

Abstract
Pseudomonas aeruginosa is an opportunistic bacterial pathogen responsible for a high incidence of acute and chronic pulmonary infection. These infections are particularly prevalent in patients with chronic obstructive pulmonary disease and cystic fibrosis: much of the morbidity and pathophysiology associated with these diseases is due to a hypersusceptibility to bacterial infection. Innate immunity, primarily through inflammatory cytokine production, cellular recruitment, and phagocytic clearance by neutrophils and macrophages, is the key to endogenous control of P. aeruginosa infection. In this review, we highlight recent advances toward understanding the innate immune response to P. aeruginosa, with a focus on the role of phagocytes in control of P. aeruginosa infection. Specifically, we summarize the cellular and molecular mechanisms of phagocytic recognition and uptake of P. aeruginosa, and how current animal models of P. aeruginosa infection reflect clinical observations in the context of phagocytic clearance of the bacteria. Several notable phenotypic changes to the bacteria are consistently observed during chronic pulmonary infections, including changes to mucoidy and flagellar motility, that likely enable or reflect their ability to persist. These traits are likewise examined in the context of how the bacteria avoid phagocytic clearance, inflammation, and sterilizing immunity.
Keywords: inflammation, lung, Pseudomonas aeruginosa, phagocytosis, clearance
pseudomonas aeruginosa is a ubiquitous gram-negative rod that grows in many environmental sites. Importantly, P. aeruginosa has evolved mechanisms to be an opportunistic extracellular pathogen of its human host. This bacterial species is responsible for acute infections commonly associated with burn wounds and invasive instrument procedures and for chronic infections typically in patients with persistent lung disease and compromised immune responses (86, 87). Conditions that permit these long-term infections are exemplified by the diseases chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF) (86, 87). The high incidences of both acute and chronic respiratory infections, observed in up to 15% of adult COPD cases and >80% of adults with CF (39, 103), have led to the endeavor to understand the underlying conditions that facilitate P. aeruginosa infection and colonization, as well as mechanisms of pathogenesis when the bacteria colonize the lungs of mammalian hosts.
Several lines of evidence support the critical importance of innate immunity and, specifically, professional phagocytic cells as a key determinant in the ability of the host to control P. aeruginosa infection. Mice, rabbits, and humans that are missing major subsets of phagocytic cells, such as neutrophils or macrophages, or that lack major innate immune response molecules, such as MyD88 (myeloid differentiation primary response gene 88), are highly susceptible to P. aeruginosa infection (6, 68, 76, 97). Additionally, patients with CF are well characterized to generate robust antibody responses against P. aeruginosa; however, this is insufficient for effective host control of infection and for sterilizing immunity (9, 115, 116). Finally, people with genetically compromised phagocyte responses, such as those with leukocyte adhesion deficiency (LAD), specific granule deficiency, or anhidrotic ectodermal dysplasia with immune deficiency have a marked predisposition to P. aeruginosa infection (6). Thus effective control and clearance of P. aeruginosa depends on phagocyte recognition, engulfment, and degradation of bacteria in a highly complex and regulated process that works in concert with other innate immune responses, such as inflammatory signals. Although the well-defined opsonin-mediated uptake pathways (5, 40, 147) apply to P. aeruginosa, the mechanisms behind P. aeruginosa uptake in the absence of effective opsonization, and how this ties into inflammatory cytokine regulation, are only beginning to be elucidated. Nonopsonic phagocytosis is a crucial mechanism for host control during the initial stages of infection, and mouse models indicate that this process may dictate colonization efficiency at the onset of P. aeruginosa challenge (4). Whereas recent reports and reviews have detailed various other components of P. aeruginosa respiratory disease, such as pulmonary edema, humoral immunity, and therapeutic avenues (10, 16, 20, 31, 32, 46, 51, 61, 73, 88, 96, 102, 110, 122), this review will spotlight the current state of knowledge in nonopsonic phagocytic recognition and response mechanisms to P. aeruginosa. Here, we will focus on mechanisms of cellular internalization and clearance of the bacteria in the context of phagocytic receptors and pathways that modulate uptake. We will highlight bacterial strategies for phagocytic evasion and discuss the known inflammatory mechanisms that alter phagocytic clearance of the bacteria.
Bacterial Invasion vs. Phagocytic Engulfment
Cellular invasion by P. aeruginosa.
A source of controversy and occasional confusion, especially when discussing phagocytic cells, is the concept of bacterial invasion vs. cellular uptake (see Fig. 1). In the first instance, bacteria actively move from an extracellular space into the host cell cytoplasm or cellular compartment, as exhibited by archetypal intracellular pathogens such as Salmonella enterica and Listeria monocytogenes (37, 119). Consistent with their lifestyle, these microorganisms have evolved mechanisms to facilitate cellular entry and then survive in the intracellular space using machinery and mechanisms specifically designed for that purpose. An example of this is the specialized secretion system (designated Salmonella pathogenicity island-2) that S. enterica Typhi utilizes to inject effector proteins across the phagosome membrane and into the host cytosol to modify the phagosome for bacterial replication (37). Another example is the hemolysin listeriolysin O, secreted by L. monocytogenes, which allows the bacterium to escape the phagocytic vacuole and thereby enter and replicate in the host cell cytoplasm (119). The demonstration by Fleiszig et al. (41) that P. aeruginosa can invade corneal and epithelial cells in vitro led to the proposal that P. aeruginosa may analogously invade host cells to facilitate replication (2, 41, 66, 67, 155, 156). Later experiments show P. aeruginosa entry into human embryonic kidney and HeLa cells, as well as bacterial traversal across corneal cell barriers in vivo (2, 63, 66, 67). A consistent theme in many of these studies is host cell membrane composition and polarity. Acute lung infection experiments by Zaas et al. (155, 156) demonstrated that lipid raft composition and the caveolin protein Cav-2 were necessary for P. aeruginosa invasion into nonphagocytic host cells, but not in alveolar macrophages. These data were later supported by evidence that demonstrated that P. aeruginosa binding affects cell polarity and subsequent membrane composition, allowing the bacteria to preferentially invade the basolateral side of Madin-Darby canine kidney cells (67). Bacterial binding may be facilitated through cellular heparan sulfate proteoglycans and N-glycans, though the role of these receptors in subsequent invasion is still fairly preliminary (17, 18, 117). These collective observations are particularly intriguing since P. aeruginosa is not known to exhibit specific adaptations for intracellular survival and, as noted below, does employ potent toxins that efficiently kill host cells. When considering bacterial nutrient acquisition and the optimal niches for persistence, a better understanding of how invasion into host cells is advantageous for the bacteria may elucidate the physiological and clinical relevance of intracellular P. aeruginosa.
Fig. 1.
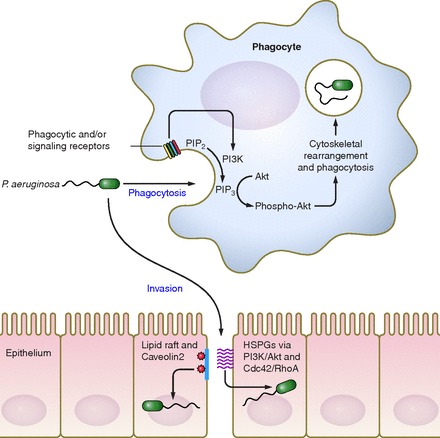
Flagellar motility is a key bacterial determinant for phagocytic susceptibility of Pseudomonas aeruginosa by both murine and human phagocytes, with motile bacteria phagocytosed ∼100-fold more efficiently than nonmotile bacteria (top). Although the cell surface phagocytic receptor(s) for P. aeruginosa require further elucidation, independent reports indicate that bacterial engulfment is actin dependent and controlled by the PI3K/Akt signaling pathway. P. aeruginosa has also been proposed to invade epithelial cells (bottom), particularly on the basolateral surfaces of these cells, through alterations in cell polarity and in breaches in the monolayer. Proposed mechanisms include invasion via cell surface lipid rafts in conjunction with the caveolin-2 protein, and via heparin sulfate proteoglycans (HSPGs). The HSPG-mediated pathway is subsequently dependent on intracellular PI3K/Akt activity, and RhoA and/or Cdc42 activation. The relationship between the HSPG and lipid raft-mediated pathways is currently unclear.
