

Abstract
Cardiac failure is associated with diminished activation of the transcription factor cyclic nucleotide regulatory element binding-protein (CREB), and heart-specific expression of a phosphorylation-deficient CREB mutant in transgenic mice [dominant negative CREB (dnCREB) mice] recapitulates the contractile phenotypes of cardiac failure (Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest 101: 2415–2426, 1998). In the present study, we demonstrated significantly elevated mortality and contractile dysfunction in female compared with male dnCREB mice. Female dnCREB mice demonstrated a 21-wk survival of only 17% compared with 67% in males (P < 0.05) and exclusively manifest decreased cardiac peroxisome proliferator-activated receptor-γ coactivator-1α and estrogen-related receptor-α content, suggesting sex-related effects on cardiac mitochondrial function. Hearts from 4-wk-old dnCREB mice of both sexes demonstrated diminished mitochondrial respiratory capacity compared with nontransgenic controls. However, by 12 wk of age, there was a significant decrease in mitochondrial density (citrate synthase activity) and deterioration of mitochondrial structure, as demonstrated by transmission electron microscopy, in female dnCREB mice, which were not found in male transgenic littermates. Subsarcolemmal mitochondria isolated from hearts of female, but not male, dnCREB mice demonstrated increased ROS accompanied by decreases in the expression/activity of the mitochondrial antioxidants MnSOD and glutathione peroxidase. These results demonstrate that heart-specific dnCREB expression results in mitochondrial respiratory dysfunction in both sexes; however, increased oxidant burden, reduced antioxidant expression, and disrupted mitochondrial structure are exacerbated by the female sex, preceding and contributing to the greater contractile morbidity and mortality. These results provide further support for the role of the CREB transcription factor in regulating mitochondrial integrity and identify a critical pathway that may contribute to sex differences in heart failure.
Keywords: oxidative stress, mitochondria, cyclic nucleotide regulatory element binding-protein, apoptosis
heart disease is frequently perceived as a disease that mainly impacts men; however, statistics do not support such a perception. Sexually divergent manifestation of heart failure (HF) has been observed in response to specific etiological factors. Of patients who survive a myocardial infarction (MI), women are more likely than men to develop HF within 6 yr (46% vs. 22%) (5). Analysis of data from the Framingham study (26) has indicated that hypertension increases the risk for developing HF relative to normotensive individuals by twofold in men but threefold in women. These observations were supported by results from Studies of Left Ventricular Dysfunction (SOLVD) Trials (31), which demonstrated that 55% of women with hypertension develop HF, whereas 39% of men with hypertension develop HF. Diabetes has been reported to be a stronger risk factor for HF in women than in men; analysis in SOLVD Trials (31) indicated that 49.3% of women but only 37.2% of men with diabetes go on to develop HF. Similarly, the Framingham Study (19) identified an eightfold increase incidence of HF in women with diabetes versus a fourfold increase in men with diabetes.
Morbidity and mortality from HF also demonstrate significant sexual dimorphism. Women who develop HF are more likely to remain symptomatic and to be more functionally limited (16, 24, 30). Women experienced less improvement 1 yr after hospitalization for HF than men (10), which may, in part, be accounted for by less-aggressive treatment of their disease (11, 12, 29, 34). Mortality from HF concomitant with diabetes is worse in women, who show a 70% increase in mortality (19). Overall, 62.4% of HF deaths since the year 2000 have occurred in women (1). Thus, while women appear to have a resilient coronary vascular phenotype, they also appear to have a “susceptible” cardiac phenotype, which is revealed subsequent to diabetes, hypertension, or MI.
Work in our laboratory has focused on the impact of the loss of signaling to the transcription factor cyclic nucleotide regulatory element-binding protein (CREB) on the cardiac failure phenotype. The important contribution of loss of CREB function, or more accurately the diminished expression of CREB-dependent genes, to the failure phenotype in humans has been supported by recent work involving microarray analysis of gene expression in the failing human heart (32). This study (32) demonstrated that diminished expression of gene targets for peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α; whose expression responds to estrogen in the hearts of rodents) and estrogen-related receptor-α (ERRα), each a primary regulator of metabolic and mitochondrial phenotypes and shown by us in rodent models to be regulated by CREB (37), have been identified as a “signature” of the failing human heart.
Our work demonstrates that loss of CREB function occurs early in the etiology of HF in a plethora of rodent models (37). The importance of CREB activity to healthy growth of the myocardium in the face of increasing energy demands has been supported by work in a mouse model expressing a phosphorylation-deficient (Ser133-to-Ala133 mutation) dominant negative (dn)CREB in a heart-specific manner (15). These transgenic dnCREB animals begin to develop dilated cardiomyopathy soon after puberty, which affects both the right ventricle and left ventricle (LV), leading to 40% mixed-sex mortality within the first 20 wk of life (15). Unlike other rodent models of cardiomyopathy studied by our group and others (37), exercise fails to improve either cardiac function or survival in the dnCREB transgenic mouse (33), implying that the ability to activate CREB function is important in exercise-induced improvements in the cardiac phenotype. The observation that dnCREB mice fail to induce physiological remodeling with exercise training is suggestive that CREB is important for physiological remodeling.
Experiments included in this report, using the dnCREB transgenic mouse model, revealed a previously unreported divergence in survival between male and female dnCREB transgenic mice. While the expression of the dnCREB gene in cardiac myocytes recapitulated the apoptotic phenotype we have previously reported in other models of HF (37) in a sex-independent manner, surprisingly, female dnCREB mice demonstrated an exaggerated susceptibility to the deleterious effects of CREB dysfunction, which specifically manifested as exacerbated mitochondrial oxidant burden and damage, diminished pump function, and subsequent decreased longevity.
MATERIALS AND METHODS
Materials.
Rabbit polyclonal antisera for the detection of Bax (Cell Signaling), active caspase-3 (BioMol), active caspase-9 (Cell Signaling), MnSOD (Upstate Cell Signaling Solutions), glutathione peroxidase-1 (GPx-1; Abcam), and uncoupling protein-3 (UCP-3; CalBiochem) were all used. Additional antisera obtained and used for detection included mouse monoclonal β-actin (Sigma Chemical), Bcl-2 (BD Transduction Laboratories), cytochrome oxidase-IV (Abcam), and cytochrome c (BD Biosciences). The anti-nuclear respiratory factor-1 (NRF-1) rabbit polyclonal antibody was the generous gift of Dr. Richard Scarpulla (Northwestern University of Medicine). All chemicals used for mitochondrial isolation were from Sigma Chemical.
Animals.
The use of animals, as well as all of the experimental interventions used in this study, received prior approval from the Institutional Animal Care and Use Committees (IACUC) of Denver Veterans Affairs Medical Center and the Health Sciences Center of University of Colorado (Denver, CO). The mouse models used were heterozygous, phosphorylation-deficient (Ser133 to Ala133) mutant CREB (dnCREB) transgenic mice (15, 33) and nontransgenic ICR outbred littermates. Animals were euthanized in accordance with the animal facility protocols using IACUC-approved (Denver Veterans Affairs Medical Center) procedures.
