

Abstract
Peroxisome proliferator-activated receptor-α (PPARα) mediates metabolic remodeling, resulting in enhanced mitochondrial and peroxisomal β-oxidation of fatty acids. In addition to the physiological stimuli of fasting and high-fat diet, PPARα is activated by the fibrate class of drugs for the treatment of dyslipidemia. Sirtuin 1 (SIRT1), an important regulator of energy homeostasis, was downregulated in fibrate-treated wild-type mice, suggesting PPARα regulation of Sirt1 gene expression. The impact of SIRT1 loss on PPARα functionality in vivo was assessed in hepatocyte-specific knockout mice that lack the deacetylase domain of SIRT1 (Sirt1ΔLiv). Knockout mice were treated with fibrates or fasted for 24 h to activate PPARα. Basal expression of the PPARα target genes Cyp4a10 and Cyp4a14 was reduced in Sirt1ΔLiv mice compared with wild-type mice. However, no difference was observed between wild-type and Sirt1ΔLiv mice in either fasting- or fibrate-mediated induction of PPARα target genes. Similar to the initial results, there was no difference in fibrate-activated PPARα gene induction. To assess the relationship between SIRT1 and PPARα in a pathophysiological setting, Sirt1ΔLiv mice were maintained on a high-fat diet for 14 wk, followed by fibrate treatment. Sirt1ΔLiv mice exhibited increased body mass compared with control mice. In the context of a high-fat diet, Sirt1ΔLiv mice did not respond to the cholesterol-lowering effects of the fibrate treatment. However, there were no significant differences in PPARα target gene expression. These results suggest that, in vivo, SIRT1 deacetylase activity does not significantly impact induced PPARα activity.
Keywords: peroxisome proliferator-activated receptor-α, sirtuin 1, fibrates
peroxisome proliferator activated receptor-α (PPARα; NR1C1) is a member of the ligand-activated nuclear receptor superfamily of transcription factors. Although expressed ubiquitously throughout the body, PPARα plays a particularly important role in lipid homeostasis by regulating genes necessary for lipid transport and fatty acid oxidation in tissues such as the liver, adipose, and muscle (19). Because the natural ligands for PPARα include fatty acids, PPARα is a sensor of nutrient stress and regulates a battery of genes necessary for the utilization of fatty acids in the generation of acetyl-CoA for ketogenesis (7). PPARα plays a critical role in the remodeling of metabolic pathways in response to fasting. This includes regulation of genes responsible for mitochondrial and peroxisomal β-oxidation (15). Studies have also shown that PPARα expression and activation is increased in response to high-fat diet (HFD) (22). A role for PPARα in lipid metabolism in humans and as a drug target was uncovered when it was discovered that the triglyceride-lowering drugs clofibrate (Atromid-S), fenofibrate (Tricor), and gemfibrozil (Lopid) were ligands for PPARα, thus sparking renewed interest in the development of high-affinity PPARα agonists (16). Many factors influence PPARα function in response to ligand activation, including posttranslational modifications such as phosphorylation, ubiquitination, SUMOylation, and acetylation.
Sirtuin 1 (SIRT1) is the mammalian NAD+-dependent deacetylase homolog of yeast Sir2p that came to prominence when it was identified as a critical determinant of aging in yeast (14). In mammals, SIRT1 serves as a sensor of nutrient status, as it is activated by increased intracellular NAD+ concentrations that are seen during fasting and calorie restriction (8). SIRT1 also has a complex role in tumorigenesis due to its involvement in DNA damage repair as well as inactivation of p53 (21, 32, 33). Since the identification of p53 as the first non-histone target of SIRT1, a large number of transcription factors involved in lipid metabolism have been identified as SIRT1 targets, including PPARγ, PPARγ coactivator-1α (PPARGC1A), sterol regulatory element-binding transcription factor 1 (SREBF1), forkhead box (FOX) transcription factors, and PPARα (12). PPARα is deacetylated by SIRT1, and hepatic loss of SIRT1 was reported to result in decreased expression of PPARα target genes involved in fatty acid oxidation in vitro (27). Additionally, cell culture experiments suggest that PPARα may also positively regulate Sirt1 expression (10).
The current study investigated the potential impact of Sirt1 disruption on the pharmacological and physiological activation of PPARα in rodent models. However, lack of hepatic Sirt1 did not significantly impact the physiological responses associated with PPARα activation induced by ligand treatment, fasting, or HFD.
MATERIALS AND METHODS
Animals and treatments.
Six- to eight-week-old wild-type (WT) or Ppara-null (Ppara−/−) mice on the 129/Sv background were described previously (18). The floxed exon 4 Sirt1 allele (Sirt1F/F) mouse line on a C57BL6/129 background (B6; 129-Sirt1tm1Ygu/J) was obtained from The Jackson Laboratory and crossed with mice carrying the albumin-cre transgene to generate hepatic-specific mutant Sirt1 (Sirt1ΔLiv) mice (37). The Sirt1ΔLiv mice express a truncated form of SIRT1 that lacks the deacetylase domain (unpublished data), similar to results obtained by another laboratory (27). A second liver-specific Sirt1 knockout mouse line (Sirt1F/F;Deng and Sirt1ΔLiv;Deng) on a 129/Sv background in which exons 5 and 6 are disrupted generates nearly complete loss of the SIRT1 protein in liver (35). Mice (6–8 wk old) were provided a grain-based diet containing 0.1% WY-14643 or 0.5% fenofibrate (FF) ad libitum. In addition, 9-mo-old (aged) control mice (Sirt1F/F) and Sirt1ΔLiv mice were treated for 5 days with the 0.1% WY-14643 diet. To study the effects of mutant SIRT1 on long-term PPARα activation, 6- to 8-wk-old mice were fed the 0.1% WY-14643 diet for 3 mo. Serum was collected from all mice in the fed state prior to euthanization. For fasting studies, food was removed from 8-wk-old mice for either 0 or 24 h starting at the onset of the light cycle. After fasting, animals were euthanized and samples collected. For HFD studies, 8-wk-old mice were placed on a 60% HFD for 14 wk (Ser3282; Bio-Serv, Frenchtown, NJ). Body weight measurements were taken weekly. After 14 wk, glucose and insulin tolerance tests were performed. Mice were then fed the standard chow diet containing 0.1% WY-14643 diet or left on the standard control grain diet for 5 days. Body composition of control groups was measured using an EchoMRI-3in1 quantitative magnetic resonance system (EchoMRI, Houston, TX). Following WY treatment, all groups were euthanized and samples collected for analysis. Mice were housed in a temperature- and light-controlled vivarium and given food and water ad libitum. All animal studies were carried out in accordance with and the approval of the National Cancer Institute Animal Care and Use Committee.
