

Abstract
Acute Helicobacter pylori infection of gastric epithelial cells induces CagA oncoprotein- and peptidoglycan (SLT)-dependent mobilization of NF-κB p50 homodimers that bind to H-K-ATPase α-subunit (HKα) promoter and repress HKα gene transcription. This process may facilitate gastric H. pylori colonization by induction of transient hypochlorhydria. We hypothesized that H. pylori also regulates HKα expression posttranscriptionally by miRNA interaction with HKα mRNA. In silico analysis of the HKα 3′ untranslated region (UTR) identified miR-1289 as a highly conserved putative HKα-regulatory miRNA. H. pylori infection of AGS cells transfected with HKα 3′ UTR-Luc reporter construct repressed luciferase activity by 70%, whereas ΔcagA or Δslt H. pylori infections partially abrogated repression. Transfection of AGS cells expressing HKα 3′ UTR-Luc construct with an oligoribonucleotide mimetic of miR-1289 induced maximal repression (54%) of UTR activity within 30 min; UTR activity was unchanged by nontargeting siRNA transfection. Gastric biopsies from patients infected with cagA+ H. pylori showed a significant increase in miR-1289 expression compared with uninfected patients or those infected with cagA− H. pylori. Finally, miR-1289 expression was necessary and sufficient to attenuate biopsy HKα protein expression in the absence of infection. Taken together, these data indicate that miR-1289 is upregulated by H. pylori in a CagA- and SLT-dependent manner and targets HKα 3′ UTR, affecting HKα mRNA translation. The sensitivity of HKα mRNA 3′ UTR to binding of miR-1289 identifies a novel regulatory mechanism of gastric acid secretion and offers new insights into mechanisms underlying transient H. pylori-induced hypochlorhydria.
Keywords: H-K-ATPase, H. pylori, microRNA, proton pump, regulation acid secretion
helicobacter pylori, a gram-negative bacterium that colonizes gastric epithelium and induces chronic inflammation, is a major risk factor for gastric cancer. Although the majority of H. pylori-infected people are asymptomatic, ∼10% develop H. pylori-related peptic ulcer disease, and ∼1% progress to atrophic gastritis, intestinal metaplasia, gastric mucosa-associated lymphoid tissue lymphoma, or gastric adenocarcinoma (23). Gastric H. pylori colonization drives the early stages of gastric carcinogenesis because virtually all infected persons have superficial gastritis, and H. pylori eradication significantly decreases gastric cancer risk in infected individuals without premalignant lesions (39). Human H. pylori infection transiently inhibits acid secretion, as shown by self-administration experiments and reports of putative and confirmed H. pylori-induced gastritis (8, 9, 16, 24). H. pylori-induced progression to gastric neoplasia is thus initiated in a transiently hypochlorhydric environment that facilitates colonization and activates proinflammatory pathways involved in development of disease (17).
Gastric acid secretion is mediated by H-K-ATPase (25) whose α-subunit (HKα) is an apical membrane protein of gastric parietal cells. The H. pylori type 4 secretion system (T4SS) protein CagL interacts with host cell α5β1 integrins (14), facilitating injection of the oncogenic bacterial protein CagA that activates NF-κB (2). We showed previously that H. pylori inhibition of HKα gene expression is dependent in part on bacterial CagA expression (27) and results from ERK 1/2-mediated NF-κB p50 homodimer binding to HKα promoter (29). We also showed that acute H. pylori infection of AGS cells causes CagL to dissociate epithelial cell ADAM17 from α5β1 integrins, activating ADAM17-dependent, NF-κB-mediated repression of HKα promoter (26). Moreover, 24 h H. pylori infection of human gastric biopsies in vitro was shown to decrease the expression of HKα mRNA and render HKα protein virtually undetectable by enzyme-linked immunosorbent assay, and 6-h infection significantly decreased biopsy acid secretory capacity (27). We also showed that ΔcagE, ΔcagM, and ΔcagL H. pylori isogenic mutants caused no HKα repression in AGS cells, confirming the need for T4SS integrity (27). Lastly, nucleotide-binding oligomerization domain receptor (Nod1) activation by a glycosylated tripeptide (GM-3) released from H. pylori peptidoglycan by soluble lytic transglycosylase (Slt) activates NF-κB (36), potentially suppressing HKα transcription. We recently reviewed this field, including evidence that acute H. pylori-induced acid inhibition is not caused by parietal cell ablation, neutrophil IL-1β secretion, or H. pylori vacuolating toxin (VacA) (31).
Given that H. pylori repressed HKα promoter activity by ∼75%, but H. pylori-infected gastric biopsies expressed virtually no HKα protein, we reasoned that H. pylori must mobilize additional cellular mechanisms to shut down acid secretion. This study tests the hypothesis that H. pylori exerts posttranscriptional control mediated by upregulation of gastric epithelial cell miRNAs that exhibit seed sequence complementarity with the HKα 3′ untranslated region (3′ UTR). MicroRNAs are 19–24 nt long evolutionarily conserved noncoding RNAs that bind to complementary sequences in the 3′ UTR of target mRNAs, blocking translation of the transcript and potentially targeting the transcript for degradation (10). As such, miRNAs are potent posttranscriptional regulators of gene expression. A growing list of miRNAs has been reported to be induced in an H. pylori cagA-dependent manner in gastric cells in vitro and in vivo (15, 20). The present study provides evidence for involvement of H. pylori-induced gastric miRNA in posttranscriptional regulation of parietal cell proton pump synthesis.
MATERIALS AND METHODS
Cells, bacteria, and reagents.