Host phagocytosis of P. aeruginosa.
In contrast to the above studies that describe a potential intracellular niche, P. aeruginosa is generally believed to be an extracellular pathogen, preferring to associate outside of host cell membranes and form extracellular biofilms (153). Entry into neutrophils and macrophages is achieved through active phagocytosis by the host cell. Indeed, this is supported by data linking phagocytosis to bacterial clearance from the host (6, 68, 76). Animal models that lack phagocytic cells quickly succumb to P. aeruginosa challenge (68, 76), and people with defects in phagocytic cell function are highly susceptible to P. aeruginosa infection (6). Thus internalization is utilized by the host for control of invasive P. aeruginosa, and so if the bacterium actively promotes cellular entry, there must be an adaptive benefit that outweighs the cost of potential phagocytic degradation. This leads to a fundamental question in regard to P. aeruginosa infections: Are invasion and phagocytosis the same process, and is this merely a semantic, rather than mechanistic, difference? Independent studies have identified that both invasion and phagocytosis rely on phosphoinositide 3-kinase (PI3K) activity in the host cell (62, 66, 67). PI3K converts the host cell membrane lipid phosphatidylinositol (4, 5) bisphosphate (PIP2) into phosphatidylinositol (3, 4, 5) triphosphate (PIP3) (154), a ligand for Akt recruitment from the cytoplasm. This phosphorylation activity is required for both P. aeruginosa invasion into nonphagocytic cells and P. aeruginosa engulfment by professional phagocytes (unpublished data). In support of this, the loss of murine PTEN (phosphatase and tensin homolog), the regulatory phosphatase that converts PIP3 back into PIP2, leads to increased phagocytosis of P. aeruginosa and in vivo clearance from the lung (60). However, secreted toxins employed by P. aeruginosa actively block the activity of certain GTPases, such as Rac1 (Ras-related C3 botulinum toxin substrate 1) and CDC42 (cell division and control protein 42) (64). The functions of these proteins are necessary for phagocytosis by macrophages (discussed below), but the GTPase activities are not necessary for P. aeruginosa internalization into epithelial cells (64). Separate reports found that P. aeruginosa activates the upstream GTPase RhoA (Ras homolog gene family, member A) in epithelial and endothelial cells, which in turn promotes internalization and vascular permeability (46, 63, 72). Therefore, targeted regulation of host GTPase activity by P. aeruginosa can both promote intracellular invasion and inhibit phagocytic ingestion.
Role of activated protein C pathway.
The RhoA pathway has also garnered recent attention in the context of activated protein C. The ratio of active Rac1 to RhoA in the lung endothelium, which is regulated by the activated protein C pathway, appears to be critical for the regulation of lung edema and protein permeability during P. aeruginosa infection (13, 22, 123). Pretreatment of animal models with activated protein C is reported to modulate paracellular permeability and lung injury caused by P. aeruginosa through inhibition of Rho [necessary for P. aeruginosa internalization into epithelial cells (64)] and activation of Rac1 [necessary for engulfment by phagocytes (80)], but it does not change neutrophil influx or inflammatory cytokine production (13, 22, 123). However, clinical trials using recombinant human protein C for treatment of severe sepsis and septic shock failed to reproducibly decrease patient mortality, and the therapy has subsequently been withdrawn from use (77). Unfortunately, the patient parameters utilized across these trials were inconsistent, making it difficult to reconcile why the treatment failed (77). Recombinant protein C efficacy may be optimized in combination with traditional antibiotic treatment and with pulmonary therapies targeted to patients presenting disorders in pulmonary coagulation. Indeed, there is speculation that patients in the early PROWESS trial, which reported a 6.1% decrease in 28-day mortality, benefitted from early antibiotic treatment and volume resuscitation, whereas patients in later trials were selected on the basis of clinical presentation of refractory shock, which may not obligatorily be linked to dysregulated coagulation and is an independent indicator of poor prognosis (77). Therefore, the data to date indicate that, for P. aeruginosa-elicited pneumonia, recombinant protein C treatment may be most beneficial if applied during the early stages of infection. Pretreatment has been shown to reduce subsequent pulmonary bleeding and edema and to promote phagocytic clearance through stimulation of Rac1 GTPases in resident alveolar macrophages (13, 123). However, exogenous protein C does not change the host inflammatory response (22), and so efficacy will likely be reduced once P. aeruginosa is established and has triggered broad bacterial-derived and immune pathology.
In summary, although invasion and engulfment are distinct cellular processes, there is significant overlap in the machinery that facilitates both activities, which in turn are influenced by both bacterial (e.g., toxins) and host (e.g., protein C) factors. P. aeruginosa may coopt cellular uptake mechanisms, evolved to facilitate bacterial clearance by professional phagocytic cells, at certain sites of infection, notably in the eye and potentially in the lungs, to traverse the initial epithelial barriers and promote its own colonization. Since P. aeruginosa has adapted to survive in a wide range of environmental niches, specific evolution to invade the human epithelium seems unlikely. Rather, it has likely evolved global mechanisms that can be regulated to promote colonization at sites that provide the most favorable growth conditions. In mammalian hosts, bacterial internalization by host cells at certain anatomical sites may be beneficial and yet must be balanced against phagocytic susceptibility. Indeed, P. aeruginosa employs multiple factors, such as exotoxins, alginate secretion, and the regulation of motility (discussed later) to evade phagocytosis during infection and likewise avoid predation by amoeba in the environment. The evolution of these factors suggests that internalization by eukaryotic cells is more often detrimental to P. aeruginosa than it is beneficial.
Receptor-Mediated Internalization of P. aeruginosa
The signaling events that govern phagocytic uptake of P. aeruginosa, primarily by neutrophils and macrophages, are complex and feature various levels of specificity. Engulfment is mediated through bacteria-to-phagocyte contact and binding, in concert with receptor-based recognition of P. aeruginosa and downstream signaling to regulate ingestion. As such, engulfment is dictated by numerous factors, including receptor expression on the phagocytes (see Table 1), as well as ligand expression and exposure on the pathogen.
Table 1.
Host cell receptors implicated in Pseudomonas aeruginosa invasion or phagocytosis
| Potential Host Receptors Interacting with P. aeruginosa | Function | Implicated in Invasion or Phagocytosis? | Reference(s) |
|---|---|---|---|
| Fcγ-receptors | Binds Fc portion of host IgG opsonins | phagocytosis | 1, 5, 40, 140 |
| Complement receptors (CD11b/CR3) | Binds host iC3b complement opsonins | phagocytosis | 57, 118 |
| Scavenger receptor A | Binds polyanionic sugars and lipoproteins | neither | 3, 30 |
| MARCO | Binds polyanionic ligands | phagocytosis | 30 |
| Toll-like receptors | Binds bacterial extracellular components (e.g., LPS, flagellin, peptidoglycan) | neither | 4, 147 |
| CFTR | N-glycan chloride ion channel | phagocytosis | 29, 109, 114, 148 |
| Caveolin proteins (Cav-1, Cav-2) | Lipid-raft (caveolae) integrated signal transduction | invasion | 155, 156 |
| Heparan sulfate proteoglycans | Binds pilin, low molecular weight OMPs, ionic residues | invasion/adherence | 17, 18, 117 |
MARCO, macrophage receptor with collagenous structure; CFTR, cystic fibrosis transmembrane conductance regulator; OMP, outer membrane protein.