Lipid analysis.
Serum chemistry analysis for the total cholesterol, triglycerides, and nonesterified free fatty acids (NEFA) was performed on fresh serum using Wako Clinical Diagnostics kits (Wako USA, Richmond, VA). For analysis of liver lipid content, 20 mg of frozen liver was homogenized in 200 μl of 50 mM Tris + 1% Triton-X 100 buffer and incubated for 1 h at room temperature with shaking. Two microliters of the suspension was used for analysis using the same Wako Clinical Diagnostic kits as those used for serum analysis.
Glucose and insulin tolerance tests.
Tolerance tests were performed on mice following 14 wk on HFD. For glucose tolerance [intraperitoneal glucose tolerance test (IPGTT)], mice were fasted overnight, weighed, and given an intraperitoneal injection of glucose (1 mg/kg body wt). Blood glucose levels were measured by tail bleed at 0, 15, 30, 60, 90, and 120 min using a Contour blood glucose meter (Bayer, Mishawaka, IN). Insulin tolerance tests (ITT) were performed after fasting mice for 6 h. Mice were weighed and given a 1 U/kg insulin injection. Blood glucose measurements were taken by tail bleed at 0, 15, 30, 45, 60, 90, and 120 min. GraphPad Prism 6 software (GraphPad Software, La Jolla, CA) was used to calculate the area under the curves.
RNA analysis.
Total RNA was isolated from frozen liver using the RNeasy mini kit (Qiagen, Valencia, CA). One microgram of RNA was reverse transcribed and used for quantitative RT-PCR analysis. Primers were designed for gene specificity and to cross exon-exon junctions using Primer-BLAST (National Center for Biotechnology Information). Sirt1 primers were designed to target recombinase excised exon 4. Results are normalized to actin gene expression. Values given are fold over control or relative expression value, where appropriate, calculated using the 2ΔCT quantitative PCR calculation method (24).
Western blot analysis.
Whole cell extracts were prepared from frozen livers using RIPA buffer. Fifty micrograms of protein was loading on 4–12% Criterion TGX Precast Gel (Bio-Rad, Hercules, CA). Following transfer to polyvinylidene difluoride membranes, blots were blocked in 5% nonfat milk, followed by overnight incubation in primary antibody at 4°C. Primary antibodies used are α-SIRT1 (07-131; Millipore, Temecula, CA) and α/β-actin (ab8227; Abcam, Cambridge, MA). Densitometry was performed on the resulting Western data using Image J (version 1.47m) image analysis software. Density measurements for SIRT1 protein levels were normalized to β-actin expression.
Immunohistochemistry.
Liver tissue was fixed in 10% phosphate-buffered formalin for 24 h and then processed in paraffin blocks. Four-micrometer sections were used for hematoxylin and eosin staining and immunohistochemistry. A rabbit anti-human ki67 antibody (ab16667; Abcam) was used for immunohistochemical detection of proliferation. For each liver section, 10 microscope fields (×200) were counted for ki67 staining and overall hepatocyte nuclei number. Proliferation index is indicated as the average percentage of ki67-positive nuclei in a liver section for each mouse.
Statistical analysis.
All results are expressed as means ± SD. Significance was determined by t-test or one-way ANOVA with Bonferroni posttest using Prism 6.0 software (GraphPad Software,). A P value of <0.05 was considered significant.
RESULTS
PPARα influences Sirt1 gene expression.
Initial concerns over the ability of fibrates to initiate hepatomegaly and hepatocellular carcinoma in rodent models prompted extensive study into the mechanism of carcinogenicity of these compounds (16). Studies using Ppara−/− knockout mice revealed the requirement for PPARα in fibrate-induced hepatocellular carcinoma in rodents (18, 23). In an effort to understand the species-specific effects of fibrates, gene expression was assessed in livers from mice treated for 5 days with FF, a PPARα- specific agonist. Among several DNA repair-associated genes that were regulated after FF treatment (unpublished data), a 30% decrease in the expression of Sirt1 was of particular interest due to a previous report of SIRT1-mediated regulation of PPARα (27). To further assess the repression of Sirt1 expression, WT and Ppara−/− mice were fed a diet containing 0.5% FF, a clinically relevant albeit less potent PPARα agonist, or 0.1% WY-14643 (WY), a highly potent PPARα agonist, for 5 days. Sirt1 expression was reduced following both FF and WY treatment (Fig. 1A). The known PPARα target gene Acox1 was included as a positive control to confirm the PPARα-dependent regulation by WY and FF. Western blot analysis confirmed reduction in SIRT1 protein (110 kDa) after both FF and WY treatment (Fig. 1B). Densitometry analysis indicates a 40–60% decrease in SIRT1 levels when normalized to β-actin expression. A liver sample from a 24-h fasted mouse was used as a positive control for SIRT1 upregulation, and Sirt1ΔLiv liver was used to confirm antibody specificity. Interestingly, Sirt1 expression was also reduced in Ppara−/− mice. Ppara disruption abrogated the FF reduction in Sirt1 expression, whereas WY treatment increased Sirt1 expression slightly in Ppara−/− mice.
Fig. 1.
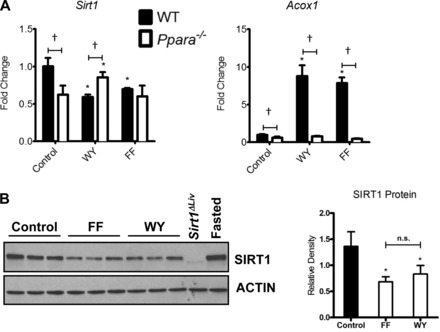
Peroxisome proliferator-activated receptor-α (PPARα) influences expression of sirtuin 1 (SIRT1). A: RT-quantitative PCR (qPCR) analysis of hepatic Sirt1 expression in wild-type (WT) and Ppara−/− mice treated for 5 days with fenofibrate (FF) or WY-14643 (WY). Expression of the known PPARα target gene Acox1 was included as a positive control. Values were normalized to β-actin and expressed as fold change over the control WT. Significant changes in expression between treated and control mice of the same genotype are indicated (*P < 0.05). Significant differences in Sirt1 expression between WT and Ppara−/− mice are indicated (†P < 0.05). B: Western blot analysis of whole cell extracts from livers of WT mice treated with FF or WY for 5 days. Extracts from fasted mice and Sirt1ΔLiv mice were used as antibody controls. Densitometry values for each SIRT1 band were normalized to β-actin expression for each lane and presented as relative density. NS, not significant.