Human gastric epithelial AGS cells (ATCC, Manassas, VA) were maintained in Ham's F12 medium containing 10% FBS as described (29). H. pylori wild-type cag PAI+ strain 7.13 and isogenic mutants were grown on Brucella broth (Difco Laboratories, Detroit, MI) plates containing 2.4% agar, 10% fetal bovine serum (Brucella-agar plates), with kanamycin (25 μg/ml) or chloramphenicol (4 μg/ml), as indicated, at 37°C by using a microaerophilic gas pack system (BD Biosciences, Sparks, MD). Nonpolar H. pylori isogenic mutants were generated by insertion of a kanamycin resistance cassette into the unique BglII restriction site of a cloned PCR product of the full-length cagA gene (HP0547), followed by transformation into H. pylori and kanamycin selection, yielding ΔcagA. Insertion of a chloramphenicol resistance cassette into the unique HindIII restriction site of a cloned PCR product of the full-length slt gene (HP0645), followed by transformation into H. pylori and chloramphenicol selection, yielded a nonpolar Δslt mutant. In addition, insertion of a chloramphenicol resistance cassette into the slt HindIII restriction site of ΔcagA H. pylori, with chloramphenicol/kanamycin selection, yielded the double-mutant ΔcagA/Δslt. Bacterial multiplicities of infection (MOI) were calculated as described (29). All reagents were of molecular biology grade.
HKα 3′ UTR-luc reporter construct, AGS cell transfection, and H. pylori infection.
RNA was extracted from a human gastric biopsy using RNA Stat-60 (Tel-Test, Friendwood, TX). cDNA was prepared with the Bio-Rad iScript cDNA synthesis kit. The HKα 3′ UTR with 5′ and 3′ XbaI restriction sites was amplified by PCR (Roche FastStart High Fidelity PCR System) by using forward and reverse primers HKα-UTRF1-ATAGTATCTAGAAGGGACGACTGCCTTCAAGCATC and HKα-UTRR1-CAGTACTCTAGATACAGCACAGTCAGCATGAGCTGGTT (Sigma-Genosys). The 446-bp PCR product (confirmed by polyacrylamide gel electrophoresis) was extracted (Roche High Pure PCR Product Purification Kit), digested with XbaI (Promega, Madison, WI), reextracted, and ligated into the XbaI site of pGL3 control vector (Promega), placing the HKα UTR at the 3′ terminus of the luciferase (Luc) reporter gene. Sequence and orientation of the ligated amplicon were verified by DNA sequencing. PCR products encompassing the 5′ half (UTR1) or the 3′ half (UTR2) of the HKα 3′ UTR were prepared and cloned into a pGL3 control vector by similar methods. Near-confluent AGS cells in Ham's F12 medium with 10% serum (1.5 × 105 cells·ml−1·well−1 in 24-well plates) were transfected with wild-type or mutated HKα 3′ UTR-Luc reporter construct and pMaxGFP for transfection efficiency control and normalization, using Fugene 6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. Transfected AGS cells were seeded in six-well plates (5 × 105 cells/well) and incubated overnight in serum-free F12 medium. After addition of fresh F12 medium, aliquots of H. pylori 7.13 wild-type strain or the isogenic mutant strains ΔcagA, Δslt, or ΔcagA/Δslt were added to the cells to a final MOI of 50 or 100. After coincubation at 37°C in a conventional 5% CO2 incubator for 4 and 24 h, the culture medium was removed and HKα 3′ UTR-reporter activities were measured and normalized as described (28).
AGS cell MicroRNA analysis by quantitative real-time RT-PCR.
AGS cells were seeded in six-well plates (5 × 105 cells/well) and incubated overnight in serum-free F12 medium. After addition of fresh F12 medium, aliquots of H. pylori 7.13 wild-type strain or the isogenic mutant strains ΔcagA, Δslt, or ΔcagA/Δslt were added to the cells to a final MOI of 50 or 100. After coincubation at 37°C in a conventional 5% CO2 incubator for between 4 and 24 h, the culture medium was removed and cellular microRNA was isolated using a High Pure miRNA isolation kit (cat. no. 05080576001, Roche Diagnostics) and reverse transcribed by using RT2 miRNA First Strand Kit (Qiagen, Santa Clarita, CA) according to manufacturers' instructions. Quantitative real-time RT-PCR analysis of AGS cell miR-1289 content was carried out with PCR oligo sets specific for hsa-mir-1289 and RNU6 (housekeeping control), each containing a pair of miRNA-specific forward and universal reverse oligos (Qiagen), TaqMan microRNA Assay Kit (Life Technologies, Baltimore, MD), and RT2 SYBR Green Fluor PCR Mastermix (Qiagen) on a Bio-Rad iCycler (10′ at 95°C, followed by 40 cycles each comprising 30″ at 95°C, 40″ at 60°C, and 30″ at 72°C).
Synthetic oligoribonucleotides.
Transfection mixes comprising Fugene 6 (Roche Diagnostics) and a 23-nucleotide synthetic oligoribonucleotide (IDT DNA Technologies, Coralville, IA) based on the sequence of hsa-mir-1289 or nontargeting siRNA (Thermo Fisher Scientific, Pittsburgh, PA) were prepared according to manufacturers' instructions and added dropwise to AGS cell transfected 24 h previously with HKα 3′ UTR-Luc reporter construct and pMaxGFP as described above, to a final oligoribonucleotide concentration of 100 nM.
Human gastric biopsies.
RNA was isolated from human gastric biopsies from uninfected patients (n = 3), cagA+ H. pylori-infected patients (n = 3), or cagA− H. pylori-infected patients (n = 3) (21) by using the miRNeasy Mini Kit (Qiagen, Venlo, Limburg) according to the manufacturer's instructions. Reverse transcriptase PCR and quantitative real-time PCR (7300 Real-Time PCR System, Applied Biosystems Life Technologies, Carlsbad, CA) were carried out according to the manufacturer's instructions. Levels of human miR-1289 expression were standardized to levels of human RNU44 expression (TaqMan, Applied Biosystems Life Technologies). Experiments were performed on three independent occasions. Data are represented as fold over uninfected controls. Student's t-test was used to determine statistical significance among the groups.