Opsonic phagocytosis.
The most elaborated mechanisms of foreign particle engulfment involve phagocytosis driven by antibody-binding of Fc-receptors and complement binding of complement receptors (CRs). Like many gram-negative pathogens, P. aeruginosa is susceptible to opsonic phagocytosis. This process has been extensively covered elsewhere (1, 5, 40, 140) and shares many of the same molecular mechanisms as in nonopsonic uptake. Briefly, receptor engagement leads to recruitment and phosphorylation of kinase signaling molecules, which enable the downstream recruitment and activation of cytoskeletal arrangement proteins such as Rho and Rac GTPases and actin-related proteins; these subsequently coordinate actin polymerization and membrane distortion at the target site (106, 147). Additional host factors, such as surfactant protein A and serum amyloid A, have also been reported to bind P. aeruginosa and facilitate phagocytosis by alveolar macrophages (52, 91). Although opsonic phagocytosis contributes to P. aeruginosa clearance, it is important to note that P. aeruginosa commonly infects sites, such as the lung, where phagocytes express relatively low levels of opsonic receptors (137).
Nonopsonic phagocytosis.
Importantly, phagocytosis of P. aeruginosa and some other bacteria also occurs when opsonization is absent or ineffective, and this is independent of Fc-receptor or CR activation (89, 106, 125, 133, 134). Nonopsonic phagocytosis likely evolved as a frontline mechanism for the immune system to recognize and respond to pathogens at the very onset of infection. Likewise, this mechanism is critical for bacterial clearance at anatomical sites of endogenously low levels of antibody and complement, such as the bronchopulmonary tree and the urinary tract (134). Although this ingestion process uses much of the same intracellular machinery as opsonization-based phagocytosis, the mechanism is not well understood. The process is further complicated by redundant receptor activity and differential expression, a dependence on the type of particle being engulfed, and promiscuous receptor binding to bacterial ligands (3, 40, 106). Early studies by Heale et al. (57) and Pollard et al. (118) suggested that the integrin CD11b (CR3) as well as CD14 mediate phagocytosis of P. aeruginosa, but the evidence indicates that this is strain dependent and seems to be highly variable on the basis of the ligand moieties and polysaccharide architecture presented by the bacteria. For example, Pollard et al. observed that a P. aeruginosa strain isolated from a CF patient was not efficiently engulfed by neutrophils isolated from a patient with LAD (deficient in CR3), yet a strain isolated from the LAD patient was efficiently engulfed by LAD phagocytes, thus pointing to the variability and specificity in phagocyte-strain interactions (118). This variability in receptor-ligand interaction for effective phagocytosis extends to other bacterial species as well. Some outer membrane proteins, such as OmpA, in virulent strains of Escherichia coli and Klebsiella pneumoniae dictate recognition and internalization by macrophages (59, 141), but not necessarily neutrophils (45), and loss of the OmpA ortholog, OprF, in P. aeruginosa may affect both association with host cells and the ability of the bacteria to ultimately form a biofilm (38, 75). Likewise, we recently showed that scavenger receptor A (SR-A) on mouse phagocytes is the major phagocytic receptor engaged by specific strains of γ-proteobacteria (e.g., DH5α E. coli), but it is not singularly necessary for uptake of the parental strain K12 E. coli, nor is it needed for uptake of PA14 P. aeruginosa (3). However, a recent report by Domingo-Gonzalez et al. (30) demonstrated that treatment of alveolar macrophages with soluble MARCO (macrophage receptor with collagenous structure), which is also a class-A scavenger receptor, inhibited phagocytosis of P. aeruginosa by ∼50%. Observations such as these highlight the complexity and heterogeneity of receptor signaling and ligand specificity during nonopsonic engulfment processes. Lastly, it is important to distinguish phagocytic uptake from bactericidal killing. For example, the loss or blockade of active CFTR (cystic fibrosis transmembrane conductance regulator) protein has been interpreted as being directly responsible for the defective clearance of P. aeruginosa by phagocytes (148) and also for epithelial cells (114), although the contribution of epithelial cell phagocytic activity to clearance of P. aeruginosa is likely minimal. Subsequent experimentation has called into question whether CFTR specifically facilitates engulfment of P. aeruginosa, since the data support an alternative explanation that CFTR activity may instead facilitate phagolysosomal degradation of the bacteria (26, 29, 109).
An additional complication in the search for a specific phagocytic receptor for P. aeruginosa is that many of the pathogen-associated molecular patterns (PAMPs) that might be expected to facilitate phagocytosis, such as lipopolysaccharide (LPS), peptidoglycan, and flagellin, also stimulate host immune signaling receptors, particularly the Toll-like receptors (TLRs) (128). The TLRs responsible for recognition of these components, particularly TLR2, TLR4, and TLR5, play a critical role in leukocyte stimulation and activation, yet these do not appear to directly facilitate ingestion (147). Additionally, the loss of MyD88, a signaling adaptor for many TLRs, does not alter the phagocytic capacity of mouse macrophages for P. aeruginosa (4). Taken together, the accumulated data indicate that the stimulus for phagocytosis during P. aeruginosa infection involves multiple mechanisms of uptake that can work independent of, or synergistically with, each other and can potentially involve the stimulation of multiple families of receptors. Thus a current and challenging avenue of investigation is to understand the underlying mechanisms of phagocytosis during host clearance of P. aeruginosa and to clarify how differential ligand expression patterns and receptor-ligand interactions between phagocytes and bacterial strains change the phagocytic dynamic, in terms of both recognition and killing.
Bacterial Strategies for Phagocytic Evasion
Although P. aeruginosa is susceptible to phagocytic engulfment and degradation, multiple tactics have been identified by which the pathogen attenuates vulnerability to phagocytosis. One such phenotypic trait is the conversion from a free-swimming, planktonic lifestyle to a biofilm-associated, sessile lifestyle (85). This transition correlates with long-standing clinical observations that P. aeruginosa downregulates flagellar motility and converts to a strong mucoid phenotype. Additionally, most P. aeruginosa strains express potent toxin secretion systems which, along with nonmotility and exopolysaccharide secretion, are believed to cumulatively contribute to phagocytic resistance and subsequent bacterial persistence.
Loss of flagellar motility by P. aeruginosa.