Preparation of heart tissue for Western blot analysis.
Frozen mouse hearts were pulverized in a liquid nitrogen-cooled mortar and pestle along with a frozen aliquot of 1× mammalian lysis buffer as previously described (37). Bradford assays were performed to determine the amount of protein in each sample extract. Samples were diluted to 1 μg protein/μl in 1× Laemmli sample buffer. Samples were then stored at −80°C until analysis.
Western blot analysis.
SDS-12% polyacrylamide gels were used for assessment, and 40 μg of the prepared protein samples were loaded per lane of the gel. Separated proteins were electrophoretically transferred to a polyvinylidene difluoride membrane and exposed to one or more of the antibodies listed above (see Materials). Alkaline phosphatase-conjugated secondary antibodies and CDP-Star (New England Biolabs) were used for antibody-antigen detection. Sample loading of proteins and outcomes were controlled by the assessment of β-actin content. Densitometric analysis was performed using QuantityOne software (Bio-Rad).
Caspase-3 activity assay.
Extracts were prepared from hearts of 12-wk-old dnCREB and nontransgenic littermates. Frozen heart tissue was powdered in a porcelain mortar and pestle cooled to the temperature of liquid nitrogen along with 450 μl of frozen extraction buffer (20 mM HEPES, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 1 mM EDTA; pH 7.5) and allowed to thaw on ice. Samples were homogenized using a Polytron tissue disruptor and centrifuged, and the supernatant (100 μg protein/assay) was used for the assessment of caspase-3/7 activity using the reagents and methods supplied in the Caspase Glo 3/7 Kit from Promega (Madison, WI). Results were expressed as relative luminescent units per 100 μg protein per hour.
Mitochondrial isolation.
Cardiac mitochondria were isolated using the procedures of Palmer et al. (28). For each mitochondrial isolation, hearts from three to five animals in each sex/genotype were pooled. Pooled hearts were finely minced in buffer A (100 mM KCl, 50 mM MOPS, 5.0 mM MgSO4, 1.0 mM EGTA, and 1.0 mM ATP; pH 7.4), and the minced tissue was transferred into fresh buffer A and homogenized in a Polytron for 3 s. The homogenate was centrifuged at 580 g at 4°C for 10 min. The supernatant was decanted for subsarcolemmal (SSL) mitochondrial isolation, and the pellet was resuspended in buffer A. The resuspended pellet was spun a second time at 580 g, and the decanted supernatant was combined with the initial supernatant for SSL mitochondrial isolation. The pellet was resuspended in buffer A supplemented with trypsin (4 mg/ml) and incubated by shaking on ice for 7 min, and an equivalent volume of buffer A containing BSA (2 mg/ml) was added to the pellet digestion to neutralize the trypsin. The digested pellet was centrifuged at 580 g for 10 min at 4°C, and the supernatant containing the interfibrillar (IF) mitochondria was decanted. Both SSL- and IF-containing supernatants were spun at 3,000 g for 10 min at 4°C, and the supernatants were discarded. Pellets were resuspended in buffer A and BSA and recentrifuged at 3,000 g for 10 min at 4°C. Pellets were resuspended in a buffer containing 100 mM KCl, 50 mM MOPS, and 0.5 mM EGTA (KME buffer) and centrifuged at 3,000 g for 10 min at 4°C to generate purified mitochondrial preparations. The mitochondria were resuspended in KME buffer, and the protein concentration of the mitochondrial suspensions was determined by a BCA protein assay (Thermo Fisher Scientific). Preparations were used immediately for the assessment of mitochondrial O2 consumption, and the remainder of the samples was frozen at −80°C for future analyses.
Assessment of MnSOD activity in whole tissue homogenates.
Whole tissue homogenates prepared from hearts of dnCREB and nontransgenic littermates were assayed for MnSOD activity using the reagents and protocols in a Superoxide Dismutase Assay Kit obtained from Cayman Chemical (Ann Arbor, MI). Assays were performed after the inclusion of 3 mM potassium cyanide in the assays, which has been shown to inhibit both Cu/Zn-SOD and extracellular SOD. The activity remaining in the presence of potassium cyanide was representative of MnSOD activity in the homogenate and was expressed as units of activity per microgram of protein.
Citrate synthase activity in whole heart extracts.
Protein extracts were prepared from hearts of 12-wk-old male and female dnCREB mice and their nontransgenic littermates, and the analysis for citrate synthase activity was performed as described in Citrate Synthase Assay Kit obtained from Sigma-Aldrich Chemicals. Samples were run in triplicate, and results are expressed as micromoles per minute per microgram of total heart protein. The sample size was four hearts in each sex/genotype group.
Assessment of mitochondrial O2 consumption.
Measurements of mitochondrial respiration were performed using a Clark-type electrode (StrathKelvin). Cardiac mitochondrial respiration (125 μg mitochondrial protein/assessment) was initiated using either malate/pyruvate or malate/palmitoyl-l-carnitine as the substrate. ADP was added to initiate state 3 respiration.
Assessment of superoxide anion and H2O2 generation by isolated mitochondria.
H2O2 production was determined via the well-established Amplex red/horseradish peroxidase method (17) using a spectrofluorometric plate reader (Molecular Devices). Assays were performed on mitochondria isolated as described above. ROS production was examined in the presence of 5 mM pyruvate/malate. Assaying intact mitochondria using this method provides a more “physiological” assessment of H2O2 release by mitochondria in the presence of endogenous matrix antioxidant enzymes (e.g., MnSOD and GPx-1) and the intact inner mitochochondrial membrane (IMM), permitting an assessment of the influence of any changes in the fatty acid composition of the IMM.
Echocardiography.
Cardiac function was assessed by two-dimensional transthoracic echocardiography using a VisualSonics Vevo 770 high-resolution ultrasound imager equipped with a 35-MHz transducer. Mice were lightly sedated with isoflurane, and heart rates were maintained above 500 beats/min. Core body temperature was maintained at 37 ± 0.5°C using a heated platform and a warming light, if necessary. Parasternal long- and short-axis B-mode videos, M-modes images (at the level of the midpapillary short axis), and pulse-wave Doppler tracings of flow across the aortic and pulmonary valves were routinely acquired. Left ventricular (LV) wall thicknesses and inner dimensions at diastole and systole were measured from the parasternal short-axis M-mode images. All measurements were made in triplicate during the expiratory phase of the respiratory cycle, and the values were averaged for each mouse. Measurements and calculations were obtained offline in a blinded mode.
Electron microscopy.
Small pieces of cardiac muscle were fixed by immersion in 2% glutaraldehyde-2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4) for 1 h at room temperature. After several brief washes in buffer, samples were postfixed in 1% osmium tetroxide-1.5% potassium ferrocyanide in cacodylate buffer for 1 h at room temperature. Samples were dehydrated through a graded series of ethanol washes, embedded in epon-araldite resin, and polymerized overnight at 60°C. Thin sections were cut and poststained with lead citrate and uranyl acetate to increase contrast. Sections were viewed and photographed using a FEI Technai G2 electron microscope at 80 kV.