The effect of synthetic miR-1289 mimetic on human gastric mucosal HKα expression was studied in gastric biopsies acquired from consenting patients (21–60 yr old) undergoing esophagogastroduodenoscopy or endoscopic ultrasound at the MUSC Digestive Disease Center (IRB protocol HR16941). Exclusion criteria included patients with positive urea breath and campylobacter-like organism tests. Four endoscopic biopsies (6–42 mg each) per patient were obtained from normal-appearing corpus mucosa on the greater curvature of the stomach. Single biopsies were incubated in individual wells of a six-well culture plate with Ham's F12, 10% fetal bovine serum culture medium (1.8 ml, 1 h, 37°C). Transfection mix was prepared by dropwise addition of X-TremeGeneHP transfection reagent (Roche Diagnostics) to a 1 μM solution of miR-1289 synthetic oligoribonucleotide in serum-free Ham's F12, to a final ratio of X-TremeGeneHP:RNA of 4 μl:1 μg. The transfection mix (200 μl) was added to biopsies and incubated with slow swirling at 37°C for 4 h. Biopsies were removed, rinsed in serum-free Ham'S F12, and stored at −20°C pending analysis by immunoblotting. Same-patient biopsies were incubated with F12 medium/X-TremeGeneHP alone or with 100 nM nontargeting siRNA instead of synthetic miR-1289 served as controls.
Immunoblotting.
Gastric biopsies transfected in vitro with synthetic oligoribonucleotide as described were washed in ice-cold buffer (50 mM Tris, pH 7.2, 5 mM EGTA) and disaggregated by use of a Potter-Elvehjem glass-Teflon homogenizer. The homogenate was centrifuged at 7,000 g for 10 min, and aliquots of supernatant (5 μg protein) were heated at 55°C for 5 min with equal volumes of SDS-PAGE sample buffer. Biopsy HKα content was assessed by immunoblotting with HK 12.18 antibody as described (32) and β-actin antibody (Sigma-Aldrich, St. Louis, MO) as a gel loading control. Immunoblots shown are representative of three replicates.
Statistical analysis.
Two-way ANOVA followed by Bonferroni posttest were used to analyze the effect of H. pylori infection on luciferase activity of AGS cells transfected with HKα 3′ UTR-Luc reporter constructs. Analyses were done with GraphPad Prism 4.0 or Microsoft Excel 2007. P values less than 0.05 were considered significant.
RESULTS
To examine whether human HKα (Genbank ATP4A; NM000704) might be regulated posttranscriptionally via miRNA interactions, we conducted an in silico analysis of human HKα 3′ UTR sequence. Analysis with miRBase 17 (www.mirbase.org/) identified eight putative miRNA candidates, including miR-1289; TargetScanHuman 5.2 (www.targetscan.org/) identified 41 poorly conserved miRNAs and one highly conserved miRNA (miR-1289); and Microcosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/) identified 55 putative miRNA candidates. None of these miRNAs have previously been associated with in vitro gastric epithelial cell H. pylori infection, in vivo gastric mucosal infection, or gastric cancer. The highly conserved eight-nucleotide match for human miR-1289 seed sequence in HKα 3′ UTR is shown in Fig. 1. Interestingly, a 5-nt sequence in the human HKα 3′ UTR (shown in blue in Fig. 1) corresponds to a putative “zipcode”-like sequence that has been implicated in targeting of mRNAs to intracellular microvesicles (3). Resting, unstimulated gastric parietal cells are characterized by dense accumulations of intracellular tubulovesicles enriched in HKα protein (30), conceiveably synthesized in situ by appropriately targeted HKα mRNA.
Fig. 1.

Human H-K-ATPase α-subunit (HKα) 3′ untranslated region (3′ UTR) sequence has a highly conserved miRNA binding site. The 462-nt sequence of 3′ UTR of human H-K-ATPase α-subunit mRNA (NCBI reference sequence NM 000704.2) is shown, together with the sequence of hsa-miR-1289 (www.mirbase.org, gray italic), aligned to show complementarity of the miRNA seed sequence (gray italic) with the putative HKα 3′ UTR binding site (boldface italic). A 5-nt sequence that together with the miR-1289 binding sequence may target mRNAs to microvesicles (3) is shown in boldface roman.
Given that the in silico analyses predicted that the miR-1289 seed sequence is complementary to a potential binding site in the HKα 3′ UTR, we assayed miR-1289 expression in H. pylori infected AGS cells and in gastric biopsies from H. pylori-infected patients by using custom primer/probe sets and quantitative real-time RT-PCR. Figure 2 shows that wild-type H. pylori infection of AGS cells (MOI = 100, 8 h) caused ∼10-fold upregulation of miR-1289 levels; ΔcagA and Δslt infections caused 1.5- and 3-fold upregulation, and ΔcagA/Δslt double isogenic mutant infection caused <2-fold upregulation. We next measured the temporal expression of miR-1289 in AGS cells following infection with wild-type H. pylori (MOI = 100). As shown in Fig. 3A, infection caused a time-dependent upregulation of miR-1289 expression culminating in a 3.6-fold increase after 6 h. Extending the assay to human tissue, we measured miR-1289 expression levels relative to an endogenously expressed small RNA, RNU44, in gastric biopsies from uninfected, cagA+, or cagA− H. pylori-infected patients. As shown in Fig. 3B, patients infected with cagA+ H. pylori strains showed a significant increase in miR-1289 expression compared with both uninfected patients or those infected with cagA− H. pylori strains, indicating that H. pylori induces miR-1289 expression in a cagA-dependent manner. These data are consistent with a putative role for miR-1289 in H. pylori-induced regulation of HKα mRNA. We sought to validate this role in a series of in vitro experiments using the AGS cell line.