The majority of laboratory and environmental P. aeruginosa strains are highly motile, utilizing a single, polar, monotrichous flagellum for swimming. Infectivity studies have found that flagellar functionality is necessary for P. aeruginosa to initially colonize the host and establish infection (8, 115). Lillehoj et al. (82, 83) demonstrated that the P. aeruginosa flagellum binds to host cell Muc1 mucin, which is produced in the upper airway endothelium, whereas later studies confirmed that flagellar motility is required for P. aeruginosa to establish stable colonization within the host (86, 105, 115). This colonization corresponds to a conversion to a biofilm phenotype in which the bacteria are typically nonmotile, since reports that examine bacterial isolates recovered from long-term, chronic respiratory disease have shown that P. aeruginosa populations in these infections have absent or downregulated flagellar gene expression (42, 85, 104). The basis for this loss of flagellar motility is likely multifaceted. For the pathogen, conversion to a sessile lifestyle can be metabolically beneficial if the bacteria are in a nutrient-rich site, and the advantages to bacterial populations living in close-knit aggregates (e.g., quorum sensing) have been well documented (126). Additionally, host immune factors may also contribute to this conversion. Neutrophil elastase, a serine proteinase secreted during inflammation, has been shown to repress expression of P. aeruginosa flagellin (132). The observed conversion to a nonmotile biofilm during chronic infection may also reflect selection for this state. P. aeruginosa that form biofilms are much more resistant to standard-of-care antibiotics used in the clinic (100, 101, 151). Additionally, the progressive recovery of nonmotile bacterial isolates may reflect selection of bacteria resistant to phagocytosis. We recently identified that flagellar motility in P. aeruginosa is a critical phagocytic activation pattern both in vitro and in vivo (4). Loss of flagellar motility, independent of the flagellum itself, provides the bacteria with a ∼100-fold increase in resistance to phagocytic uptake by macrophages, neutrophils, and dendritic cells (see Fig. 1) (4). This is independent of other bacterial virulence factors or host opsonization and is due to bacterial activation of the host cell PI3K/Akt signaling pathway specifically by swimming motility (Ref. 84 and Lovewell et al., unpublished data). Flagellar swimming motility results in cellular Akt activation, which in turn regulates phagocytic recognition in the host cell and subsequent phagocytosis of the bacteria (unpublished data). This is supported by separate studies illustrating that the PI3K/Akt pathway is necessary for both TLR-based activation and bacterial internalization into mammalian cells in various models of P. aeruginosa infection (49, 60, 66, 67). Furthermore, deletion of PTEN phosphatase, an enzyme which has the opposite activity of PI3K and dephosphorylates PIP3, leads to increased phagocytic activity of P. aeruginosa in alveolar macrophages (60). How the host response to bacterial motility is transduced through the plasma membrane is unknown, but we speculate that it may reflect the ability of swimming bacteria to induce receptor clustering or a membrane perturbation. Regardless, it is clear that the downregulation of flagellar motility, which is observed in chronic lung infections, directly contributes to P. aeruginosa persistence by providing the pathogen a potent means of phagocytic evasion.
P. aeruginosa exopolysaccharide secretion.
A second phenotype commonly observed in chronic P. aeruginosa infections is exopolysaccharide secretion (EPS). P. aeruginosa contains a number of genetic loci that can generate or modify exopolysaccharides (psl locus), alginate production (alg and muc loci), and a mucoidy phenotype (98, 107, 127). These gene products elicit the EPS matrix and canonical biofilms commonly associated with P. aeruginosa establishment in the lungs and elsewhere. Importantly, genetic analyses have also identified that expression of some of these gene products provides for phagocytic resistance (19, 74, 108). During initial host colonization, nonmucoid strains expressing the polysaccharide Psl effectively limit deposition of complement molecules C3, C5, and C7, presumably by hindering access of the complement to the bacterial outer membrane (98). This provides the bacteria with measurable resistance to CR-mediated phagocytosis and an early advantage against the innate immune system (98). Moreover, once P. aeruginosa has established persistent infection in the host, it converts to an increasingly mucoid phenotype, characterized by the upregulation of alginate production (87, 92, 93). Leid et al. (81) and others have illustrated that mucoid strains and biofilm-associated P. aeruginosa resist phagocyte activation and uptake in both opsonized and nonopsonized conditions, supporting an initial finding by Eftekhar and Speert (35) that the phagocytic defect in alginate-positive strains can be rescued by treatment with alginase to degrade the mucoid EPS. Unfortunately, the direct mechanism of alginate-based phagocytic inhibition remains unclear, since conflicting interpretations suggest that alginate may sterically inhibit bacteria-to-phagocyte binding and influence engulfment processes (108), or alternatively it may inhibit GTPase activation of signal transduction pathways (69), which are believed to contribute to P. aeruginosa engulfment. Again, although many of the underlying mechanisms remain to be elucidated, it is clear that P. aeruginosa has evolved strategies to limit the host phagocytic response at the onset of infection (Psl production), during establishment within the host (progressive loss of flagellar motility), and postcolonization (alginate production).
P. aeruginosa type-III secretion system.
Although EPS production by bacterial pathogens has been well documented in many systems, secretion of anti-phagocytic molecules by P. aeruginosa is not limited to sugars. P. aeruginosa has a fully functional type-III secretion system (T3SS) by which it can inject effector proteins ExoT, ExoS, ExoY, and ExoU directly into host cells (12, 36, 79, 120). Relative expression of these factors is strain dependent, with different P. aeruginosa strains exhibiting different combinations of effectors (55, 129). ExoY is a nonspecific cyclase that increases vascular permeability but is not directly cytotoxic, whereas ExoU is a potent phospholipase that compromises mammalian cell membranes (36, 54). Although neither of these bacterial proteins has directly been shown to affect phagocytic functionality, the toxins ExoT and ExoS have been shown to limit uptake of P. aeruginosa by macrophages (54). The inhibitory mechanism of these two effectors is based on targeting host Rho, Rac, and CDC42 GTPases. Mechanistically, these host G proteins regulate various cellular functions including cytoskeletal organization and their activity is necessary for P. aeruginosa internalization. When injected into host cells, ExoT and ExoS act as GTPase-activating proteins that provide for the loss of GTP-bound Rac1 and CDC42 in macrophages (47, 63, 143). Thus these toxins shift the equilibrium of host GTPases into the inactive, GDP-bound form, functionally locking out the major regulators of cytoskeleton rearrangement and hindering phagocytic capability (27, 47). Additionally, these exotoxins can induce host cell death, particularly in epithelial cells, which promotes tissue damage and limits cellular immune responses (36). During P. aeruginosa-induced respiratory disease and infections, these effectors contribute to persistence by attenuation of ingestion and clearance by phagocytes and also facilitate cell death, thereby exacerbating infection severity.
P. aeruginosa inhibition of PAR2.
Another virulence factor of note that has recently been identified to functionally inhibit host phagocytosis is the elastolytic metalloproteinase LasB (bacterial elastase, not to be confused with neutrophil elastase), which cleaves host protease-activated receptors such as PAR2 (34). PAR2 signaling is described to regulate host inflammatory responses, particularly in the lung, and a recent paper by Moraes et al. (99) demonstrated that loss of PAR2 on neutrophils significantly decreased phagocytic uptake of P. aeruginosa through an undefined mechanism. Activation of PAR2 has been reported to recruit phagocytic machinery, such as MAP kinases, Rho-Rac GTPases, and actin (136), and the protein could therefore could be acting as a direct phagocytic receptor. Conversely, stimulation could simply lead to indirect immunomodulatory effects that activate phagocytes for enhanced phagocytic capability. Regardless, the capacity for PAR2 as a regulator of phagocytic uptake for P. aeruginosa remains intriguing, especially when considering that expression of elastase by virulent strains of P. aeruginosa could potentially modulate both inflammatory responses and phagocytosis through PAR2 inhibition.
Taken together, it is apparent that P. aeruginosa utilizes toxin secretion in concert with alginate production and alterations in motility to limit effective host responses. These observations highlight the adaptations P. aeruginosa has developed to enhance colonization and persistence in human hosts, and consequently the recent mechanistic findings are now clarifying the bases of the bacterial phenotypes documented in chronic pulmonary infections.