Statistical analyses.
Data obtained from the experiments detailed here were compared using the most appropriate single (Students t-tests) and multivariate analyses (ANOVA) suited to the particular experiments and a priori hypotheses to detect significant differences and any interaction(s) between the selected variables. A significance level of P < 0.05 was chosen, with Bonferroni corrections for multiple comparisons where appropriate. Survival was assessed with Peto's log-rank test for multiple survival curves; P values of ≤0.05 were considered statistically significant for this analysis.
RESULTS
Expression of dnCREB transgene in the heart leads to accelerated mortality in female mice relative to male dnCREB mice.
Cohorts of dnCREB mice and nontransgenic littermates were examined for mortality over a 21-wk period, which was sufficient to allow for 50% mortality of dnCREB transgenic mice based on previously published Kaplan-Meier analyses on combined survival data in dnCREB transgenic mice of both sexes (15, 33). Segregation of our results by sex revealed an accelerated rate of mortality in female dnCREB transgenic mice, which was statistically significant compared with male dnCREB transgenic mice and nontransgenic littermates of both sexes (P ≤ 0.05; Fig. 1).
Fig. 1.
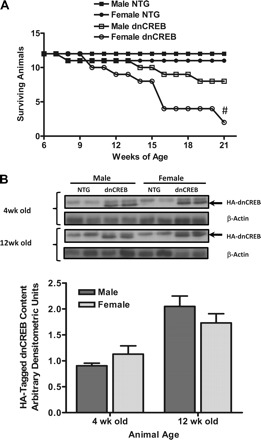
Kaplan-Meier survival curve demonstrating increased mortality in female dominant negative cyclic nucleotide regulatory element-binding protein (dnCREB) transgenic mice without differences in dnCREB transgene expression. A: a cohort of dnCREB transgenic mice and their nontransgenic (NTG) littermates (n = 12 for each sex/genotype group) were assessed over 21 wk for survival. #Statistical significance vs. all groups (P < 0.05). B: Western blot analysis hemagglutinin (HA)-tagged dnCREB protein content performed on 30 μg of total protein extract prepared from hearts of 4- and 12-wk-old male and female dnCREB transgenic mice and NTG littermates. No statistically significant differences were observed (P < 0.05).
Expression of the dnCREB transgene in the hearts of male and female mice was examined to ascertain whether differential dnCREB protein content might account for the differences in survival between male and female mice. Western blots for the hemagglutinin tag on the expressed dnCREB protein in protein extracts prepared from dnCREB transgenic mice at 4 and 12 wk of age indicated that no significant differences between the sexes were seen in dnCREB protein content when normalized to β-actin (Fig. 1B). Expression of the dnCREB transgene product was not found to significantly alter the content of endogenous CREB protein in the heart between the sexes (Supplemental Material, Supplemental Fig. 1),1 consistent with previously published results (15). The data from Western blot analysis from hearts of dnCREB mice showed significant variability and revealed a possible trend for increased endogenous CREB content in hearts of some dnCREB mice; however, the relative abundance of the dnCREB transgene product likely precludes any significant impact of a modest increase in endogenous CREB protein on CREB-mediated gene expression.
Expression of dnCREB transgene in the heart leads to contractile dysfunction in both sexes, which is exacerbated in female mice relative to male mice.
To assess whether greater contractile dysfunction might underlie the accelerated mortality of female dnCREB mice, male and female dnCREB transgenic mice were examined at 8 wk of age for contractile function and cardiac morphometry by echocardiography. Representative M-mode echocardiographs from each experimental group are shown in Fig. 2. Subsequent analysis from this imaging (Table 1) indicated that while both male and female mice showed greater end-systolic LV dimensions, decreased fractional shortening, and impaired stroke volume and cardiac output compared with nontransgenic littermate controls, the magnitude of these deleterious changes was exacerbated by female sex. Female dnCREB transgenic mice also manifested greater heart rates than nontransgenic controls and male dnCREB mice, which we believe is a compensatory response to maintain cardiac output in the face of a severely compromised stroke volume.
Fig. 2.
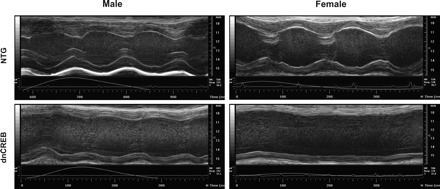
Representative images from M-mode echocardiograph analysis of cardiac structure and contractile function in hearts of 8-wk-old dnCREB mice and their NTG littermates. Quanitified outcomes from analyses of echocardiograms are shown in Table 1.
Table 1.
M-mode, B-mode, and Doppler echocardiographic data from dnCREB and NTG mice
| LV Internal Diameter at Diastole, mm | LV Internal Diameter at Systole, mm | Fractional Shortening, % | Stroke Volume, μl | Heart Rate, beats/min | Cardiac Output, ml/min | |
|---|---|---|---|---|---|---|
| Male | ||||||
| NTG | 4.26 ± 0.11 | 2.60 ± 0.15 | 39.14 ± 2.17 | 41.09 ± 2.19 | 533 ± 16 | 21.84 ± 1.19 |
| dnCREB | 4.61 ± 0.17 | 3.72 ± 0.22* | 19.40 ± 1.23* | 36.82 ± 1.06* | 541 ± 38.2 | 17.43 ± 2.54* |
| Female | ||||||
| NTG | 4.10 ± 0.10 | 2.50 ± 0.05 | 39.80 ± 1.35 | 45.25 ± 1.79 | 525 ± 10 | 23.76 ± 1.03 |
| dnCREB | 4.39 ± 0.29 | 4.02 ± 0.36*† | 8.79 ± 2.48*† | 22.45 ± 1.60*† | 659 ± 64*† | 14.61 ± 1.32*† |
Values are means ± SE; n = 4 animals/group. Animals were 8 wk of age.
NTG, nontransgenic; dnCREB, heart-specific Ser133-to-Ala133 (dominant negative) cyclic nucleotide regulatory element-binding protein; LV, left ventricular.
Statistical significance vs. same-sex NTG littermates;
statistical significance vs. opposite-sex dnCREB mice (P < 0.05).
Transgenic, heart-specific expression of dnCREB results in increases in markers of apoptosis after pubescence.
We assessed whether sex-specific changes in the rates of cardiac cell apoptosis were contributing to the increased morbidity and mortality of female dnCREB mice relative to male dnCREB mice. Hearts were removed from male and female transgenic animals expressing dnCREB in a heart-specific manner as well as nontransgenic littermates at 12 wk of age, and proteins were extracted. Western blot analysis of markers of apoptosis indicated that hearts from dnCREB transgenic animals showed decreases in the Bcl-2-to-Bax ratio and increases in active cleavage products of both caspase-9 and caspase-3 (Fig. 3, A and B), consistent with an increased risk for apoptosis in both sexes of dnCREB mice. No changes in these markers of susceptibility to apoptosis were noted before puberty (Supplemental Fig. 2).
Fig. 3.