Fig. 2.
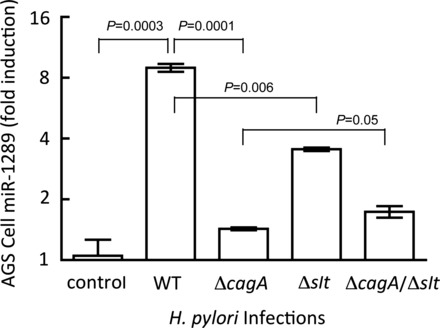
Wild-type (WT) Helicobacter pylori induces miR-1289 in AGS cells in a CagA- and Slt-dependent manner. AGS cell content of miR-1289 in response to H. pylori infection [8 h, multiplicities of infection (MOI) = 100] was measured by quantitative real-time RT-PCR (RT-qPCR) using a custom primer/probe set. Values are means ± SE (n = 3).
Fig. 3.
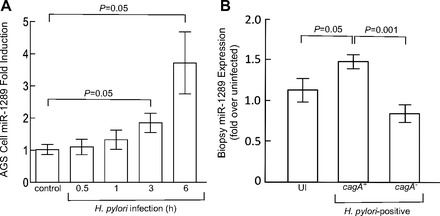
A: H. pylori infection of AGS cells upregulates miR-1289 expression. AGS cells were mock infected or infected with wild-type H. pylori (MOI = 100, for the indicated times) and the cellular content of miR-1289 was measured by RT-qPCR. Values are means ± SE (n = 3). B: patients infected with a cagA+ strain of H. pylori expression significantly higher levels of gastric mucosal miR-1289. RNA was isolated from human gastric biopsies from uninfected (UI) patients or cagA+ or cagA− H. pylori-infected patients. MiR-1289 expression relative to an endogenously expressed small RNA, RNU44, was measured by RT-qPCR. Values are means ± SE (n = 3).
An HKα 3′ UTR luciferase reporter plasmid was created by cloning the 446 bp HKα 3′ UTR into the pGL3 control vector (Promega), placing the HKα 3′ UTR at the 3′ terminus of the luciferase reporter gene. Figure 4 shows that H. pylori infection of AGS cells transfected with the pGL3 control vector repressed luciferase activity by ∼42%, suggesting susceptibility of Luc 3′ UTR to interactions with H. pylori-induced AGS cell miRNAs. In the absence of H. pylori infection, addition of HKα 3′ UTR to the pGL3 control vector repressed luciferase activity by ∼70%, indicating increased susceptibility to AGS cell miRNAs conferred by HKα sequence; following H. pylori infection, luciferase activity was repressed by a further 50%. The data show that the presence of HKα 3′ UTR during infection caused overall repression of luciferase activity by ∼85%, consistent with the hypothesis that H. pylori infection upregulates a subset of AGS cell miRNAs that target the HKα 3′ UTR, thereby affecting luciferase production. Increasing the MOI of H. pylori infection of transfected AGS cells from 50 to 100 caused more robust repression of HKα 3′ UTR-dependent luciferase expression, allowing us to examine the role of bacterial cagA and slt genes in this repression. As shown in Fig. 5, wild-type H. pylori infection at an MOI of 100 repressed HKα 3′ UTR-Luc activity by 70%, whereas infection with ΔcagA or Δslt isogenic mutants repressed activity by only 30 and 50%, respectively. Infection with a double ΔcagA/Δslt isogenic mutant did not return Luc activity to control levels.
Fig. 4.
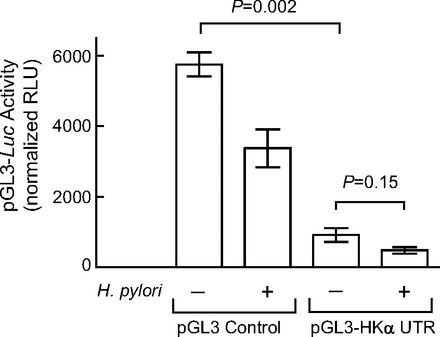
AGS cell H. pylori infection regulates translation mediated by HKα 3′ UTR. AGS cells transfected with the pGL3 control vector or with HKα 3′ UTR-pGL3 Luc reporter construct were infected with wild-type H. pylori (MOI = 50, 6 h), and luciferase activity in cell lysates was measured by luminometry. Values are means ± SE (n = 3). RLU, relative light units.
Fig. 5.
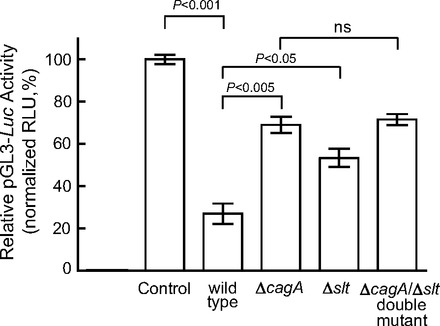
ΔcagA and Δslt isogenic mutants repress HKα 3′ UTR significantly less than wild-type. AGS cells transfected with HKα 3′ UTR-pGL3 Luc reporter construct were infected with wild-type H. pylori, ΔcagA, Δslt or ΔcagA/Δslt isogenic mutant H. pylori (MOI = 100, 6 h), and luciferase activity in cell lysates was measured by luminometry. Values are means ± SE (n = 3); ns, not significant.