Models of Phagocytosis of P. aeruginosa
P. aeruginosa can infect human hosts through multiple routes. Acute infections are commonly found in COPD-based pulmonary disease, burn wounds, and keratitis of the eye, and with invasive instrument procedures such as bronchial tubes, catheters, and intravenous needles (86). Chronic P. aeruginosa infection, particularly in the lungs, has been extensively studied and is usually limited to immune-compromised patients, and the associated bacterial pneumonia in patients with CF is often the ultimate cause of mortality (48, 87, 104). Since the host phagocytic response is believed to be a key determinant in the subsequent pathology, multiple in vitro and animal models of phagocytic cell responses to P. aeruginosa, with various strengths and weaknesses, have been developed to help elucidate how this pathogen interacts with this critical arm of the innate immune system.
In vitro models of phagocytosis.
Labeling of P. aeruginosa with fluorescence and antibiotic-resistance genes is the basis for many of the classic phagocytic assays. These assays are typically assessed by either microscopy or antibiotic protection following coincubation with phagocytic cells derived from the lung or elsewhere. These in vitro assays are well suited to provide information on P. aeruginosa host-phagocyte interactions and have been extensively used to contribute to our understanding of the mechanisms of binding and the downstream effects leading to ingestion, both through bacterial adhesins such as pili and flagellin and through cellular integrins, scavenger receptors, and opsonic receptors. Much of our understanding of phagocytic components and regulation involved in P. aeruginosa infection, as well as the aforementioned bacterial mechanisms of phagocyte evasion, also come from standard coincubation experiments. These continue to be the easiest means to tease apart the basic biology of P. aeruginosa-phagocyte interactions, but unfortunately these experiments are also limited. During P. aeruginosa infection of the lungs or at other sites, bacteria interact with highly heterogeneous cell populations that cannot be completely reproduced in vitro. Similarly, the dynamic interactions between humoral components, such as opsonins, proteinases, and extracellular fibers (e.g., neutrophil NETS) and pathogens make comprehensive in vitro modeling very challenging. Concomitantly, extracellular factors expressed by P. aeruginosa, such as the previously mentioned mucoid phenotype, are not static and can change on the basis of environmental factors. Therefore, modeling phagocytosis in vitro is useful in elucidating basic mechanisms but is limited, as all in vitro experiments are, in terms of scope and translation to natural disease.
Murine models of phagocytosis.
The more clinically relevant in vivo models of infection have also proven useful in understanding many various P. aeruginosa interactions, although there continue to be many challenges with execution and interpretation of the phagocytosis and bacterial clearance experiments. Mice, and to a lesser extent rats, infected with P. aeruginosa via the lung (inhalation/aspiration), eye (corneal scratch), and peritoneum (direct injection) are the most widely used models. A major strength of these murine models is that mice display a robust acute response as measured by neutrophil and macrophage cellularity and proinflammatory cytokine production in a matter of hours postinfection (111, 149, 150). When mice are challenged with a sublethal dose, phagocytes will rapidly clear the commonly used laboratory strains of P. aeruginosa, whereas increasing dosages of bacteria lead to exacerbation of bacterial sepsis, cytokine storm, and increased host lethality, but not necessarily chronic infection (68, 111, 139, 149). As such, these murine infections are well suited models for acute pneumonia and instrument-based and wound-associated infections. A recent advance in assessment of in vivo phagocytosis, which was subsequently adapted for use with Borrelia burgdorferi (56), was the use of a modified gentamicin-protection assay employed to quantify phagocytosis of P. aeruginosa in both the murine lung and peritoneal cavity over a short, single-dose infection period (4). This assay is useful in modeling initial phagocytic and innate immune responses but is a poor representation of long-term exposure to P. aeruginosa, since the infection is typically resolved by mice within 48 h.
Zebrafish models of phagocytosis.
Like the murine model, the newly emerging zebrafish model of P. aeruginosa infection is an exciting tool for studying host-pathogen interactions in acute infections. Zebrafish can be genetically manipulated, are susceptible to P. aeruginosa infection, and rely on innate immune mechanisms for control of bacterial infections. Thus, with the advent of transparent fish lines that express fluorescent neutrophils and macrophages, investigators have recently been able to perform intravital imaging of P. aeruginosa interactions with phagocytes in real time (15). Moreover, the immune cell profile in zebrafish embryos can be manipulated through morpholino-based gene regulation tools, allowing investigators to tease apart the relative spatiotemporal contribution of various effector cells during P. aeruginosa infection (23). These advances provide a powerful tool for probing the dynamics of P. aeruginosa-host cell interactions, trafficking, and phagocytosis in vivo. Two major limitations of this model are the disparities in anatomy (e.g., lungs) and that, as in murine infections, P. aeruginosa does not chronically infect zebrafish and the model does not effectively replicate persistent human disease.
Models of phagocytosis in chronic infections.
Studies of chronic P. aeruginosa infection, especially in the context of CF, are some of the most pursued avenues in P. aeruginosa research. CF is a genetic disorder caused by a mutation in the CFTR gene, which codes for an ion transport protein. In humans, loss of CFTR function leads to numerous physiological defects, most notably defective mucociliary clearance in the lungs and the buildup of mucus and alveolar fluid (121). This allows for eventual chronic colonization by P. aeruginosa, leading to inflammation and fatal pneumonia (121). Modeling the disease in murine hosts is complicated by the well-documented observation that mice with a deletion in the CFTR gene do not recapitulate human CF disease, which includes susceptibility to chronic P. aeruginosa colonization (50, 65). Therefore, although acute infections in mice retain a high degree of relevance for P. aeruginosa in humans, including in vivo experiments of phagocytic clearance, a major risk factor in chronic P. aeruginosa infection is not well represented in classic murine models.
A potential workaround in mice is the agarose-bead model of P. aeruginosa infection. Developed by Cash et al. (21) and modified for mice by Starke et al. (135), the protocol utilizes bacteria embedded in small agarose polymer plugs. The polymer physically retards bacterial clearance in vivo and allows for a longer, more chroniclike infection. This model has been used to examine various immune and drug responses to persistent P. aeruginosa in vivo, such as mortality and cytokine induction between CF-related mouse genotypes (58, 145). However, the use of the beads inhibits phagocyte association with bacteria and does not enable bacterial properties, such as motility, binding, and adherence, that influence interactions with host cells and therefore initiation and persistence of infection (58, 135). Also, the scope and duration of infection is on the order of days and weeks, and longer infection studies require reinfection of the mice (58, 135). Thus the model is still limited in clinical relevance to natural chronic P. aeruginosa infection in humans, especially in terms of phagocytic clearance.
The recently developed porcine model of CF and, to a degree, the recently reported ferret model have many advantages in studying lung disease in vivo, including pneumonia associated with P. aeruginosa infection (65, 124, 142). Mutation of the CFTR gene in pigs and ferrets leads to pathology more closely resembling that observed in humans (65). Specific to P. aeruginosa engulfment, several reports have identified impaired bacterial clearance from CFTR-deficient pigs (113, 124, 138). So far the evidence indicates that this is not due to inherent phagocytic deficits conferred by loss of CFTR, but rather due to impaired bacterial killing elicited from misregulated pH of the alveolar fluid and insufficient oxidation of the lysosomal compartment in the alveolar phagocytes (113, 124). This is supported by corresponding in vitro work using murine and human cftr−/− phagocytes that demonstrate that CFTR does not play a role in the actual engulfment process but instead contributes to phagolysosomal degradation by modulation of compartmental ion gradients (26, 29, 109). Thus it is likely that the chronic P. aeruginosa colonization in cftr−/− pigs, as well as human patients, is due to multiple contributing factors such as impaired mucociliary clearance, phagocytic evasion mechanisms of the bacteria, and inhibition of bactericidal activity in alveolar phagocytes. In both the pig and ferret models, the eventual development and availability of genetic and immunological tools for these animals will hopefully better delineate the in vivo contribution of bacterial motility, alginate production, and toxin secretion to phagocytosis and pathogen clearance in the context of CF.