The role of SIRT1 in PPARα activation in mice.
SIRT1 was shown to interact with PPARα and positively regulate PPARα ligand activation in primary hepatocytes (27). Given the potential reciprocal regulation between SIRT1 and PPARα, the effect of hepatic SIRT1 inactivation on PPARα activation by WY was investigated in vivo. Mice carrying the floxed exon 4 of Sirt1 (Sirt1F/F) were crossed with albumin-cre recombinase mice to generate hepatocyte-specific Sirt1 mutant mice (Sirt1ΔLiv) that express a truncated form of SIRT1 lacking the deacetylase domain. Sirt1ΔLiv mice have elevated serum cholesterol levels compared with the floxed control mice (Fig. 2A), in accord with findings in a similarly generated hepatocyte-specific Sirt1 knockout mouse (27). Young (6–8 wk old) male mice were treated for 5 days with the PPARα agonists WY and FF. PPARα activation is known to cause hepatomegaly in mice, as evidenced by the increased liver/body mass ratio (Fig. 2A). There was no significant difference in liver body/mass ratio between floxed and Sirt1ΔLiv mice after WY treatment. Sirt1ΔLiv mice treated with FF did have a slightly lower liver/body mass ratio. PPARα ligand treatment resulted in the expected lowering of serum lipid levels, and there was no difference between genotypes in this response (Fig. 2A). Liver lipid content was also measured, and no difference was observed between genotypes in regard to hepatic fibrate response (Fig. 2B).
Fig. 2.
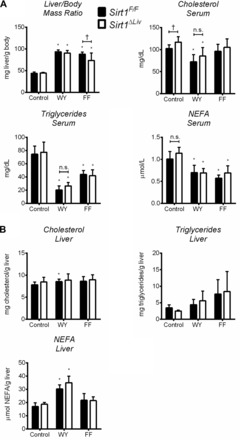
SIRT1 does not influence physiological response to short-term PPARα agonist treatment. WT (Sirt1F/F) and liver-specific Sirt1 mutant (Sirt1ΔLiv) mice were treated with WY or FF for 5 days. A: liver/body mass ratio, as well as serum triglyceride, cholesterol, and nonesterified fatty acid (NEFA) concentrations, was measured. B: hepatic concentrations of total triglycerides, cholesterol, and NEFA. Significant differences between control and WY or FF treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
Next, the impact of mutant SIRT1 on PPARα target gene expression was examined. The basal expression of Cyp4a10, Cyp4a14, and c-Myc was reduced in Sirt1ΔLiv mice (Fig. 3). However, despite this impact on basal gene expression, there was no influence of SIRT1 mutation on WY or FF induction of mRNAs encoded by the PPARα target genes Acox, Cyp4a10, Cyp4a14, Cpt2, Mcad, L-Fabp, and Pdk4 assessed in addition to the indirect target c-Myc. In addition, Foxo3 and Ppargc1a (PGC-1α) expression was equally downregulated after fibrate treatment in both Sirt1F/F and Sirt1ΔLiv mice.
Fig. 3.
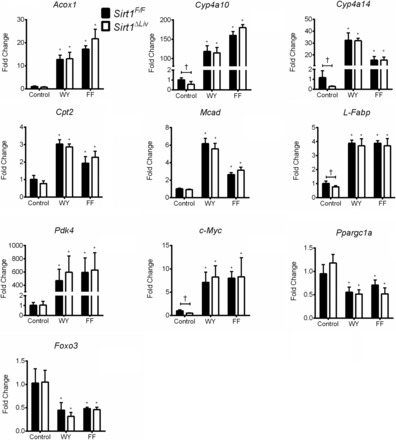
Expression of PPARα target genes after short-term PPARα agonist treatment. Sirt1F/F and Sirt1ΔLiv mice were treated with WY or fenofibrate for 5 days. RT-qPCR analysis was performed on liver samples, and values were normalized to β-actin and expressed as fold change from Sirt1F/F mice. Significant differences between control and WY or FF treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
SIRT1 function is closely associated with the aging process in yeast, C. elegans, Drosophila, and mammals (12). Additionally, the effects of Sirt1 mutations or genetic knockouts on lipid metabolism were reported to be more pronounced when older mice (>6 mo of age) were studied (28, 35). To examine the possibility that the role of hepatic SIRT1 in aging could influence PPARα activation, 9-mo-old Sirt1F/F and Sirt1ΔLiv mice were treated for 5 days with WY. Aged Sirt1ΔLiv mice have a significantly more pronounced increase in serum cholesterol and triglyceride concentrations (Fig. 4A). However, in agreement with the results in young mice, there was no significant difference between Sirt1F/F and Sirt1ΔLiv mice in liver/body mass ratio or serum lipid levels after WY treatment (Fig. 4A). Notably, there was an increase in liver NEFA levels in the Sirt1ΔLiv mice treated with WY compared with their Sirt1F/F counterparts (Fig. 4B). PPARα target gene expression was also analyzed in the aged mice (data not shown). Although no differences in basal expression between the genotypes were observed in the aged mice, there was a statistically significant reduction in the induction of Cyp4a14 from 35-fold in Sirt1F/F to 25-fold in Sirt1ΔLiv.
Fig. 4.
Aged Sirt1ΔLiv mice have similar physiological responses to WY as young mice. Nine-month-old Sirt1F/F and Sirt1ΔLiv mice were treated with WY for 5 days. A: liver/body mass ratio and serum cholesterol, triglyceride, and NEFA levels. B: hepatic concentrations of triglycerides, cholesterol, and NEFA. Significant differences between control and WY-14643 treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
The role of SIRT1 in fibrate-induced hepatic proliferation.