We extended this analysis by examining the roles of the 5′ or 3′ halves of the HKα 3′ UTR. Two additional Luc reporter constructs containing either the 5′ half (UTR1) or the 3′ half (UTR2) of the HKα 3′ UTR were prepared. UTR2 contains the eight-nucleotide sequence complementary to the miR-1289 seed sequence. These constructs were transfected into AGS cells followed by infection with H. pylori wild-type, ΔcagA, or Δslt isogenic mutants. Figure 6 shows that infection of UTR1-expressing AGS cells induced a moderate repression of luciferase activity, with no dependence on bacterial CagA or Slt expression. In contrast, infection of UTR2-expressing AGS cells induced a robust suppression of luciferase activity, with significant dependence on bacterial CagA and Slt expression. These data are consistent with location of the miR-1289 binding site in the 3′ half of the HKα 3′ UTR. As before, double-mutant infection did not return luciferase activity to control values, suggesting that factors other than CagA and Slt may play roles in acid inhibition.
Fig. 6.
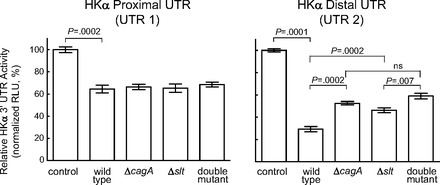
HKα proximal UTR activity is less sensitive to H. pylori infection than distal UTR. AGS cells transfected with HKα 3′ UTR-pGL3 Luc reporter constructs expressing either the proximal or distal halves of the HKα 3′ UTR were infected with wild-type H. pylori, ΔcagA, Δslt, or ΔcagA/Δslt isogenic mutant H. pylori (MOI = 100, 6 h), and luciferase activity in cell lysates was measured by luminometry. Values are means ± SE (n = 3).
To assess the functional consequences of H. pylori-induced miR-1289 upregulation in terms of HKα 3 UTR-mediated regulation of messenger translation, AGS cells were transfected with the human HKα UTR-Luc construct and then transfected 24 h later with a synthetic 23-nucleotide oligoribonucleotide mimic of miR-1289. As shown in Fig. 7A, HKα 3′ UTR activity was maximally repressed (54%) within 30 min of miR-1289 transfection, whereas transfection with nontargeting siRNA had no effect on HKα 3′ UTR activity. Lastly, human gastric corpus biopsies were transfected for 4 h with the miR-1289 mimic, and biopsy lysates were then probed by Western blot using the HKα-specific monoclonal antibody HK 12.18. Figure 7B shows that miR-1289 transfection induced significant attenuation of HKα expression, whereas nontargeting siRNA transfection was without effect on HKα expression. Quantitation of these Western blot data confirmed the synthetic miR-1289-induced suppression of HKα protein expression in human gastric biopsies (Fig. 7C).
Fig. 7.
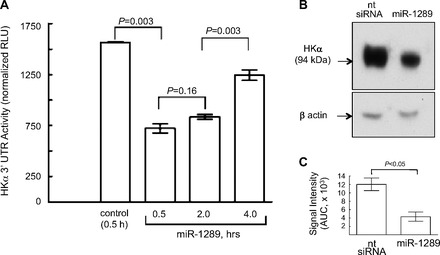
A miR-1289 mimic suppresses HKα 3′ UTR-Luc activity and HKα protein expression. A: AGS cells were transfected with the HKα 3′ UTR-Luc construct and then transfected with a synthetic 23-nucleotide oligoribonucleotide mimetic of miR-1289. Values are means ± SE (n = 3). B: human gastric corpus biopsies were transfected with the miR-1289 mimetic oligonucleotide, and biopsy lysates were analyzed by Western blot using monoclonal antibody HK 12.18 specific for HKα protein. β-Actin signal served as a gel loading control. The immunoblot image shown is representative of 3 replicate experiments. C: Image J quantitation of HKα-specific gastric biopsy lysate immunoblots as described in B (n = 3). AUC, area under the curve.
Taken together, these data indicate that H. pylori infection upregulates a gastric epithelial cell micro RNA miR-1289 that targets and blocks protein translation, and that CagA and Slt activity are implicated in miR-1289 upregulation specific for HKα suppression. The data specifically implicate miR-1289 with a highly conserved distal binding site on HKα UTR as playing a role in repressing HKα mRNA translation.
DISCUSSION
As a potentially pathogenic bacterium and as a neutralophile, H. pylori faces significant challenges in colonizing the gastric mucosa, an environment adapted to sterilizing ingested microorganisms by exposure to concentrated hydrochloric acid. The evolutionary success of H. pylori in this ecological niche, attested to by at least 60,000 years of cohabitation with humans, depends on a spectrum of phenotypic strategies that protect the bacterium as it seeks shelter below the gastric mucus in close proximity to the apical surfaces and intercellular junctions of gastric epithelial cells. Flagellar activity, spiral morphology, and pH-dependent reduction of mucus viscoelasticity facilitate bacterial penetration of the mucus layer (5), and proton-gated cytoplasmic membrane urea channels provide substrate for bacterial urease generating ammonium ions that efficiently buffer gastric HCl permeating the periplasmic space (33, 37). The less acidic microenvironment on the surface of HCO3-secreting surface epithelial cells provides opportunities for bacterial delivery of virulence factors that modify host cell biology and physiology to favor bacterial persistence in this niche. Such modifications include rearrangement of cytoskeletal actin and loosening of intercellular architecture allowing bacterial access to epithelial basolateral membranes. H. pylori has been shown to interact with α5β1 integrins on these membranes, promoting delivery of the oncogenic protein CagA through the bacterial T4SS into host cells (14). We have previously shown that this interaction also activates ADAM17, a metalloprotease complexed with α5β1 integrins, with concomitant release of the EGF receptor ligand HB-EGF and activation of EGFR-dependent signaling pathways within the cells (26).