Ties between phagocytosis and inflammation.
The host inflammatory response to P. aeruginosa is intimately connected to the phagocytic clearance of the bacteria. P. aeruginosa expresses a variety of PAMPs (e.g., LPS, flagellin) that lead to a robust inflammatory response. This response is critical for control of the bacterial infection through recruitment and activation of phagocytic cells. The inflammatory response has to be tightly regulated, however, since lack of an efficient inflammatory response can lead to reduced infiltration of phagocytes, resulting in reduced bacterial clearance via phagocytosis and other bactericidal mechanisms (71). Conversely, disproportionate inflammation can damage the host tissue due to excessive infiltration and activation of phagocytes and in some cases may even be coupled to reduced bacterial clearance (24, 130). Although there is certainly evidence that alveolar macrophages are direct contributors to bacterial clearance through phagocytosis (4, 60), they are also essential for mounting early inflammatory responses and recruitment of neutrophils for subsequent bacterial uptake and clearance. In support of this, depletion of alveolar macrophages prior to intratracheal instillation of bacteria in an acute model of infection led to delayed bacterial clearance kinetics, likely as a consequence of impaired neutrophil influx. (71).
PRRs and PAMPs in P. aeruginosa-induced inflammation.
P. aeruginosa expresses potent TLR2, TLR4, TLR5, and TLR9 agonists, and the TLR4-dependent inflammatory response to LPS, in particular, is critical to clear infection (78, 131). Since the loss of MyD88-dependent responses leads to lower inflammatory cytokine production and reduced bacterial clearance in an acute infection model (131), many of the early studies focused on the roles of TNF-α and (to a lesser degree) IL-6. Interestingly, the magnitude of the TNF-α response to P. aeruginosa is modulated by the phagocytic class-A scavenger receptor (SR-A or MSR1), with loss of SR-A expression leading to hyperinflammation (3). This was initially hypothesized to be due to impaired clearance of the bacteria through loss of a direct receptor-bacteria interaction. However, genetic analyses have revealed that SR-A is not required for phagocytosis of P. aeruginosa (3, 30). Rather, SR-A deficiency leads to hyperinflammation in response to infection due to impaired clearance of LPS and thereby through modulation of TLR4-driven proinflammatory responses (3). The complete role of TNF-α in bacterial clearance, lung injury, and repair is complex and is not fully understood (78). The data from murine models on the role of TNF-α are inconsistent owing to differences in the models and bacterial strains employed (78); however, recent evidence from normal and CF airway epithelial cells indicates that TNF-α promotes epithelial wound closure but has adverse effects on cell proliferation (90).
Inflammasome activation during P. aeruginosa infection.
An emergent mechanism for the relationship between inflammation and phagocytic response to P. aeruginosa focuses on the role of the cytokine IL-1β. IL-1β is increased in the sputum and bronchoalveolar lavage fluid of CF patients with P. aeruginosa infections (14, 53). Likewise, clearance of P. aeruginosa with antibiotics in children is correlated with reduced IL-1β production (33). As discussed previously, P. aeruginosa uses its T3SS to inject effector proteins into host cells (54). Yet along with effector proteins, potent P. aeruginosa PAMPs such as flagellin, the T3SS rod-protein PscI, and pilin can also be translocated through the T3SS, which leads to cellular activation of the NLRC4 (NLR family, CARD domain 4) inflammasome and the release of IL-1β (and IL-18) (7, 44, 94, 95, 144). The fundamental mechanisms underlying NLRC4-dependent release of IL-1β in response to P. aeruginosa has recently been the subject of several excellent reviews (25, 43, 112); here we will focus on the role of the elicited IL-1β in relation to subsequent bacterial phagocytosis, killing, and clearance.
IL-1β is predominantly produced by macrophages and, in response to P. aeruginosa, is dependent on the NLRC4 inflammasome. In the early phases of infection, IL-1β is sensed by resident epithelial cells through the IL-1 receptor (IL-1R) leading to the upregulation of other neutrophil chemoattractants such as macrophage inflammatory protein 2 (MIP-2) and KC/IL-8 (70, 146). An effective immune response depends on the early recruitment of neutrophils through these chemokines, since blockade of CXCR (chemokine C-X-C motif receptor)-dependent signaling leads to reduced neutrophil cellularity and bacterial clearance (146). Although cleavage of monomeric flagellin in the environment by the bacterial protease AprA is thought to limit TLR5-dependent responses (11), recent evidence suggests that the TLR5-MyD88 axis also plays an important role in bacterial phagocytosis and killing in alveolar macrophages through an IL-1R-dependent pathway in response to swimming-competent P. aeruginosa (28).
The use of genetic knockout mice and Anakinra (IL-1R antagonist effective on both humans and mice) to elucidate the bactericidal mechanisms involved has indicated that IL-1R signaling is crucial to control bacterial growth in an acute lung infection model (97, 152) but could lead to deleterious effects on the host during the later phases of infection (24, 130). Independent studies by Cohen and Prince (24) and Schultz et al. (130) demonstrated the role of IL-1-driven pathology during the early course of infection, leading to delayed clearance of bacteria from the murine lungs. It should be noted that these results are in the context of an acute P. aeruginosa infection in IL-1R−/− mice, which may have compensatory or interrelated mechanisms (such as reduced autophagic degradation of inflammatory cytokines) leading to excessive inflammation and delayed bacterial clearance. Although Schultz et al. pharmacologically blocked IL-1R signaling in wild-type (WT) mice and obtained similar data compared with the IL-1R−/− experiments, the differences in bacterial survival in the lungs of WT mice and IL-1R−/− mice were dependent on the dose of infection as well as the time at which bacterial survival was measured (130). Another explanation for the reduced bacterial burden in the lungs of IL-1R−/− mice compared with the WT controls may be that the excessive inflammation elicited by P. aeruginosa could have deleterious effects on the WT host, thereby providing the bacteria with a niche to survive and multiply. This could account for the differences seen in bacterial burden in WT and IL-1R−/− mice at later time points [18 or 24 h postinfection (hpi)] (24, 130). In contrast, IL-1R−/− mice displayed reduced neutrophil influx and bacterial clearance compared with WT mice at 4 hpi (152). Similarly, pharmacological blockade of IL-1R signaling in a mouse model where MyD88 expression was restricted to airway epithelial cells led to reduced neutrophil recruitment and reduced bacterial clearance from the lung up to 24 hpi (97). In this model, only airway epithelial cells could signal through IL-1R owing to the presence of MyD88, whereas IL-1R signaling was deficient in the hematopoietic compartment. Cumulatively, these data indicate that IL-1β is required for the early clearance of P. aeruginosa and can lead to the secretion of IL-1R-dependent chemokines such as KC/IL-8 and MIP-2 via epithelial cells to control infection through recruitment of neutrophils (97, 152). Thus potent IL-1β response to P. aeruginosa is pivotal since these models demonstrate the protective as well as the deleterious effects downstream of IL-1R signaling.