In rodents, long-term PPARα activation leads to hepatocyte proliferation and eventually hepatocellular carcinoma in a PPARα-dependent manner (23). SIRT1 was proposed initially to be a tumor promoter due to its inhibitory effects on p53 function. However, in vivo mouse tumorigenesis models indicate that SIRT1 activity is actually tumor suppressive (21, 33, 34). Additionally, c-Myc gene expression, one component of WY-induced hepatocyte proliferation, was slightly reduced in young Sirt1ΔLiv control mice (Fig. 3) (30). To examine a potential role for SIRT1 in PPARα-mediated fibrate-induced hepatocyte proliferation, Sirt1F/F and Sirt1ΔLiv mice were treated with WY for 3 mo. As was seen with short-term WY treatment, there was no significant difference in serum or lipid chemistries between Sirt1F/F and Sirt1ΔLiv mice (Fig. 5, A and B). Similarly, induction of seven PPARα target genes and c-Myc mRNA was not affected by the SIRT1 mutation (data not shown). To assess hepatocyte proliferation, immunohistochemical staining for the proliferation marker Ki67 was performed on the livers (Fig. 6A). There was no statistically significant difference in Ki67 staining between Sirt1F/F and Sirt1ΔLiv mice treated with WY for 3 mo (Fig. 6B).
Fig. 5.
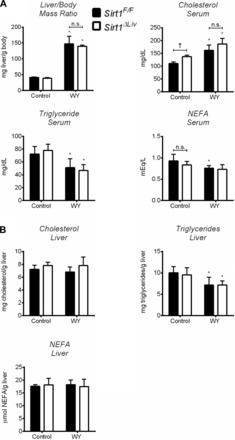
SIRT1 does not influence physiological response to long-term PPARα agonist treatment. Sirt1F/F and Sirt1ΔLiv mice were treated with WY for 3 mo. A: liver/body mass ratio and serum triglyceride, cholesterol, and NEFA concentrations. B: hepatic concentrations of total triglycerides, cholesterol, and NEFAs. Significant differences between control and WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
Fig. 6.
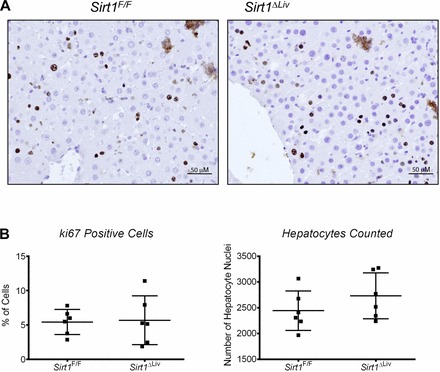
Hepatocyte proliferation after long-term PPARα treatment. Sirt1F/F and Sirt1ΔLiv mice were treated with WY for 3 mo. Immunohistochemistry was performed on formalin-fixed paraffin-embedded liver tissue sections for ki67, a marker of proliferation. A: representative images (×200) for ki67 staining in Sirt1F/F and Sirt1ΔLiv mice. B: %Ki67-positive cells counted for each mouse. No statistically significant difference in %ki67-positive cells or total nuclei was observed.
Complete loss of hepatic SIRT1 reproduces results obtained with the deacetylase-deficient SIRT1 mice.
Although Sirt1ΔLiv mice have slightly reduced basal expression of several PPARα target genes, there was no effect of this SIRT1 mutation on fibrate-induced gene expression. To examine the possibility that domains other than the deacetylase domain of SIRT1 are critical for ligand-activated PPARα function, a second hepatocyte-specific Sirt1 knockout mouse strain was tested. The Sirt1ΔLiv;Deng mouse is a hepatocyte-specific Sirt1 knockout in which exons 5 and 6 were disrupted, resulting in nearly complete loss of the entire SIRT1 protein (35). Basally, there was no statistically significant difference in serum lipid concentrations between 6- and 8-wk-old Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice (Fig. 7A). Serum triglyceride levels are reported to be elevated at 9 mo of age, but earlier time points were not reported (35). However, liver triglyceride content was elevated in Sirt1ΔLiv;Deng mice at 6–8 wk of age, consistent with the previous report (Fig. 7B). Following 5 days of WY treatment, there was no difference in serum triglyceride and cholesterol levels between Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice (Fig. 7A). Serum NEFA levels were slightly elevated in Sirt1ΔLiv;Deng mice after WY treatment compared with control floxed mice. There was no difference in liver lipid accumulation after WY treatment between Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice (Fig. 7B). Finally, PPARα target gene expression was assessed. Unlike Sirt1ΔLiv mice, there was no statistically significant difference in basal expression of PPARα target genes between Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice (Fig. 8). Only Pdk4 induction was reduced in Sirt1ΔLiv;Deng mice compared with flox after 5 days of WY treatment. The difference in basal regulation of PPARα target genes may be related to differences in strain background between Sirt1ΔLiv mice on a C57BL6 background and the Sirt1ΔLiv;Deng mice on a 129 background.
Fig. 7.
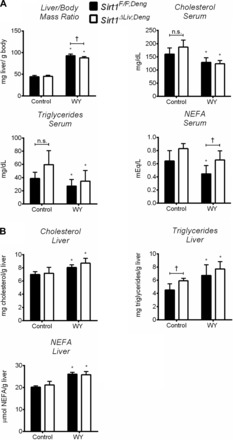
Complete deletion of SIRT1 protein does not influence physiological response to short-term PPARα agonist treatment. WT (Sirt1F/F;Deng) and liver-specific Sirt1 knockout (Sirt1ΔLiv;Deng) mice were treated with WY for 5 days. A: liver/body mass ratio and serum triglyceride, cholesterol, and NEFA concentrations. B: hepatic concentrations of total triglycerides, cholesterol, and NEFA. Significant differences between control and WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice are indicated (†P < 0.05).
Fig. 8.
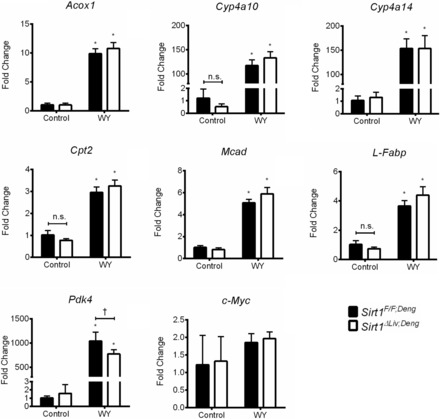
No change in induction of PPARα target genes in the absence of SIRT1. Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice were treated with WY for 5 days. RT-qPCR analysis was performed on liver samples, and values were normalized to β-actin and expressed as fold change from Sirt1F/F mice. Significant differences between control and WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F;Deng and Sirt1ΔLiv;Deng mice are indicated (†P < 0.05).