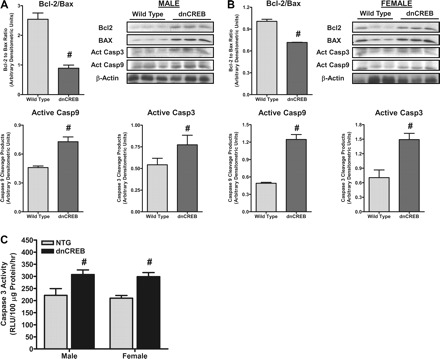
A proapoptotic phenotype manifests in hearts of 12-wk-old dnCREB transgenic mice of both sexes. A and B: Western blot analysis of protein extracts from hearts of 12-wk-old dnCREB transgenic male (A) and female (B) mice demonstrated significant changes in the Bcl-2-to-Bax ratio in both male or female dnCREB transgenic mice relative to their NTG littermates. Significant increases in the active cleavage products of caspase (Casp)-9 and caspase 3, relative to NTG littermates, were seen in hearts of dnCREB mice of both sexes. C: caspase-3 activity, as assessed in extracts from hearts of 12-wk-old dnCREB mice and their NTG littermates, indicated a significant increase in hearts of dnCREB mice of both sexes, consistent with the outcomes of markers of apoptosis shown in A. RLU, relative luminescent units. n = 6 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
Assessment of caspase-3 activity was also performed in extracts prepared from hearts of 12-wk-old dnCREB and nontransgenic littermates. The results indicated increased caspase-3 activity in hearts of both male and female dnCREB mice (Fig. 3C), consistent with the results from the Western blot analysis of markers of apoptosis.
Female-specific loss of mitochondrial protein expression with heart-specific expression of a dnCREB transgene.
As mitochondrial dysfunction has been shown to contribute to the initiation and progression of cardiac dysfunction, we examined the contents of proteins crucial for the regulation of mitochondrial gene expression in the nucleus and critical protein markers of mitochondrial function for sex-related differences. Western blot analysis of protein extracts from hearts of dnCREB and nontransgenic animals at 12 wk of age demonstrated that hearts from female dnCREB mice manifested significant reductions in PGC-1α (Fig. 4A) as well as Nrf-1 protein content, cytochrome c, and cytochrome oxidase-IV (Fig. 4C), a crucial component of complex IV of the electron transport chain involved in oxidative phosphorylation. Interestingly, hearts from male dnCREB transgenic mice failed to show reductions in PGC-1α, Nrf-1, or cytochrome oxidase-IV protein content (Fig. 4B). The sex-dependent divergence in the contents of PGC-1α and Nrf-1 were observed as early as 4 wk of age (Supplemental Fig. 3).
Fig. 4.
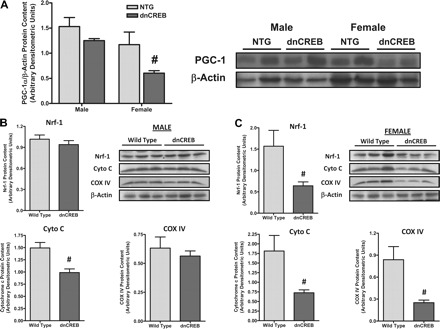
Expression of the dnCREB transgene in hearts of transgenic mice leads to female sex-specific decreases in the content of nuclear transcriptional modulators peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), nuclear respiratory factor-1 (Nrf-1), and proteins of the mitochondrial electron transport chain. A: Western blot analysis of protein extracts from male and female 12-wk-old dnCREB transgenic mice demonstrated significantly diminished PGC-1α protein content in hearts of female dnCREB transgenic mice, with no differences being observed between other sex/genotype experimental groups. B and C: further Western Blot analysis indicated decreases in Nrf-1, cytochrome c (Cyto C), and cytochrome oxidase-4 (COX IV) proteins in protein extracts from female dnCREB mice (C) but not male dnCREB mice (B) relative to NTG littermates. Representative images of Western Blots are shown; statistical analyses were performed using n = 6 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
The decrease observed in the protein content of PGC-1α (Fig. 4A) in hearts of female dnCREB mice was accompanied by decreases in the protein content of ERRα at both 4 and 12 wk of age (Fig. 5A). As PGC-1α and ERRα are primary drivers of mitochondrial biogenesis (3, 20), citrate synthase activity, a classic marker of mitochondrial content, was assessed in extracts prepared from whole hearts of 12-wk-old dnCREB mice and their nontransgenic littermates. The results indicated a reduction in citrate synthase activity only in extracts from female dnCREB mouse hearts (Fig. 5B), consistent with a reduced mitochondrial density in hearts of female dnCREB mice.
Fig. 5.
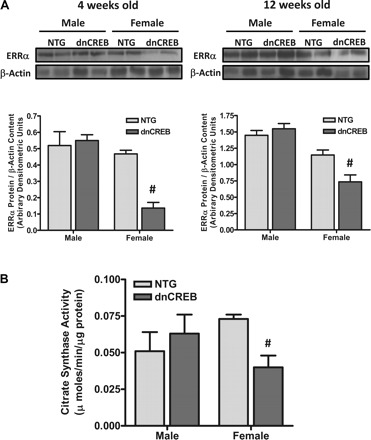
Estrogen-related receptor-α (ERRα) content, a primary determinant of mitochondrial content and function, and citrate synthase activity (a marker of mitochondrial density) are diminished in hearts of only female dnCREB transgenic mice. A: Western blot analyses were performed on hearts of 4- and 12-wk-old male and female dnCREB transgenic mice and their NTG littermates. Blots were sequentially probed for ERRα and β-actin to control for protein loading. Representative blots are shown for n = 4 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05). B: protein extracts were prepared from hearts of 12-wk-old male and female dnCREB mice and their NTG littermates, which were assessed for citrate synthase activity. n = 4 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
Diminished respiratory capacity in mitochondria isolated from dnCREB transgenic mice.
The results of the Western blot analyses implied that mitochondrial dysfunction may be expected, concurrent with the diminished contents of cytochrome c and cytochrome oxidase-4, in hearts of dnCREB mice. We hypothesized that the loss of expression of important proteins in the electron transport complex in the mitochondria of female dnCREB mice (cytochrome c and cytochrome oxidase-IV) would lead to diminished respiratory capacity in these mitochondria as well. Mitochondria were isolated from hearts of 4-wk-old dnCREB transgenic mice and their nontransgenic littermates and were assessed for mitochondrial O2 consumption using a Clark electrode respirometry. Yields of mitochondria from these preparations were consistent, with the exception of a significantly decreased recovery of mitochondria in the SSL subpopulation from the hearts of female dnCREB mice (Table 2).
Table 2.
Mitochondrial protein yields from isolations performed from hearts of dnCREB and NTG mice for assessments of mitochondrial respiration
| Mitochondrial Protein Yield, mg mitochondrial protein/g heart wet wt |
||
|---|---|---|
| Interfibrillar mitochondrial subpopulation | Subsarcolemmal mitochondrial subpopulation | |
| Male | ||
| NTG | 4.58 ± 0.31 | 4.34 ± 0.44 |
| dnCREB | 4.03 ± 0.43 | 4.91 ± 0.32 |
| Female | ||
| NTG | 4.29 ± 0.26 | 5.14 ± 0.38 |
| dnCREB | 4.28 ± 0.31 | 3.37 ± 0.30*† |
Values are means ± SE; n = 3 animals/group (experimental replications). Animals were 8 wk of age.