An additional layer of complexity was recently revealed that ties in to the ability of P. aeruginosa to evade the host innate immune response. IL-1β production in response to P. aeruginosa is independent of phagocytosis but requires both bacterial contact with host phagocytes and a functional T3SS (111). However, phagocytic evasion by P. aeruginosa through loss of flagellar motility is conferred, in part, by reduced bacterial interaction with the phagocyte cell surface (84). This results in reduced in vitro and in vivo IL-1β responses to nonmotile P. aeruginosa and is correlated with neutrophil recruitment and subsequent bacterial clearance (111). Thus loss of bacterial flagellar expression or motility likely confers an advantage to the bacteria and enables chronic infection through evasion of phagocytosis by resident cells and, through minimization of the IL-1β response, a synergistic amelioration of subsequent IL-1β-dependent bactericidal responses including recruitment and activation of additional phagocytes.
The threshold of IL-1β required for clearance of bacteria through recruitment of phagocytes without causation of excessive damage to host tissue is currently not well understood. In the case of chronic infection, understanding the kinetics of IL-1R signaling and bacterial clearance will be critical since excessive or reduced IL-1β production could both lead to increased bacterial survival, albeit through different signaling mechanisms. Finally, understanding the pivotal effects of the magnitude of IL-1β in response to bacterial load on epithelial and hematopoietic cells, as well as subsequent chemokine release and the downstream effector responses, will be essential toward gaining a more comprehensive understanding of the extent to which different cell types dictate bacterial clearance vs. pathology.
Summary
The interactions between host and pathogen are often complex and dynamic, even when simplified to a particular immunological response to a specific bacteria. Although phagocytosis is a critical process within the innate repertoire of defenses against P. aeruginosa in the lung and elsewhere, understanding how it is regulated has not been simple. Care must be taken to differentiate between bacterial invasion and host internalization, because the two processes can share similar components and outcomes but are operationally different, especially in terms of pathogenesis. Likewise, the cellular structures and receptors that govern phagocytosis of P. aeruginosa, and how these interact with pathogen effectors and virulence factors, will need to be further elucidated before we can fully understand why phagocytosis is such an important factor in P. aeruginosa infection and, mechanistically, how it is initiated and regulated both by the host and the pathogen. Lastly, effective clearance of P. aeruginosa must be considered in the context of inflammatory responses. Although uptake and inflammation are not necessarily dependent on each other, they can influence the efficacy of one another and are indirectly linked as the major mechanisms of control, and successful pathogen clearance requires both processes to work synergistically. Current and emerging models of P. aeruginosa disease, especially in the context of chronic pulmonary infection, will provide further insights into how effective phagocytic clearance during infection is achieved.
REFERENCES
- 1. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17: 593–623, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Alarcon I, Tam C, Mun JJ, LeDue J, Evans DJ, Fleiszig SM. Factors impacting corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Invest Ophthalmol Vis Sci 52: 1368–1377, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amiel E, Acker JL, Collins RM, Berwin B. Uncoupling scavenger receptor A-mediated phagocytosis of bacteria from endotoxic shock resistance. Infect Immun 77: 4567–4573, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amiel E, Lovewell RR, O'Toole GA, Hogan DA, Berwin B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect Immun 78: 2937–2945, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J Exp Med 171: 1333–1345, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev 16: 597–621, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arlehamn CS, Evans TJ. Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol 13: 388–401, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arora SK, Neely AN, Blair B, Lory S, Ramphal R. Role of motility and flagellin glycosylation in the pathogenesis of Pseudomonas aeruginosa burn wound infections. Infect Immun 73: 4395–4398, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baltimore RS, Mitchell M. Immunologic investigations of mucoid strains of Pseudomonas aeruginosa: comparison of susceptibility to opsonic antibody in mucoid and nonmucoid strains. J Infect Dis 141: 238–247, 1980 [DOI] [PubMed] [Google Scholar]
- 10. Baniak N, Luan X, Grunow A, Machen TE, Ianowski JP. The cytokines interleukin-1β and tumor necrosis factor-α stimulate CFTR-mediated fluid secretion by swine airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 303: L327–L333, 2012 [DOI] [PubMed] [Google Scholar]
- 11. Bardoel BW, van Kessel KP, van Strijp JA, Milder FJ. Inhibition of Pseudomonas aeruginosa virulence: characterization of the AprA-AprI interface and species selectivity. J Mol Biol 415: 573–583, 2011 [DOI] [PubMed] [Google Scholar]
- 12. Ben Haj Khalifa A, Moissenet D, Vu Thien H, Khedher M. Virulence factors in Pseudomonas aeruginosa: mechanisms and modes of regulation. Ann Biol Clin (Paris) 69: 393–403, 2011 [DOI] [PubMed] [Google Scholar]
- 13. Bir N, Lafargue M, Howard M, Goolaerts A, Roux J, Carles M, Cohen MJ, Iles KE, Fernandez JA, Griffin JH, Pittet JF. Cytoprotective-selective activated protein C attenuates Pseudomonas aeruginosa-induced lung injury in mice. Am J Respir Cell Mol Biol 45: 632–641, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 152: 2111–2118, 1995 [DOI] [PubMed] [Google Scholar]
- 15. Brannon MK, Davis JM, Mathias JR, Hall CJ, Emerson JC, Crosier PS, Huttenlocher A, Ramakrishnan L, Moskowitz SM. Pseudomonas aeruginosa Type III secretion system interacts with phagocytes to modulate systemic infection of zebrafish embryos. Cell Microbiol 11: 755–768, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bucior I, Abbott J, Song Y, Matthay MA, Engel JN. Sugar administration is an effective adjunctive therapy in the treatment of Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 305: L352–L363, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bucior I, Mostov K, Engel JN. Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect Immun 78: 939–953, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog 8: e1002616, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cabral DA, Loh BA, Speert DP. Mucoid Pseudomonas aeruginosa resists nonopsonic phagocytosis by human neutrophils and macrophages. Pediatr Res 22: 429–431, 1987 [DOI] [PubMed] [Google Scholar]
- 20. Campodonico VL, Gadjeva M, Paradis-Bleau C, Uluer A, Pier GB. Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol Med 14: 120–133, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cash HA, Woods DE, McCullough B, Johanson WG, Jr, Bass JA. A rat model of chronic respiratory infection with Pseudomonas aeruginosa. Am Rev Respir Dis 119: 453–459, 1979 [DOI] [PubMed] [Google Scholar]
- 22. Choi G, Hofstra JJ, Roelofs JJ, Florquin S, Bresser P, Levi M, van der Poll T, Schultz MJ. Recombinant human activated protein C inhibits local and systemic activation of coagulation without influencing inflammation during Pseudomonas aeruginosa pneumonia in rats. Crit Care Med 35: 1362–1368, 2007 [DOI] [PubMed] [Google Scholar]
- 23. Clatworthy AE, Lee JS, Leibman M, Kostun Z, Davidson AJ, Hung DT. Pseudomonas aeruginosa infection of zebrafish involves both host and pathogen determinants. Infect Immun 77: 1293–1303, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 123: 1630–1637, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggio MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, Ascenzioni F. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 6: e19970, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng Q, Barbieri JT. Modulation of host cell endocytosis by the type III cytotoxin, Pseudomonas ExoS. Traffic 9: 1948–1957, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Descamps D, Le Gars M, Balloy V, Barbier D, Maschalidi S, Tohme M, Chignard M, Ramphal R, Manoury B, Sallenave JM. Toll-like receptor 5 (TLR5), IL-1beta secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc Natl Acad Sci USA 109: 1619–1624, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8: 933–944, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. J Immunol 190: 5809–5817, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doring G, Pier GB. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 26: 1011–1024, 2008 [DOI] [PubMed] [Google Scholar]