SIRT1 deacetylase activity and PPARα activation during fasting.
To address the relationship between SIRT1 and PPARα in a physiologically relevant setting, Sirt1F/F and Sirt1ΔLiv mice were fasted for 24 h. Serum and liver lipid levels were analyzed following fasting (Fig. 9, A and B, respectively). As expected, there was a significant increase in liver triglycerides in both groups. Liver cholesterol and NEFA levels, as well as serum triglycerides, did not change in response to fasting in either group. There was a slight but significant increase in liver/body mass ratio in WT but not Sirt1 mutants. There was also a slight increase in serum cholesterol levels in SIRT1 mutants and an increase in serum NEFA for both groups after fasting. NEFA levels were shown previously to increase after fasting, and NEFA concentrations are elevated further in the absence of PPARα activity (15). If SIRT1 was responsible for regulating PPARα activity, then the lack of functional SIRT1 would be expected to further contribute to elevated serum NEFAs. A number of known PPARα target genes were positively regulated after fasting, including Acox1, Cpt2, Mcad, L-Fabp, Pdk1, and cMyc, confirming PPARα activation in response to fasting (Fig. 10). However, there was no discernable difference between Sirt1F/F and Sirt1ΔLiv expression levels. Cyp4a10 and Cyp4a14 were also upregulated in response to fasting, and in accord with our previous observations, basal levels were significantly lower in SIRT1 mutant animals. Furthermore, although both were upregulated in response to fasting conditions, the level of upregulation was slightly lower than in WT animals. Foxo3 and Ppargc1a expression was equally downregulated after fasting in both Sirt1F/F and Sirt1ΔLiv mice (Fig. 10). Foxo4 expression was also downregulated after fasting, but only in Sirt1ΔLiv (data not shown).
Fig. 9.
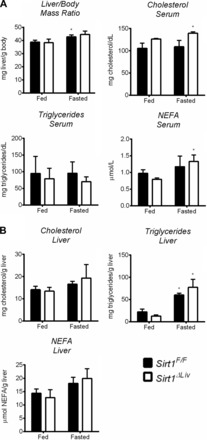
Deacetylase activity of SIRT1 has minor influence on physiological response to fasting. WT (Sirt1F/F) and liver-specific Sirt1 mutant (Sirt1ΔLiv) mice were fasted for 24 h. A: liver/body mass ratio was measured as well as serum triglyceride, cholesterol, and NEFA concentrations. B: hepatic concentrations of total triglycerides, cholesterol, and NEFA. Significant differences between fed and fasted mice are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
Fig. 10.
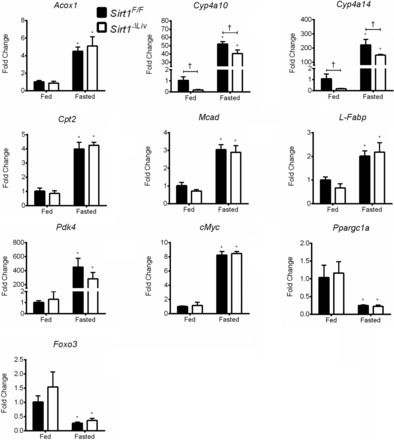
Expression of PPARα target genes after fasting. Sirt1F/F and Sirt1ΔLiv mice were fasted for 24 h. RT-qPCR analysis was performed on liver samples, and values were normalized to β-actin and expressed as fold change from Sirt1F/F mice. Significant differences between control and WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
Effects of SIRT1 on PPARα activation during HFD-induced obesity.
SIRT1 expression is protective against metabolic syndrome. Transgenic mice overexpressing SIRT1 are resistant to HFD-induced metabolic damage (25). Conversely, studies have shown that mice lacking SIRT1 are more susceptible to obesity and insulin resistance (20). PPARα has also been shown to play an important role during chronic high-fat feeding. Mice on HFD have increased expression of PPARα and PPARα-dependent increases in target genes involved in fatty acid oxidation (22).
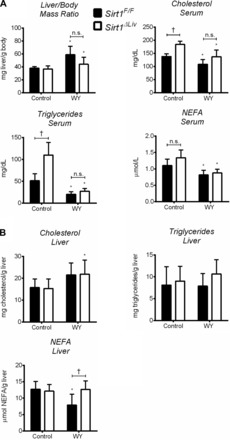
To further elucidate the relationship between SIRT1 and PPARα, Sirt1F/F and Sirt1ΔLiv mice were maintained on a HFD containing 60% fat for 14 wk and then switched to either the WY-containing diet or the control grain diet for 5 days. Both Sirt1F/F and Sirt1ΔLiv gained significant weight over the course of the study (Fig. 11A). However, there was not a statistically significant difference in percent weight gain between the two groups (Fig. 11B). Sirt1ΔLiv gained significantly more mass than WT animals, and EchoMRI measurements of control vs. SIRT1 mutants revealed that the difference in mass was the result of increased fat (Fig. 11C). Lean mass of both groups was not significantly changed. IPGTT and ITT were performed on each group of animals after 14 wk on HFD (Fig. 11, D and E). Although there was not a statistically significant difference between groups for either test, it appeared as though the mutant animals trended toward being glucose insensitive and insulin resistant, which is similar to previously published data (27).
Fig. 11.
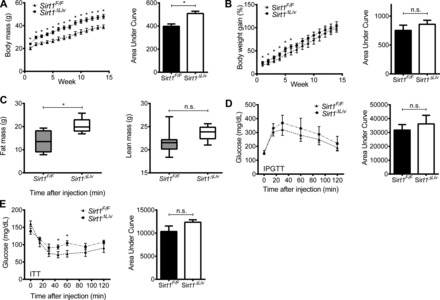
Effect of hepatic SIRT1 function on high-fat diet (HFD)-induced weight gain and glucose homeostasis. WT (Sirt1F/F) and liver-specific Sirt1 mutant (Sirt1ΔLiv) mice were fed a 60% HFD for 14 wk. A and B: body mass (A) and %weight gain (B) were measured weekly for Sirt1F/F and Sirt1ΔLiv mice on HFD. C: body composition was quantified using EchoMRI at the end of the study. D and E: glucose (IPGTT; D) and insulin tolerance tests (ITT; E) after 14 wk on HFD. Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (*P < 0.05).