Statistical significance vs. the same mitochondrial subpopulation in same-sex NTG littermates;
statistical significance vs. the same mitochondrial subpopulation in opposite-sex dnCREB mice (P < 0.05).
The results indicated that before puberty and before the onset of overt contractile dysfunction, SSL mitochondria isolated from hearts of dnCREB transgenic animals of both sexes demonstrated significantly diminished state 3 respiration in the presence of both carbohydrate (pyruvate + malate) and fat (palmitoyl-l-carnitine) substrates (Fig. 6). When succinate was provided as a fat substrate instead of palmitoyl-l-carnitine, similar results were seen as with palmitoyl-l-carnitine in IF and SSL mitochondria (data not shown). It should also be noted that IF mitochondria isolated from hearts of female, but not male, dnCREB transgenic animals demonstrated diminished state 3 respiration with both carbohydrate and fat substrates (Fig. 6).
Fig. 6.
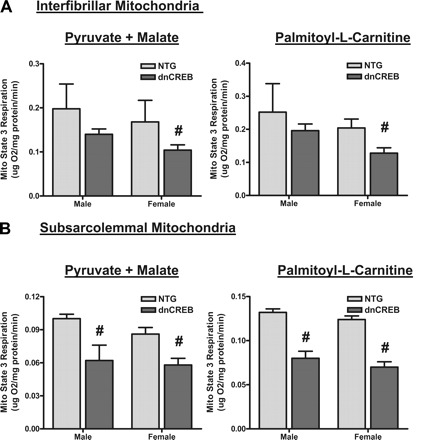
Mitochondria isolated from 4-wk-old dnCREB transgenic mice show dysfunction in mitochondrial state 3 respiration with both carbohydrate and lipid substrates. Mitochondria were isolated from five pooled hearts of 4-wk-old dnCREB mice and their NTG littermates and fractionated into subsarcolemmal (SSL; B) and interfibrillar (IF; A) subpopulations. Fractionated mitochondria were immediately assessed for state 3 mitochondrial respiration as described in materials and methods. Results from three separate experiments were used for statistical analysis. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
Expression of dnCREB transgene in the heart leads to a disruption of mitochondrial morphology in female mice.
We examined mitochondrial structure in hearts from dnCREB transgenic mice and their nontransgenic counterparts by transmission electron microscopy. Mitochondria observed in sections prepared from hearts of female dnCREB (Fig. 7D) demonstrated significant swelling and disruption of the mitochondrial matrix and cristae compared with the cardiac mitochondrial morphology observed in both male and female nontransgenic mice (Fig. 7, A and C, respectively). While some disruption of the normal mitochondrial phenotype was observed in hearts of male dnCREB mice (Fig. 7B), it did not appear as severe as that observed in mitochondria in hearts of female dnCREB mice. These observations were consistent with more pronounced oxidant damage to mitochondria in hearts of female dnCREB transgenic mice relative to their male counterparts.
Fig. 7.
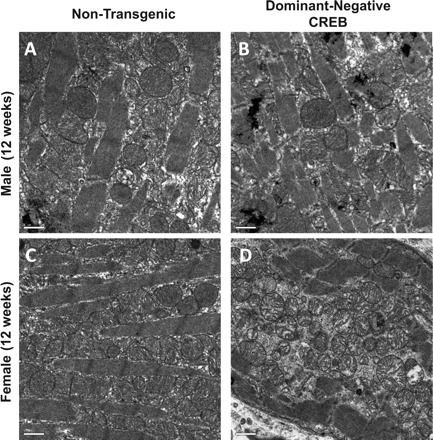
Deterioration of mitochondrial structure and morphology are observed in hearts of female (D), but not male (B), 12-wk-old dnCREB transgenic mice compared with hearts of female (C) and male (A) NTG hearts. Heart tissue was prepared from transmission electron microscopy as described in materials and methods and photographed. Sections from four hearts per sex/genotype group were examined with a minimum of six fields per section; photographs are representative of observations within hearts of that sex/genotype group.
Mitochondrial dysfunction is accompanied by increased oxidant generation and oxidant damage to mitochondrial proteins.
We next asked whether the mitochondrial oxidant damage in female dnCREB hearts was accompanied by differences in the mitochondrial generation of ROS. Oxidant generation by intact mitochondria prepared from hearts of dnCREB transgenic mice and nontransgenic littermates was assessed using an Amplex red assay as previously described (9). Significantly increased levels of ROS generation were observed from cardiac mitochondria of female, but not male, dnCREB transgenic mice at 4 wk of age (in SSL mitochondria; Fig. 8A) and 12 wk of age (in both IF and SSL mitochondrial populations; Fig. 8B).
Fig. 8.
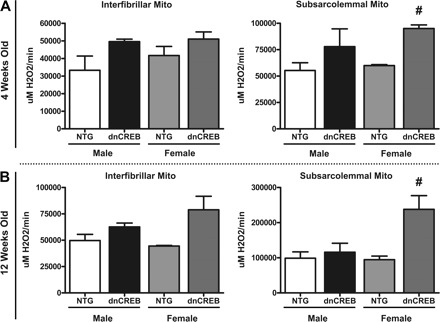
Increase ROS generation by SSL mitochondria from hearts of female, but not male, 4- (A) and 12-wk-old (B) dnCREB transgenic mice. Mitochondria were isolated and fractionated into subpopulations as in Fig. 5. Isolated mitochondria were assessed for ROS generation using the Amplex red method as described in materials and methods. Hearts of 4-wk-old dnCREB mice and NTG littermates were pooled (5 hearts/preparation), and three replications of the experiment were performed. Hearts from 12-wk-old mice were prepared separately. n = 4 for each sex/gender experimental group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
Diminished mitochondrial oxidant defense with dnCREB expression in the heart which is more severe in female transgenic mice.
The expression of two CREB-dependent, mitochondrial oxidant defense proteins, UCP-3 and MnSOD, was assessed in heart protein extracts prepared from 4- and 12-wk-old dnCREB mice and their nontransgenic littermates. Examination of whole heart extracts indicated that MnSOD content was significantly diminished only in hearts of female dnCREB mice at both 4 wk of age (Fig. 9A) and 12 wk of age (Fig. 9B). UCP-3 protein content was assessed by Western blot analysis and found to be significantly decreased only in hearts of female dnCREB transgenic mice before puberty but decreased in hearts from both male and female dnCREB transgenic mice at 12 wk of age (Supplemental Fig. 4).
Fig. 9.