In the context of bacterial adaptation to survival in the acidic gastric environment, another acute bacterial intervention in host physiology has been reported, namely, suppression of gastric acidification within a few days of infection (8, 16, 18, 24). Such repression would clearly facilitate gastric colonization, favoring survival of microorganisms with ability to interfere with parietal cell acid secretion. H. pylori acutely activates calcitonin gene-related peptide sensory neurons in rat fundic mucosa, stimulating somatostatin and inhibiting histamine secretion, and thereby inhibiting acid secretion (42). However, the bacterial factor(s) underlying this inhibition remain to be identified. We have shown that H. pylori infection of gastric epithelial cells, human gastric biopsies, and gerbil stomachs is associated with transcriptional repression of the gene encoding the α-subunit (HKα) of H-K-ATPase, the parietal cell apical membrane enzyme that mediates gastric acid secretion (4, 27). Gastric mucosal cells infected with cagA+ H. pylori exhibit significantly less expression of H-K-ATPase by immunohistochemistry and by immunoblotting with α-subunit-specific antibody (27). CagA-deficient H. pylori only partially repressed HKα transcription in cultured AGS cells, suggesting that factors in addition to CagA may be involved in HKα transcription, and failed to attenuate HKα protein expression in human gastric biopsies (27). Notably, patients infected with CagA+ H. pylori strains are at higher risk of peptic ulcer or gastric cancer than those infected with CagA− strains (35, 41). We have also reported the mechanistic basis for bacterial transcriptional repression of HKα, namely mobilization of an NF-κB p50 homodimer to the nucleus where it interacts with an HKα promoter binding site, resulting in transcriptional inhibition (29).
The present study provides evidence for a novel mechanism of H. pylori-induced repression of gastric acidification. This evidence is consistent with posttranscriptional regulation of HKα gene expression that is mediated by bacterial upregulation of the gastric epithelial cell microRNA miR-1289. Thus H. pylori infection of AGS cells was found to cause a time-dependent upregulation (up to 10-fold increase) of cellular miR-1289 content, whereas infection with cagA-deficient, slt-deficient, or cagA/slt-deficient isogenic mutants caused less than threefold miR-1289 upregulation, indicating a role for both H. pylori virulence factors in mobilizing miR-1289. The seed sequence of miR-1289 is complementary to a highly conserved eight-nucleotide sequence in the 3′ half of the human HKα 3′ UTR. Measurement of luciferase expression in AGS cells transfected with Luc-HKα 3′ UTR reporter constructs showed that H. pylori intervention in mRNA translation was dependent on bacterial cagA and slt gene expression and on the presence of HKα 3′ UTR sequence. Direct evidence for miR-1289-mediated intervention in HKα 3′ UTR regulation of mRNA translation was obtained by AGS cell cotransfections with a Luc-HKα 3′ UTR reporter construct and a synthetic miR-1289-based oligoribonucleotide. Luciferase activity was found to be significantly repressed by the miRNA mimetic, as was HKα protein expression in human gastric biopsies similarly transfected with the miR-1289 analog.
These findings introduce the possibility that in addition to known mechanisms of gastric acid secretory regulation, which include neural and hormonal secretagogue stimulation, transcriptional control of H-K-ATPase gene expression, recruitment of latent proton pump subunits to the parietal cell secretory canalicular membrane by fusion of cytoplasmic tubulovesicles, coupling to apical membrane potassium channels, and endocytic recovery of apical membrane, there is also posttranscriptional control of HKα mRNA translation mediated by parietal cell microRNAs whose expression levels are subject to physiological constraints. MicroRNA-mediated disruption and degradation of HKα mRNA would limit targeting and insertion of de novo synthesized HKα protein into tubulovesicular membranes, and because negative surface charge density of such tubulovesicles promotes their fusion with one another and the secretory membrane (6), loss of the predominant integral membrane proteins of tubulovesicles (H-K-ATPase α- and β-subunits) would retard fusion and hence active acid secretion.
A bacterial etiology of gastric acid suppression would require H. pylori to gain access to parietal cells deep in the gastric glands, and then penetrate the epithelial tight junctions into the basolateral domain where α5β1 integrins are displayed. In fact, a considerable literature documents ubiquitous distribution of H. pylori in the gastric intraepithelial, intercellular, and stromal spaces in patients with dyspepsia or gastric cancer (12, 13, 19, 22, 34). H. pylori-mediated epithelial penetration has also been extensively studied and relies on CagA interactions with components of focal adhesions and intercellular tight and adherens junctions (1, 38, 40) and HtrA-induced E-cadherin cleavage in epithelial cells that permeabilizes the gastric epithelial barrier (11). Thus H. pylori infection of the gastric mucosa triggers a spectrum of mechanisms that bring the bacteria in close proximity to parietal cell receptors whose function is then subverted to disrupt normal acid secretion.
Our finding in this study that HKα mRNA translation appears to be downregulated by H. pylori-induced gastric epithelial cell miR-1289 is consistent with our earlier study of HKα mRNA and protein expression in H. pylori-infected human gastric biopsies (27). We showed that wild-type H. pylori infection of biopsies suppressed HKα mRNA expression, but infection with H. pylori isogenic mutants deficient in the entire cag pathogenicity island (PAI), cagA, cagM, cagE, cagL, or slt did not (27). In fact, infection with Δslt H. pylori increased HKα mRNA 3.5-fold, compared with mock-infected same-patient controls. Wild-type H. pylori infection also caused disappearance of the HKα protein subunit from biopsy microsomal membrane fractions (27). The subunit was readily detected by immunoblotting in mock-infected biopsies and in biopsies infected with ΔcagPAI, ΔcagA, ΔcagM, ΔcagE, or ΔcagL H. pylori isogenic mutants. Biopsies infected with wild-type H. pylori strains or with an H. pylori isogenic mutant deficient in the vacuolating toxin gene vacA expressed virtually no HKα protein. These data emphasized that an intact H. pylori T4SS, and secretion of CagA and Slt-dependent peptidoglycan fragments, are essential for H. pylori-induced HKα gene repression.