After 14 wk on HFD, Sirt1F/F and Sirt1ΔLiv animals were switched to the WY diet or left on the HFD diet for 5 days. Sirt1ΔLiv exhibited significantly higher serum cholesterol levels than WT animals after WY treatment (Fig. 12A). Differences in serum cholesterol levels have been reported previously in conventional SIRT1 heterozygous knockout (Sirt1+/−) animals on HFD (36). Serum triglyceride and NEFA levels did not change between groups. HFD serum and liver triglyceride levels were three- and 10-fold higher, respectively, than the concentrations we observed previously in mice on regular diet (Fig. 2, A and B). Surprisingly, we did not see a significant drop in serum triglycerides after WY treatment. It is possible that prolonged WY treatment is required for effective reduction of triglyceride levels in obese mice. There was also no discernible difference in liver cholesterol or triglyceride concentrations (Fig. 12B). WY treatment did cause a significant increase in liver NEFA levels in both WT and mutant animals. Gene expression analysis showed a significant increase in PPARα target gene expression, although fold inductions were considerably lower compared with young mice fed a regular chow diet (Figs. 3 and 13). Acox1, Cyp4a14, and L-Fabp expression was significantly elevated in WY-treated Sirt1ΔLiv animals. Cyp4a10 and cMyc expression was increased in both Sirt1F/F and Sirt1ΔLiv mice after WY treatment. Cpt2 expression was diminished slightly in WY-treated Sirt1ΔLiv compared with Sirt1F/F mice. The levels of Mcad, Pdk4, Foxo3, and Ppargc1 did not respond to WY treatment after HFD (Fig. 13).
Fig. 12.
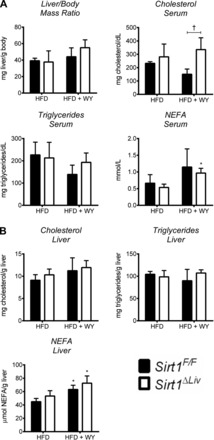
Deacetylase activity of SIRT1 has minor influence on physiological response to PPARα activation after 14 wk on HFD. WT (Sirt1F/F) and liver-specific Sirt1 mutant (Sirt1ΔLiv) mice were fed 60% HFD for 14 wk and then switched to WY diet or control diet for 5 days. A: liver/body mass ratio was measured as well as serum triglyceride, cholesterol, and NEFA concentrations. B: hepatic concentrations of total triglycerides, cholesterol, and NEFA. Significant differences between HFD and HFD with WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
Fig. 13.
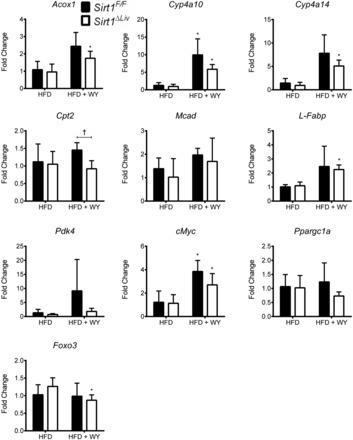
Expression of PPARα target and SIRT1-associated genes after fasting. Sirt1F/F and Sirt1ΔLiv mice were fed 60% HFD for 14 wk and then switched to WY diet or HFD for 5 days. RT-qPCR analysis was performed on liver samples, and values were normalized to β-actin and expressed as fold change from Sirt1F/F mice. Significant differences between HFD or HFD with WY treatment are indicated (*P < 0.05). Significant differences between Sirt1F/F and Sirt1ΔLiv mice are indicated (†P < 0.05).
DISCUSSION
The fibrate class of drugs has been used clinically for more than 40 years to treat hypertriglyceridemia. These drugs leverage PPARα regulation of key lipid transport and oxidation pathways to lower circulating triglyceride levels and improve HDL cholesterol numbers. There is a complex, interactive network of factors that interact with PPARα to achieve these physiological results, including the retinoid X receptor, various histone acetylases (i.e., p160/SRC and CBP/p300), and histone deacetylases (i.e., NCoR and SMRT) as well as coactivators like PPARGC1A (PGC-1α). Given the potential regulation of Sirt1 by PPARα identified in this work as well as a previous report of SIRT1 regulation of PPARα function, it seemed likely that loss of SIRT1 deacetylase activity would have a significant impact on the physiological response to PPARα activation in vivo (27). To test this hypothesis, hepatocyte-specific SIRT1 mutant (Sirt1ΔLiv) mice were generated, in which the deacetylase domain of SIRT1 was deleted. These mice recapitulated the increased serum cholesterol levels and lowered the basal expression of several PPARα target genes seen in a previously generated hepatocyte-specific SIRT1 mutant mouse line (27). However, despite various treatment conditions, the loss of SIRT1 had no significant impact on the physiological response to PPARα activation via fasting or fibrate treatment during control or HFD-fed conditions in mice, as revealed by serum and liver lipid levels. However, there was a slight reduction in the liver/body mass ratio after fibrate treatment in Sirt1ΔLiv mice compared with Sirt1F/F mice, suggesting an effect of SIRT1 on fibrate-induced hepatomegaly. In rodents, prolonged PPARα activation induces hepatomegaly, hepatocyte proliferation, and eventually hepatocarcinoma. Furthermore, SIRT1 deacetylates p53, resulting in lowered p53 activity, which plays a role in cell proliferation. To assess the potential impact of SIRT1 mutation on fibrate-induced hepatocyte proliferation, mice were treated for 3 mo with WY. However, after prolonged PPARα activation, no difference was observed between genotypes with regard to ki67 staining, a marker of cell proliferation.