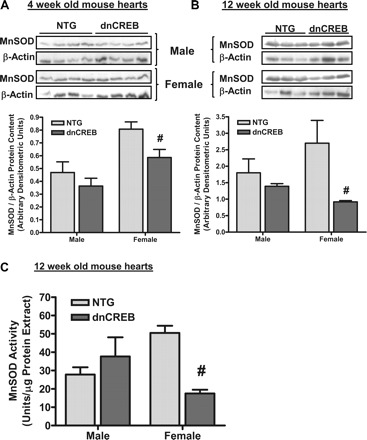
Content and activity of MnSOD are decreased in hearts of female, but not male, dnCREB transgenic mice. Protein extracts prepared from hearts of 4-wk-old (A) and 12-wk-old (B) dnCREB mice and their NTG littermates were subjected to Western blot analysis for the protein contents of MnSOD and β-actin. Data are expressed as the ratio of MnSOD relative to β-actin in the same sample. n = 6 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05). C: SOD activity in extracts from hearts of 12-wk-old dnCREB mice and their NTG littermates was assessed using a commercially available kit (Cayman Chemical, Ann Arbor, MI). Assays were performed after the inclusion of 3 mM potassium cyanide in the assays, which has been shown to inhibit both Cu/Zn-SOD and extracellular SOD. n = 3 for each sex/genotype group. #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
To assess whether changes in MnSOD protein content were indicative of changes in MnSOD activity, analysis of potassium cyanide-insensitive SOD activity (reflective of the activity of MnSOD) was performed on extracts prepared from hearts of 12-wk-old dnCREB mice and their nontransgenic littermates. The results indicated that MnSOD activity was significantly reduced in only those heart extracts prepared from 12-wk-old female dnCREB mice (Fig. 9C), similar to results obtained from the assessment of MnSOD content.
Western blot analysis of proteins extracted from isolated mitochondria demonstrated the diminished MnSOD protein content in hearts of female dnCREB mice was the result of decreased MnSOD content specifically in the SSL subpopulation of mitochondrial in females (Fig. 10A). This specific decrease in MnSOD content in only SSL mitochondria in hearts of only female dnCREB mice was accompanied by decreased GPx-1 protein content in this same mitochondrial subpopulation (Fig. 10B). This observation is consistent with our results demonstrating increased ROS generation by this subpopulation of mitochondria from hearts of female dnCREB mice (Fig. 8). We speculate that the reduced GPx-1 content concurrent with the diminished MnSOD content observed in the SSL subpopulation of female dnCREB cardiac mitochondria may result in decreased H2O2 conversion to H2O, with the net result being increased H2O2 release despite diminished MnSOD content.
Fig. 10.
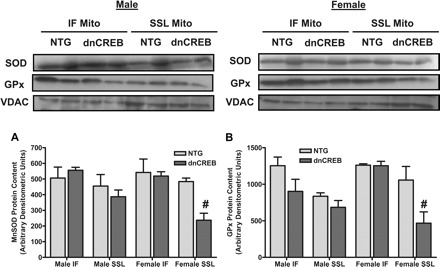
Diminished content of mitochondrial oxidant defense proteins MnSOD and glutathione peroxidase (GPx) are observed only in SSL mitochondria in hearts of female dnCREB mice. Proteins were extracted from SSL and IF mitochondria isolated from 12-wk-old male (A) and female (B) dnCREB mice and their NTG littermates. Western Blot analysis was performed on 35 μg of mitochondrial protein by serial probing on the same unstripped blot to assess the content of MnSOD, GPx-1 proteins, and voltage-dependent anion channel (VDAC)/porin as a loading control. Loading of mitochondrial protein in each sample was assessed by probing for VDAC/porin, the results of which indicated equal loading within SSL and IF sample groups. Note that the contents of MnSOD and GPx-1 were not subsequently corrected for VDAC content. n = 3 separate mitochondrial preparations (3 hearts/preparation). #Statistical significance relative to same-sex NTG littermates (P ≤ 0.05).
DISCUSSION
We have previously demonstrated an association between CREB activation, mitochondrial function, and apoptosis in pathological and physiological cardiac remodeling. Our previous study (37) left unanswered whether loss of CREB transcription factor signaling was responsible for defects in mitochondrial function. The results reported here directly examine this question by testing whether heart-specific expression of a phosphorylation-deficient CREB on cardiac function at weaning and after the physiological remodeling of puberty affected mitochondrial function. Interestingly, the mitochondrial abnormalities were most prominent in female dnCREB mice and correlated with greater cardiac dysfunction and mortality. This exaggerated pathology in female dnCREB mice is particularly interesting as the majority of HF models involving genetically modified mice have demonstrated more profound morbidity associated with the male sex (14).
The increased mortality observed during the first 20 wk of life in female dnCREB mice raises the question of whether male dnCREB mice manifest resistance to the impact of the dnCREB transgene on the heart or if the progression of the pathology is delayed in male animals. Comparison of the survival data from our work (Fig. 1A) with previously published survival data from the group who created the dnCREB transgenic mouse model (15, 33) might lead one to conclude that female mortality accounts for the majority of the deaths reported in these studies during the first 20 wk of life, assuming an equal distribution between the sexes in these studies (59% survival in a mixed-sex population of dnCREB mice in Ref. 15). As such, one could speculate that male dnCREB mice either 1) manifest a substantial delay of onset for mortality from the cardiac expression of the dnCREB transgene or 2) fail to manifest the same degree of pathology and subsequent mortality as female dnCREB mice as a result of expression of the phosphorylation-deficient CREB mutant in the heart.
With regard to which of these two possibilities is more likely, our data demonstrated that male dnCREB mice do manifest 1) diminished SSL mitochondrial O2 consumption at 4 wk of age (Fig. 6) and 2) diminished contractile function at 8 wk of age (Table 1), a time that significantly precedes the time of onset of dysfunction described for other transgenic models of pathological cardiac remodeling (3, 20). As such, male dnCREB mice manifest similar, yet less severe, morbidity concurrent with expression of the dnCREB transgene, which likely would result in increased mortality. Indeed, our survival data (Fig. 1A) demonstrated that male dnCREB mice manifest a trend toward increased mortality (33% mortality over 19 wk) relative to their nontransgenic littermates, which initiates in a similar timeframe as mortality in female dnCREB mice. The trend in male dnCREB mortality may have achieved statistical significance relative to nontransgenic mice with a larger sample size or a longer duration of the experiment. That said, the rate of mortality in the male dnCREB mice shown in Fig. 1A was significantly less than the mortality in female dnCREB mice. We interpret the contractile function and survival data as being consistent with sex impacting the severity of the pathological response of the heart to dnCREB transgene expression.
Evidence in the literature, including studies in humans and in rodent models of HF, supports a cardioprotective role for estrogen through estrogen receptor-dependent mechanisms (for a review, see Ref. 27). In studies of aortic stenosis (7), hypertension, aging, or with familial hypertrophic cardiomyopathies (6), female patients demonstrate remodeling with predominantly supernormal contractility and increased wall thickness, which contrasted with subnormal contractility, chamber dilation, and wall thinning observed in male patients. However, as discussed in the Introduction, HF of specific etiologies, such as MI (5), hypertension (25, 31), and diabetes (19), manifests with greater morbidity and mortality in females compared with males. Likewise, in patients with coronary artery disease and severely reduced LV ejection fractions, women demonstrated worse outcomes than men irrespective of the myocardial scar burden from infarct (23). All these examples support the notion that circumstances exist where the cardioprotective impact of female sex may be compromised. Mechanisms could be proposed in which loss of relative estrogenic tone in cardiac myocytes (subsequent to a shift in the relative abundance of sex steroid receptors) concurrent with loss of CREB function could reveal a susceptible phenotype in the hearts of females. Preliminary data in the dnCREB mouse model demonstrated a shift in sex steroid receptor abundance in only the hearts of female dnCREB mice, with a loss of estrogen receptor content and an increase in androgen receptor content being observed (data not show).