The involvement of bacterial CagA and Slt secretion in upregulation of gastric epithelial cell miR-1289, as shown in the present study, adds yet another perturbation of host cell function attributable to these factors, specifically the disruption of posttranscriptional processing of a gene whose expression is essential to gastric parietal cell acid secretion. Effective though this mechanism might be in inhibiting acid secretion and facilitating gastric H. pylori colonization, HKα protein disappearance 24 h postinfection cannot be attributed solely to cessation of HKα gene transcription and translation because the half-life of existing HKα-subunits is 48 h (7). We hypothesize that H. pylori infection induces unscheduled proteasomal degradation of HKα-subunit, which would rapidly deplete parietal cells of acid secretory capacity, rendering the otherwise hostile acidic environment more hospitable to H. pylori during its transit through the stomach. Consistent with this hypothesis is our observation that histamine-stimulated, SCH28080-sensitive gastric biopsy H+ secretion was markedly inhibited by 2 h coincubation with wild-type H. pylori (27).
An alternative, or complementary, mechanism accounting for HKα depopulation of gastric parietal cell microsomes may involve a recently reported property of miR-1289 binding sites. More than 20 mRNAs whose 3′ UTRs express a miR-1289 binding site have been reported to be enriched in microvesicles derived from human primary glioblastoma multiforme cells and from melanoma cells (3). These binding sites are part of a 25-nt sequence that contains a CTGCC core domain on a stem-loop structure, an interaction that orchestrates transfer of the mRNAs into microvesicles. The HKα 3′ UTR also includes a 5-nt CTGCC sequence (Fig. 1), raising the possibility that, in concert with the miR-1289 binding site sequence, a zipcode-like signal is generated that directs HKα mRNA to cytoplasmic vesicles in parietal cells. Such targeting would ensure delivery of nascent HKα-subunits to intracellular tubulovesicles in preparation for their fusion with the rudimentary secretory canalicular apical membrane and activation of proton pumping into the glandular lumen. Future mutational studies of the HKα 3′ UTR should establish the role of UTR microsequences in targeting of proton pump subunits to cellular secretory compartments.
In summary, this study indicates that H. pylori infection upregulates a gastric epithelial cell miRNA miR-1289 with a seed sequence complementary to a highly conserved binding site in the distal half of the HKα 3′ UTR. Delivery of the H. pylori virulence factors CagA and SLT, previously shown to repress HKα transcription, is shown to be implicated in upregulation of miR-1289, which causes attenuation of HKα protein production. The sensitivity of HKα mRNA 3′ UTR to binding of gastric epithelial cell miRNA upregulated by H. pylori offers new insights into the molecular mechanisms underlying the transient hypochlorhydria induced by acute H. pylori infection.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.-M.Z., J.L.B., W.S.A., S.B., R.M.P.J., and A.J.S. interpreted results of experiments; Y.-M.Z., J.M.N., C.E.H., J.L.B., W.S.A., S.B., R.M.P.J., and A.J.S. edited and revised manuscript; Y.-M.Z., J.M.N., C.E.H., J.L.B., W.S.A., S.B., R.M.P.J., and A.J.S. approved final version of manuscript; J.M.N., C.E.H., S.B., R.M.P.J., and A.J.S. performed experiments; J.M.N., C.E.H., J.L.B., W.S.A., and A.J.S. analyzed data; J.M.N., C.E.H., and A.J.S. prepared figures; W.S.A. and A.J.S. conception and design of research; A.J.S. drafted manuscript.
ACKNOWLEDGMENTS
We thank Joe Romagnuolo, Todd Dantzler, and Mark Payne, Gastroenterology Division, Department of Medicine, MUSC for provision of gastric endoscopic biopsies, and Christine Davis, Digestive Disease Center, MUSC for clinical coordination. This study was supported by National Institutes of Health Grants DK064371 to A. J. Smolka; DK 58587, CA 77955, and CA 116087 to R. M. Peek, Jr.; and the German Science Foundation CRC-796 (Project B10) to S. Backert. W. S. Argraves and J. L. Barth are supported by South Carolina COBRE for Cardiovascular Disease (NIH/NIGMS GM103342), South Carolina IDeA Networks of Biomedical Research Excellence (NIH/NIGMS GM103499), and the MUSC University Research Resource Facility program.
