1986 [DOI] [PubMed] [Google Scholar]
- 13. Fu GS, Meissner A, Simon R. Repolarization dispersion and sudden cardiac death in patients with impaired left ventricular function. Eur Heart J 18: 281–289, 1997 [DOI] [PubMed] [Google Scholar]
- 14. Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582: 675–693, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaborit N, Varro A, Le Bouter S, Szuts V, Escande D, Nattel S, Demolombe S. Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J Mol Cell Cardiol 49: 639–646, 2010 [DOI] [PubMed] [Google Scholar]
- 16. Gavrilescu S, Luca C. Right ventricular monophasic action potentials in patients with long QT syndrome. Br Heart J 40: 1014–1018, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, Schuessler RB, Moazami N, Efimov IR. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation 125: 1835–1847, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res 106: 981–991, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res 61: 70–76, 1987 [DOI] [PubMed] [Google Scholar]
- 20. Huffaker R, Lamp ST, Weiss JN, Kogan B. Intracellular calcium cycling, early afterdepolarizations, and reentry in simulated long QT syndrome. Heart Rhythm 1: 441–448, 2004 [DOI] [PubMed] [Google Scholar]
- 21. Huffaker RB, Weiss JN, Kogan B. Effects of early afterdepolarizations on reentry in cardiac tissue: a simulation study. Am J Physiol Heart Circ Physiol 292: H3089–H3102, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Kawara T, Derksen R, de Groot JR, Coronel R, Tasseron S, Linnenbank AC, Hauer RN, Kirkels H, Janse MJ, de Bakker JM. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation 104: 3069–3075, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Koumi S, Backer CL, Arentzen CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation 92: 164–174, 1995 [DOI] [PubMed] [Google Scholar]
- 24. Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol 283: H1031–H1041, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Li GR, Lau CP, Leung TK, Nattel S. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1: 460–468, 2004 [DOI] [PubMed] [Google Scholar]
- 26. Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG, Efimov IR. Transmural heterogeneity and remodeling of ventricular excitation-contraction coupling in human heart failure. Circulation 123: 1881–1890, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mantravadi R, Gabris B, Liu T, Choi BR, de Groat WC, Ng GA, Salama G. Autonomic nerve stimulation reverses ventricular repolarization sequence in rabbit hearts. Circ Res 100: e72–e80, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol 562: 223–234, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meinertz T, Hofmann T, Kasper W, Treese N, Bechtold H, Stienen U, Pop T, Leitner ER, Andresen D, Meyer J. Significance of ventricular arrhythmias in idiopathic dilated cardiomyopathy. Am J Cardiol 53: 902–907, 1984 [DOI] [PubMed] [Google Scholar]
- 30. Morgan JM, Cunningham D, Rowland E. Dispersion of monophasic action potential duration: demonstrable in humans after premature ventricular extrastimulation but not in steady state. J Am Coll Cardiol 19: 1244–1253, 1992 [DOI] [PubMed] [Google Scholar]
- 31. Pinsky DJ, Sciacca RR, Steinberg JS. QT dispersion as a marker of risk in patients awaiting heart transplantation. J Am Coll Cardiol 29: 1576–1584, 1997 [DOI] [PubMed] [Google Scholar]
- 32. Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation 102: 1664–1670, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Saumarez RC, Slade AK, Grace AA, Sadoul N, Camm AJ, McKenna WJ. The significance of paced electrogram fractionation in hypertrophic cardiomyopathy. A prospective study. Circulation 91: 2762–2768, 1995 [DOI] [PubMed] [Google Scholar]
- 34. Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 90: 2534–2539, 1994 [DOI] [PubMed] [Google Scholar]
- 35. Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 42: 270–283, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klopping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res 28: 1547–1554, 1994 [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Cheng J, Joyner RW, Wagner MB, Hill JA. Remodeling of early-phase repolarization: a mechanism of abnormal impulse conduction in heart failure. Circulation 113: 1849–1856, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamada KA, Rogers JG, Sundset R, Steinberg TH, Saffitz JE. Up-regulation of connexin45 in heart failure. J Cardiovasc Electrophysiol 14: 1205–1212, 2003 [DOI] [PubMed] [Google Scholar]