Further confirming the physiological data, there was no significant effect of SIRT1 mutation on the induction of PPARα target genes, although basal expression of the Cyp4a genes was generally repressed in Sirt1ΔLiv mice at 6–8 wk, 3 mo, and 9 mo of age. Moreover, Cyp4a10 and Cyp4a14 induction after fasting was reduced in Sirt1ΔLiv mice compared with floxed control animals. The consistency in repression of Cyp4a genes may be an indication of a distinct PPARα transcriptional complex present on these promoters compared with other target genes such as Pdk4 and Acox1. Interestingly, 5-aminoimidazole-4-carboxyamide-ribonucleoside (AICAR), an adenosine analog activator of AMP-dependent protein kinase (AMPK), was shown to induce Cyp4a10 and Cyp4a14 gene expression independent of AMPK activation through indirect activation of PPARα in primary hepatocytes (2). Although that mechanism of Cyp4a induction was shown to be AMPK independent, AICAR activation of AMPK leads to increased NAD+, which in turn activates SIRT1 activity, providing an additional potential mechanism of Cyp4a gene regulation that may exist in vivo (3).
The possibility that SIRT1 plays a structural role for anchoring or recruiting of other factors to the PPARα transcriptional complex was investigated using hepatocyte-specific knockout mice lacking the entire SIRT1 protein (Sirt1ΔLiv;Deng). Treatment of Sirt1ΔLiv;Deng mice for 5 days with WY resulted in similar findings, as obtained with the SIRT1 deacetylase mutant mice generated in this study (Sirt1ΔLiv). WY-treated Sirt1ΔLiv;Deng exhibited an increase of PPARα target gene expression and liver/body mass ratios as well as a decrease in serum triglycerides, which confirms PPARα activation. There were some slight variations between Sirt1ΔLiv;Deng and Sirt1F/F mice that may be attributed to differences in the background strains. The background strain of the mice (e.g., 129 vs. C57B6) is the most likely contributor to minor differences in serum and hepatic lipid levels and gene expression responses between the two Sirt1 knockout mouse lines. Indeed, it has been shown that 129 mice absorb cholesterol more efficiently than C57B6 mice and that C57B6 mice have higher activity levels of SREBP-1c and SCD-1 (1, 13). The lack of Cyp4a10 and Cyp4a14 basal downregulation in Sirt1ΔLiv;Deng suggests that a factor important for Cyp4a10 and Cyp4a14 basal expression is differentially expressed between 129 and C57B6 mice. SREBP-1c is a tempting candidate, as it is a direct target of SIRT1 deacetylase activity (26); however, SIRT1 inactivation leads to increased SREBP-1c activity and thus is not likely to be responsible for the loss of Cyp4a10 and Cyp4a14 in Sirt1ΔLiv mice. Thus, the basal regulation of Cyp4a10 and Cyp4a14 involves unique factors not associated with other PPARα target genes.
SIRT1 activity is known to play an important role in energy homeostasis, and overexpression protects against metabolic syndrome. Conversely, studies have shown that SIRT1 knockout mice are more susceptible to HFD-induced obesity. PPARα is a major regulator of hepatic lipid metabolism. As such, PPARα expression and activation are significantly increased under a HFD (22). To elucidate the relationship between SIRT1 and PPARα in a pathophysiological setting, Sirt1F/F and Sirt1ΔLiv mice were given a HFD containing 60% fat for 14 wk and then transferred to either a WY-containing diet or a standard control grain diet for 5 days. Results indicated that SIRT1 mutant mice gained significantly more weight than the wild-type animals. MRI results revealed that the difference in weight was due to an increase in fat and not lean mass. This data correspond with previous studies investigating mice lacking functional SIRT1 (27, 36). Glucose homeostasis in SIRT1 mutant mice was not significantly different from wild-type counterparts, as assessed by glucose and insulin tolerance tests. There was a trend suggesting glucose intolerance and insulin resistance, but the results failed to reach statistical significance. A corresponding trend toward glucose intolerance was observed in a previous study using a similar SIRT1 mouse model after 11 wk on HFD (27). In addition, that study did not observe any differences in serum or liver triglycerides and NEFAs between wild-type and liver-specific SIRT1 knockout mice on HFD. Lipid levels in serum and liver were mostly unaffected by the absence of SIRT1 activity in the present study. Serum cholesterol levels were significantly elevated in Sirt1ΔLiv mice compared with Sirt1F/F after 5 days of WY treatment, whereas the previous study noted a significant increase in liver but not serum cholesterol (27). PPARα activators are known to reduce adiposity in rodent models of diabetes and obesity by increasing fatty acid oxidation and lipolysis (9, 17). Because HFD increased fat mass, particularly in the Sirt1ΔLiv mice, the liberation of lipids from the adipose tissue may confound the serum lipid profiles. In accord with the above fibrate and fasting studies, SIRT1 activity did not appear to significantly affect PPARα-mediated gene regulation after HFD or HFD with WY treatment. HFD increased the basal expression of most of the PPARα target genes, thereby reducing the fold change induction with WY treatment and possibly obscuring the previously observed reduction in basal gene expression of Cyp4a10 and Cyp4a14. The lipid profile and gene expression results may be confounded by the change in diet from HFD to a lower-fat grain diet containing WY. However, given the numerous conditions tested in this study, the lack of an effect of SIRT1 mutation on PPARα activation is not related to the change in the basal diet during the HFD study.
In contrast to the minimal impact SIRT1 has on ligand-activated PPARα transcriptional function, we demonstrate a reduction in SIRT1 mRNA and protein levels after fibrate treatment. In times of energy stress, as occurs during fasting, intracellular nicotinamide adenine dinucleotide levels (NAD+) rise. Because SIRT1 deacetylase activity is dependent on NAD+ availability, increased NAD+ levels correlate with increased SIRT1 activity (11). In addition to regulating enzymatic activity, NAD+ levels serve as a transcriptional signal. Indeed, Sirt1 gene expression was shown to be negatively regulated by HIC1/CTBP in conjunction with lowered NAD+ intracellular levels (5). However, activation of PPARα by fibrates leads to increased hepatic NAD+ levels as well as increased urinary excretion of NAD+ metabolites (31). This suggests that PPARα activation would lead to increased SIRT1 activity rather than repression of Sirt1 transcription.