The sex differences in mortality in the dnCREB line may be at least partly explained by significant differences in mitochondrial phenotypes. Mitochondrial morphology in female dnCREB mice, in particular, was noted for oxidative damage to these organelles (Fig. 7). Furthermore, ROS release from intact cardiac SSL mitochondria of female dnCREB mice was far greater than either male dnCREB mice or either sex of nontransgenic control (Fig. 8), implying that a diminished ability to neutralize ROS may exacerbate the accumulation and impact of ROS in this subpopulation of intact mitochondria. A study (2) of human atrial cardiac tissue from diabetic patients showed a remarkably similar metabolic and oxidative phenotype to that which we describe in the dnCREB mouse heart, demonstrating mitochondrial impairments in the maximal capacity to oxidize fatty acids and glutamate, yet increased mitochondrial H2O2 emission.
We believe that both increased ROS production (likely from the electron transport chain) and diminished capacity to neutralize ROS (due to diminished MnSOD and GPx-1) in SSL mitochondria contribute to the increased oxidant burden in the hearts of female dnCREB mice. Mitochondrial damage was associated with a loss of MnSOD and GPx-1 in dnCREB mice (Figs. 9 and 10). Hearts of female dnCREB mice had a diminished MnSOD content of 30–70% in SSL mitochondria, which is consistent with the cardiac phenotype reported by others in mice harboring a single-allele knockout (KO) of SOD2. Single-allele SOD2 KO in the heart leads to a 50% reduction in MnSOD protein content (4) and a 40–50% reduction in MnSOD activity (34, 36), which is accompanied by contractile dysfunction (4), diminished mitochondrial respiratory capacity (36), and mitochondrial oxidative damage in the heart (21, 34). In contrast, overexpression of MnSOD in cardiac mitochondria of transgenic mice imparts resistance to mitochondrial damage and subsequent cardiac dysfunction resulting from the administration of the antiretroviral drug zidovudine (21), stressing the relative importance of this antioxidant in maintaining mitochondrial homeostasis.
We observe a significant decrease in cytochrome c protein content in dnCREB mice of both sexes at 4 wk of age; this is consistent with the previously reported role of CREB in regulating cytochrome c gene expression (18). The observed decrease in cytchrome c protein content may impact both metabolic capacity and efficiency in the hearts of dnCREB mice. Consistent with this reduced content of cytochrome c, we observed decreased mitochondrial O2 consumption with either carbohydrate (malate + pyruvate) or fat (palmitoyl-l-carnitine) substrates in both SSL and IF mitochondrial populations isolated from hearts of both sexes of dnCREB transgenic mice (Fig. 6). Diminished content of cytochrome c has also been linked to increased generation of oxidant species by supermolecular complexes of the electron transport chain (8, 23, 25). Our observation of accelerated ROS release by SSL mitochondria in the hearts of female dnCREB mice may reflect increased ROS generation by the electron transport chain due to a specific loss of cytochrome c, which is not accounted for as the result of a concomitant attenuation of mitochondrial oxidant defense mechanisms.
The results reported here are consistent with CREB dysfunction as a primary factor in mitochondrial dysfunction and oxidative stress with HF. This potent effect of dnCREB on cardiac function manifests at 4 wk of age, and the mitochondrial dysfunction and subsequent oxidant burden increase with age and the physiological remodeling of puberty. An exaggerated female susceptibility to the deleterious effects of CREB dysfunction is also reported, manifest in part as an enhanced mitochondrial oxidant generation concurrently with impaired mitochondrial oxidant defense. This phenotype in hearts of female dnCREB mice leads to increased mitochondrial damage and dysfunction, decreased pump function, and decreased longevity relative to their male counterparts.
Mitochondrial dysfunction, which was observed as early as 4 wk of age, precedes the development of a proapoptotic phenotype in hearts of dnCREB mice. Decreases in the Bcl-2-to-Bax ratio and activating cleavage of caspase-3 were not observed at the 4-wk time point in the hearts of dnCREB mice (Supplemental Fig. 2) but were significantly modified at 12 wk of age and were accompanied by increases in caspase-3 activity (Fig. 3). As such, we believe that mitochondrial dysfunction lies upstream of the onset of apoptosis in the sequellae of events that contribute to contractile dysfunction and accelerated mortality in dnCREB mice.
It is important to note that both sexes of dnCREB mice manifested a degree of contractile dysfunction (Fig. 2 and Table 1). However, female dnCREB mice demonstrated a more pronounced contractile impairment. While diminished mitochondrial production of ATP and increased oxidant burden subsequent to mitochondrial inefficiency likely contribute to the development of contractile dysfunction in the hearts of dnCREB transgenic mice, evidence would seem to indicate that the loss of CREB function may also impact contractile function through other mechanisms. As IF mitochondria from only the female dnCREB mice demonstrated impaired metabolic function (Fig. 6), this may indicate that some degree of the contractile dysfunction in both sexes is a direct effect of CREB dysfunction, while the greater contractile impairment observed in female dnCREB hearts is linked to an additional metabolic challenge.
Comparisons between the phenotypes of the dnCREB mouse model and the cardiac phenotypes of PGC-1α KO (3) and ERRα KO (20) mice add further support to additional nonmetabolic contributions to contractile dysfunction in the hearts of dnCREB mice. While loss of CREB function in the dnCREB mouse model leads to diminished PGC-1α gene expression and ERRα gene expression, the severity and speed of onset of the dnCREB pathological phenotype in the heart is much greater than that observed in hearts of ERRα KO (20) and PGC-1α KO mice (3). PGC-1α KO mice show no echocardiographic evidence of contractile dysfunction (3), and ERRα KO mice manifest contractile and hemodynamic characteristics that are indistinguishable from wild-type control mice (20) at 3 mo of age, a month later than dnCREB mice manifest significant contractile dysfunction (Table 1). It should be noted that diminished expression of genes in the downstream programs of PGC-1α and ERRα, including cytochrome oxidase-IV (Fig. 4 and Supplemental Fig. 3), UCP-3 (Supplemental Fig. 4), and pyruvate dehydrogenase-4 (data not shown), accompany the diminished expression of PGC-1α and ERRα seen in female dnCREB hearts. These results would appear to be consistent with diminished activity in female dnCREB hearts of gene expression pathways involving these two coactivators (for a review, see Ref. 21), which are involved in the positive regulation of mitochondrial biogenesis. Thus, while CREB's impact upon the myocardial phenotype likely involves transcriptional modulation requiring PGC-1α and ERRα, CREB has an impact on a broader array of critical cardiac phenotypes and likely regulates aspects of cardiac biology that extend beyond mitochondrial function.