REFERENCES
- 1.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300: 1430–1434, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol 10: 1573–1581, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, Fan JB, Breakefield XO, Saydam O. miR-1289 and “zipcode”-like sequence enrich mRNAs in microvesicles. Mol Ther Nucleic Acids 1: e10, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bott CJ, Hammond CE, Lewin DN, Ebert PT, Romero-Gallo J, Peek RM, Smolka AJ. Acute H. pylori infection of gerbils downregulates H-K-ATPase expression in a cagL-dependent manner (Abstract). Gastroenterology 138: A642, 2010 [Google Scholar]
- 5.Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, Ewoldt RH, McKinley GH, So P, Erramilli S, Bansil R. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci USA 106: 14321–14326, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duman JG, Lee E, Lee GY, Singh G, Forte JG. Membrane fusion correlates with surface charge in exocytic vesicles. Biochemistry 43: 7924–7939, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Gedda K, Scott D, Besancon M, Lorentzon P, Sachs G. Turnover of the gastric H+,K+-adenosine triphosphatase alpha subunit and its effect on inhibition of rat gastric acid secretion. Gastroenterology 109: 1134–1141, 1995 [DOI] [PubMed] [Google Scholar]
- 8.Graham DY, Alpert LC, Smith JL, Yoshimura HH. Iatrogenic Campylobacter pylori infection is a cause of epidemic achlorhydria. Am J Gastroenterol 83: 974–980, 1988 [PubMed] [Google Scholar]
- 9.Harford WV, Barnett C, Lee E, Perez-Perez G, Blaser MJ, Peterson WL. Acute gastritis with hypochlorhydria: report of 35 cases with long term follow up. Gut 47: 467–472, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522–531, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Hoy B, Lower M, Weydig C, Carra G, Tegtmeyer N, Geppert T, Schroder P, Sewald N, Backert S, Schneider G, Wessler S. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep 11: 798–804, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito T, Kobayashi D, Uchida K, Takemura T, Nagaoka S, Kobayashi I, Yokoyama T, Ishige I, Ishige Y, Ishida N, Furukawa A, Muraoka H, Ikeda S, Sekine M, Ando N, Suzuki Y, Yamada T, Suzuki T, Eishi Y. Helicobacter pylori invades the gastric mucosa and translocates to the gastric lymph nodes. Lab Invest 88: 664–681, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Ko GH, Kang SM, Kim YK, Lee JH, Park CK, Youn HS, Baik SC, Cho MJ, Lee WK, Rhee KH. Invasiveness of Helicobacter pylori into human gastric mucosa. Helicobacter 4: 77–81, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449: 862–866, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Li N, Xu X, Xiao B, Zhu ED, Li BS, Liu Z, Tang B, Zou QM, Liang HP, Mao XH. H. pylori related proinflammatory cytokines contribute to the induction of miR-146a in human gastric epithelial cells. Mol Biol Rep 39: 4655–4661, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Marshall BJ. Helicobacter pylori in peptic ulcer: have Koch's postulates been fulfilled? Ann Med 27: 565–568, 1995 [DOI] [PubMed] [Google Scholar]
- 17.Merchant JL. Inflammation, atrophy, gastric cancer: connecting the molecular dots. Gastroenterology 129: 1079–1082, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol 82: 192–199, 1987 [PubMed] [Google Scholar]
- 19.Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 132: 1009–1023, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Noto JM, Piazuelo MB, Chaturvedi R, Bartel CA, Thatcher EJ, Delgado A, Romero-Gallo J, Wilson KT, Correa P, Patton JG, Peek RM., Jr Strain-specific suppression of microRNA-320 by carcinogenic Helicobacter pylori promotes expression of the anti-apoptotic protein, Mcl-1. Am J Physiol Gastrointest Liver Physiol 305: G786–G796, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peek RM, Jr, Blaser MJ, Mays DJ, Forsyth MH, Cover TL, Song SY, Krishna U, Pietenpol JA. Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res 59: 6124–6131, 1999 [PubMed] [Google Scholar]
- 22.Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism? A review. FEMS Immunol Med Microbiol 36: 117–126, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10: 403–414, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramsey EJ, Carey KV, Peterson WL, Jackson JJ, Murphy FK, Read NW, Taylor KB, Trier JS, Fordtran JS. Epidemic gastritis with hypochlorhydria. Gastroenterology 76: 1449–1457, 1979 [PubMed] [Google Scholar]
- 25.Sachs G. The gastric proton pump: the H+, K+-ATPase. In: Physiology of the Gastrointestinal Tract (2nd ed.), edited by Johnson LR. New York: Raven, 1987, p. 865–881 [Google Scholar]
- 26.Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology 139: 239–248, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha A, Hammond CE, Beeson C, Peek RM, Smolka AJ. Helicobacter pylori represses proton pump expression and inhibits acid secretion in human gastric mucosa. Gut 59: 874–881, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saha A, Hammond CE, Gooz M, Smolka AJ. IL-1β modulation of H,K-ATPase α-subunit gene transcription in Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol 292: G1055–G1061, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Saha A, Hammond CE, Trojanowska M, Smolka AJ. Helicobacter pylori-induced H,K-ATPase α-subunit gene repression is mediated by NF-κB p50 homodimer promoter binding. Am J Physiol Gastrointest Liver Physiol 294: G795–G807, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Smolka A, Helander HF, Sachs G. Monoclonal antibodies against gastric H+ + K+ ATPase. Am J Physiol Gastrointest Liver Physiol 245: G589–G596, 1983 [DOI] [PubMed] [Google Scholar]
- 31.Smolka AJ, Backert S. How Helicobacter pylori infection controls gastric acid secretion. J Gastroenterol 47: 609–618, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Smolka AJ, Larsen KA, Hammond CE. Location of a cytoplasmic epitope for monoclonal antibody HK 12.18 on H,K-ATPase alpha subunit. Biochem Biophys Res Commun 273: 942–947, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Strugatsky D, McNulty R, Munson K, Chen CK, Soltis SM, Sachs G, Luecke H. Structure of the proton-gated urea channel from the gastric pathogen Helicobacter pylori. Nature 493: 255–258, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki K, Kokai Y, Sawada N, Takakuwa R, Kuwahara K, Isogai E, Isogai H, Mori M. SS1 Helicobacter pylori disrupts the paracellular barrier of the gastric mucosa and leads to neutrophilic gastritis in mice. Virchows Arch 440: 318–324, 2002 [DOI] [PubMed] [Google Scholar]
- 35.van Doorn LJ, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, de Boer W, Quint W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology 115: 58–66, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5: 1166–1174, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Weeks DL, Eskandari S, Scott DR, Sachs G. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287: 482–485, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Wessler S, Backert S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol 16: 397–405, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, Lai KC, Hu WH, Yuen ST, Leung SY, Fong DY, Ho J, Ching CK, Chen JS. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA 291: 187–194, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Wroblewski LE, Shen L, Ogden S, Romero-Gallo J, Lapierre LA, Israel DA, Turner JR, Peek RM., Jr Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 136: 236–246, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 123: 414–424, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Zaki M, Coudron PE, McCuen RW, Harrington L, Chu S, Schubert ML. H. pylori acutely inhibits gastric secretion by activating CGRP sensory neurons coupled to stimulation of somatostatin and inhibition of histamine secretion. Am J Physiol Gastrointest Liver Physiol 304: G715–G722, 2013 [DOI] [PubMed] [Google Scholar]