Adding to the perplexing nature of PPARα-mediated regulation of Sirt1 expression was the finding that the basal level of Sirt1 mRNA was also repressed in Ppara−/− mice (Fig. 1A). Interestingly, studies have reported an increase in SIRT1 and SIRT3 protein levels after fasting, another situation where PPARα activation is expected to occur, underscoring the complexity of these regulatory networks (4). Given the contrasting data between synthetically activated PPARα and genetically deleted PPARα expression on SIRT1 expression, the question of off-target effects of fibrates arises. Indeed, numerous reports on the PPARα-independent effects of fenofibrate on nonhepatocyte cells utilizing the Ppara−/− mice exist (6, 29, 38). Sirt1 expression increased slightly in Ppara−/− mice treated with WY, indicating that WY may impact Sirt1 expression in a PPARα-independent manner. In contrast, the effects of fenofibrate on Sirt1 expression were completely abolished in Ppara−/− mice, suggesting that fenofibrate's actions on SIRT1 expression are completely dependent on PPARα.
In summary, this study revealed that PPARα regulates Sirt1 expression. This implicates PPARα in the posttranslational regulation of a myriad of factors deacetylated by SIRT1, including histones 1–3, PPARGC1A, FOXO1–4, liver X receptor, sterol regulatory element binding factor-1, and PPARα, adding to the already significant influence that PPARα activation has on lipid metabolism. A previous study showed that PPARα-mediated gene regulation in response to WY was SIRT1 dependent in primary hepatocytes (27). However, the present work does not support a major role for SIRT1 activity with regard to PPARα-mediated gene expression or lipid metabolism in vivo in response to fibrates. Although no significant impact of SIRT1 on PPARα activation by fibrates was demonstrated, there is consistent evidence to suggest that SIRT1 is involved in the basal expression of Cyp4a genes.
GRANTS
This work was funded by the intramural research program at the National Cancer Institute (J. A. Bonzo, C. Brocker, C. Jiang, and F. J. Gonzalez) and the National Institute of Diabetes and Digestive and Kidney Diseases (R. -H. Wang and C. -X. Deng). J. A. Bonzo and C. Brocker were additionally supported by the Postdoctoral Research Associate program through the National Institute of General Medical Sciences.
DISCLOSURES
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
J.A.B., C.-X.D., and F.J.G. conception and design of research; J.A.B., C.B., C.J., and R.-H.W. performed experiments; J.A.B. analyzed data; J.A.B. and F.J.G. interpreted results of experiments; J.A.B. prepared figures; J.A.B. drafted manuscript; J.A.B., C.B., and F.J.G. edited and revised manuscript; J.A.B., C.B., C.J., R.-H.W., C.-X.D., and F.J.G. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for J. A. Bonzo: Primary & Stem Cell Systems, Life Technologies, Frederick, MD.
REFERENCES
- 1.Biddinger SB, Almind K, Miyazaki M, Kokkotou E, Ntambi JM, Kahn CR. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes 54: 1314–1323, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bumpus NN, Johnson EF. 5-Aminoimidazole-4-carboxyamide-ribonucleoside (AICAR)-stimulated hepatic expression of Cyp4a10, Cyp4a14, Cyp4a31, and Other peroxisome proliferator-activated receptor α-responsive mouse genes Is AICAR 5′-monophosphate-dependent and AMP-activated protein kinase-independent. J Pharmacol Exp Ther 339: 886–895, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caton PW, Holness MJ, Bishop-Bailey D, Sugden MC. PPARalpha-LXR as a novel metabolostatic signalling axis in skeletal muscle that acts to optimize substrate selection in response to nutrient status. Biochem J 437: 521–530, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123: 437–448, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Duhaney TA, Cui L, Rude MK, Lebrasseur NK, Ngoy S, De Silva DS, Siwik DA, Liao R, Sam F. Peroxisome proliferator-activated receptor alpha-independent actions of fenofibrate exacerbates left ventricular dilation and fibrosis in chronic pressure overload. Hypertension 49: 1084–1094, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA 94: 4312–4317, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guarente L. Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol 76: 81–90, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Guzmán M, Lo Verme J, Fu J, Oveisi F, Blázquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-alpha). J Biol Chem 279: 27849–27854, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S, Shimeno H, Soeda S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol Cell Biochem 339: 285–292, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jolley CD, Dietschy JM, Turley SD. Genetic differences in cholesterol absorption in 129/Sv and C57BL/6 mice: effect on cholesterol responsiveness. Am J Physiol Gastrointest Liver Physiol 276: G1117–G1124, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13: 2570–2580, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 103: 1489–1498, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol 30: 894–899, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JY, Hashizaki H, Goto T, Sakamoto T, Takahashi N, Kawada T. Activation of peroxisome proliferator-activated receptor-alpha enhances fatty acid oxidation in human adipocytes. Biochem Biophys Res Commun 407: 818–822, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15: 3012–3022, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J Clin Invest 116: 571–580, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol 5: 367–373, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107: 137–148, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Patsouris D, Reddy JK, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 147: 1508–1516, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 18: 2029–2033, 1997 [DOI] [PubMed] [Google Scholar]
- 24.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA 105: 9793–9798, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, Kemper JK. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 285: 33959–33970, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9: 327–338, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purushotham A, Xu Q, Lu J, Foley JF, Yan X, Kim DH, Kemper JK, Li X. Hepatic deletion of SIRT1 decreases hepatocyte nuclear factor 1α/farnesoid X receptor signaling and induces formation of cholesterol gallstones in mice. Mol Cell Biol 32: 1226–1236, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selim E, Frkanec JT, Cunard R. Fibrates upregulate TRB3 in lymphocytes independent of PPAR alpha by augmenting CCAAT/enhancer-binding protein beta (C/EBP beta) expression. Mol Immunol 44: 1218–1229, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol 27: 4238–4247, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin M, Ohnishi M, Iguchi S, Sano K, Umezawa C. Peroxisome-proliferator regulates key enzymes of the tryptophan-NAD+ pathway. Toxicol Appl Pharmacol 158: 71–80, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Vaziri H, Dessain SK, Eaton EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-Dependent p53 Deacetylase. Cell 107: 149–159, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng ZM, Appella E, Wang XW, Ried T, Deng CX. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14: 312–323, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Lahusen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 32: 11–20, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang RH, Li C, Deng CX. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci 6: 682–690, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu F, Gao Z, Zhang J, Rivera CA, Yin J, Weng J, Ye J. Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: a role of lipid mobilization and inflammation. Endocrinology 151: 2504–2514, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA 96: 7324–7329, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamashita M. PPARalpha/gamma-Independent Effects of PPARalpha/gamma Ligands on Cysteinyl Leukotriene Production in Mast Cells. PPAR Res 2008: 293538, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]