The mechanism(s) underlying this exaggerated impact of CREB dysfunction on the cardiac phenotype by the female sex is likely manifest by combined defects in mitochondrial function and antioxidant defense. The interactions between dnCREB and estrogen's effect on the mitochondria remain to be elucidated but may have clinical relevance to the accelerated damage observed in female versus male patients with established heart disease of vascular etiology. Full delineation of these effects will require further investigation.
GRANTS
This work was funded by a Veterans Administration Merit Award (to P. A. Watson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
The authors thank Dorothy Dill, Dr. Kelly Ambler, Dr. Adam Chicco, and Bifeng Gao for the technical expertise in electron microscopy, echocardiography, mitochondrial isolation, and analysis and microarray analysis, respectively. The authors also thank Dr. Jed Friedman and Dr. Jane Reusch for intellectual input.
Footnotes
Supplemental Material for this article is available at the American Journal of Physiology-Heart and Circulatory Physiology website.
REFERENCES
- 1. American Heart Association. Heart Disease and Stroke Statistics–2003. Update. Dallas, TX: American Heart Association, 2002 [Google Scholar]
- 2. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 54: 1891–1898, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arany Z, Huamei H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Spiegelman BM. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab 1: 259–271, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Asimakis GK, Lick S, Patterson C. Postischemic recovery of contracile function is impaired in SOD2+/− but not SOD1+/− mouse hearts. Circulation 105: 981–986, 2001 [DOI] [PubMed] [Google Scholar]
- 5. Bello N, Mosca L. Epidemiology of coronary heart disease in women. Prog Cardiovasc Dis 46: 287–295, 2004 [DOI] [PubMed] [Google Scholar]
- 6. Berko BA. Gender-related differences in cardiomyopathy. Cardiovasc Clin 19: 285–300, 1989 [PubMed] [Google Scholar]
- 7. Carroll JD, Carroll EP, Feldman T, Ward DM, Lang RM, McGaughey D, Karp RB. Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation 86: 1099–1107, 1992 [DOI] [PubMed] [Google Scholar]
- 8. Chen Q, Moghaddas S, Hoppel CL, Lefnesky EJ. Ischemic defects in the electron transport chain increase production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294: C460–C466, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria. J Biol Chem 278: 36027–36031, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Chin MH, Goldman L. Gender differences in 1-year survival and quality of life among patients with congestive heart failure. Med Care 36: 1033–1046, 1998 [DOI] [PubMed] [Google Scholar]
- 11. Clarke KW, Gray D, Hampton JR. Evidence of inadequate investigation and treatment of patients with heart failure. Br Heart J 71: 584–587, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clinical Quality Improvement Network Investigators Mortality risk and patterns of practice in 4606 acute care patients with CHF: the relative importance of age, sex and medical therapy. Arch Intern Med 156: 1669–1673, 1996 [PubMed] [Google Scholar]
- 13. Douglas PS. Cardiovascular Health and Disease in Women (2nd ed.) New York: Saunders, 2002, p. 426–444 [Google Scholar]
- 14. Du XJ. Gender modulates cardiac phenotype development in genetically modified mice. Cardiovasc Res 63: 510–519, 2004. [DOI] [PubMed] [Google Scholar]
- 15. Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest 101: 2415–2426, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friedman MM. Gender differences in the health related quality of life of older adults with heart failure. Heart Lung 32: 320–327, 2003 [DOI] [PubMed] [Google Scholar]
- 17. Gusdon AM, Chen J, Votyakova TV, Mathews CE. Chapter 24 Quantification, localization, and tissue specificities of mouse mitochondrial reactive oxygen species production. Methods Enzymol 456: 439–457, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herzig RP, Scacco S, Scarpulla RC. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J Biol Chem 275: 13134–13141, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 22: 6A–13A, 1993 [DOI] [PubMed] [Google Scholar]
- 20. Huss JM, Imahashi KI, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERRα is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab 6: 25–37, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Huss HM, Kelly DP. Nuclear receptor signaling and cardiac energetic. Circ Res 95: 568–578, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Kohler JJ, Cucoranu I, Fields E, Green E, He S, Hoying A, Russ R, Abuin A, Johnson D, Hosseini SH, Raper CM, Lewis W. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab Invest 89: 782–790, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kushnareva Y, Murphy AN, Andreyev A. Complex 1-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J 368: 545–553, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon DH, Halley CM, Popovic ZB, Carrigan TP, Zysek V, Setser R, Schoenhagen P, Flamm SD, Starling RC, Desai MY. Gender differences in survival in patients with severe left ventricular dysfunction despite similar extent of myocardial scar measured on cardiac magnetic resonance. Eur J Heart Fail 11: 937–944, 2009 [DOI] [PubMed] [Google Scholar]
- 25. Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol 33: 37–47, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Levy D, Larson MG, Ramachandran SV, Kannel WB, Ho KKL. The progression from hypertension to heart failure. J Am Coll Cardiol 275: 1557–1562, 1996 [PubMed] [Google Scholar]
- 27. Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res 75: 478–486, 2007 [DOI] [PubMed] [Google Scholar]
- 28. Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252: 8731–8739, 1977 [PubMed] [Google Scholar]
- 29. Philbin EF, DiSalvo TG. Influence of race and gender on care process, resource use, and outcomes in congestive heart failure. Am J Cardiol 82: 76–81, 1998 [DOI] [PubMed] [Google Scholar]
- 30. Riedinger MS, Dracup KA, Brecht ML, Padilla G, Sarna L, Ganz PA. Quality of life in patients with heart failure: do gender differences exist? Heart Lung 30: 105–116, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Shindler DM, Kostis JB, Yusuf S, Quinones MA, Pitt B, Stewart D, Pinkett T, Ghali JK, Wilson AC. Diabetes mellitus: a predictor of morbidity and mortality in the Studies of Left Ventricular Dysfunction (SOLVD) Trials and Registry. Am J Cardiol 77: 1017–1020, 1996 [DOI] [PubMed] [Google Scholar]
- 32. Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JL. PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol 46: 201–212, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spencer KT, Collins K, Korcarz C, Fentzke R, Lang RM, Leiden JM. Effects of exercise training on LV performance and mortality in a murine model of dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 279: H210–H215, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Straasburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A, Wenk J, Peters T, Wlaschek M, Krieg T, Hafner M, Kumin A, Werner S, Muller W, Scharffetter-Kochanek K. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med 38: 1458–1470, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Tabassome S, Mary-Krause M, Funk-Brentano C, Jaillon P. Sex differences in the prognosis of CHF. Results from CIBIS-II. Circulation 103: 375–380, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol 281: H1422–H1432, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Watson PA, Reusch JE, McCune SA, Leinwand LA, Luckey SW, Konhilas JP, et al. Restoration of CREB function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. Am J Physiol Heart Circ Physiol 293: H246–H259, 2007 [DOI] [PubMed] [Google Scholar]