- 32. dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol 303: L627–L633, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Douglas TA, Brennan S, Gard S, Berry L, Gangell C, Stick SM, Clements BS, Sly PD. Acquisition and eradication of P. aeruginosa in young children with cystic fibrosis. Eur Respir J 33: 305–311, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 32: 411–419, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Eftekhar F, Speert DP. Alginase treatment of mucoid Pseudomonas aeruginosa enhances phagocytosis by human monocyte-derived macrophages. Infect Immun 56: 2788–2793, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Engel J, Balachandran P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol 12: 61–66, 2009 [DOI] [PubMed] [Google Scholar]
- 37. Figueira R, Holden DW. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158: 1147–1161, 2012 [DOI] [PubMed] [Google Scholar]
- 38. Fito-Boncompte L, Chapalain A, Bouffartigues E, Chaker H, Lesouhaitier O, Gicquel G, Bazire A, Madi A, Connil N, Veron W, Taupin L, Toussaint B, Cornelis P, Wei Q, Shioya K, Deziel E, Feuilloley MG, Orange N, Dufour A, Chevalier S. Full virulence of Pseudomonas aeruginosa requires OprF. Infect Immun 79: 1176–1186, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. FitzSimmons SC. The changing epidemiology of cystic fibrosis. J Pediatr 122: 1–9, 1993 [DOI] [PubMed] [Google Scholar]
- 40. Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol 7: 61–98, 2012 [DOI] [PubMed] [Google Scholar]
- 41. Fleiszig SM, Evans DJ, Do N, Vallas V, Shin S, Mostov KE. Epithelial cell polarity affects susceptibility to Pseudomonas aeruginosa invasion and cytotoxicity. Infect Immun 65: 2861–2867, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10: 841–851, 2012 [DOI] [PubMed] [Google Scholar]
- 43. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol 37: 3030–3039, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Fu H, Belaaouaj AA, Dahlgren C, Bylund J. Outer membrane protein A deficient Escherichia coli activates neutrophils to produce superoxide and shows increased susceptibility to antibacterial peptides. Microbes Infect 5: 781–788, 2003 [DOI] [PubMed] [Google Scholar]
- 46. Ganter MT, Roux J, Su G, Lynch SV, Deutschman CS, Weiss YG, Christiaans SC, Myazawa B, Kipnis E, Wiener-Kronish JP, Howard M, Pittet JF. Role of small GTPases and alphavbeta5 integrin in Pseudomonas aeruginosa-induced increase in lung endothelial permeability. Am J Respir Cell Mol Biol 40: 108–118, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel JN. The arginine finger domain of ExoT contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect Immun 68: 7100–7113, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168: 918–951, 2003 [DOI] [PubMed] [Google Scholar]
- 49. Gomez MI, Prince A. Airway epithelial cell signaling in response to bacterial pathogens. Pediatr Pulmonol 43: 11–19, 2008 [DOI] [PubMed] [Google Scholar]
- 50. Gosselin D, Stevenson MM, Cowley EA, Griesenbach U, Eidelman DH, Boule M, Tam MF, Kent G, Skamene E, Tsui LC, Radzioch D. Impaired ability of Cftr knockout mice to control lung infection with Pseudomonas aeruginosa. Am J Respir Crit Care Med 157: 1253–1262, 1998 [DOI] [PubMed] [Google Scholar]
- 51. Hampton TH, Ballok AE, Bomberger JM, Rutkowski MR, Barnaby R, Coutermarsh B, Conejo-Garcia JR, O'Toole GA, Stanton BA. Does the F508-CFTR mutation induce a proinflammatory response in human airway epithelial cells? Am J Physiol Lung Cell Mol Physiol 303: L509–L518, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hari-Dass R, Shah C, Meyer DJ, Raynes JG. Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem 280: 18562–18567, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Doring G. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 11: 363–382, 2012 [DOI] [PubMed] [Google Scholar]
- 54. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7: 654–665, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hauser AR, Cobb E, Bodi M, Mariscal D, Valles J, Engel JN, Rello J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit Care Med 30: 521–528, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Hawley KL, Olson CM, Jr, Iglesias-Pedraz JM, Navasa N, Cervantes JL, Caimano MJ, Izadi H, Ingalls RR, Pal U, Salazar JC, Radolf JD, Anguita J. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci USA 109: 1228–1232, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heale JP, Pollard AJ, Stokes RW, Simpson D, Tsang A, Massing B, Speert DP. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J Infect Dis 183: 1214–1220, 2001 [DOI] [PubMed] [Google Scholar]
- 58. Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest 100: 2810–2815, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsieh PF, Liu JY, Pan YJ, Wu MC, Lin TL, Huang YT, Wang JT. Klebsiella pneumoniae peptidoglycan-associated lipoprotein and murein lipoprotein contribute to serum resistance, antiphagocytosis, and proinflammatory cytokine stimulation. J Infect Dis 208: 1580–1589, 2013 [DOI] [PubMed] [Google Scholar]
- 60. Hubbard LL, Wilke CA, White ES, Moore BB. PTEN limits alveolar macrophage function against Pseudomonas aeruginosa after bone marrow transplantation. Am J Respir Cell Mol Biol 45: 1050–1058, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hunt WR, Zughaier SM, Guentert DE, Shenep MA, Koval M, McCarty NA, Hansen JM. Hyperglycemia impedes lung bacterial clearance in a murine model of cystic fibrosis-related diabetes. Am J Physiol Lung Cell Mol Physiol 306: L43–L49, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kannan S, Audet A, Huang H, Chen LJ, Wu M. Cholesterol-rich membrane rafts and Lyn are involved in phagocytosis during Pseudomonas aeruginosa infection. J Immunol 180: 2396–2408, 2008 [DOI] [PubMed] [Google Scholar]
- 63. Kazmierczak BI, Engel JN. Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun 70: 2198–2205, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kazmierczak BI, Jou TS, Mostov K, Engel JN. Rho GTPase activity modulates Pseudomonas aeruginosa internalization by epithelial cells. Cell Microbiol 3: 85–98, 2001 [DOI] [PubMed] [Google Scholar]
- 65. Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med 17: 478–483, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kierbel A, Gassama-Diagne A, Mostov K, Engel JN. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol Biol Cell 16: 2577–2585, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kierbel A, Gassama-Diagne A, Rocha C, Radoshevich L, Olson J, Mostov K, Engel J. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J Cell Biol 177: 21–27, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77: 5300–5310, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Konig B, Friedl P, Pedersen SS, Konig W. Alginate—its role in neutrophil responses and signal transduction towards mucoid Pseudomonas aeruginosa bacteria. Int Arch Allergy Immunol 99: 98–106, 1992 [DOI] [PubMed] [Google Scholar]
- 70. Konstan MW, Davis PB. Pharmacological approaches for the discovery and development of new anti-inflammatory agents for the treatment of cystic fibrosis. Adv Drug Deliv Rev 54: 1409–1423, 2002 [DOI] [PubMed] [Google Scholar]
- 71. Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J, Sawa T. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun 66: 3164–3169, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krall R, Schmidt G, Aktories K, Barbieri JT. Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect Immun 68: 6066–6068, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Krasnodembskaya A, Samarani G, Song Y, Zhuo H, Su X, Lee JW, Gupta N, Petrini M, Matthay MA. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol 302: L1003–L1013, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krieg DP, Helmke RJ, German VF, Mangos JA. Resistance of mucoid Pseudomonas aeruginosa to nonopsonic phagocytosis by alveolar macrophages in vitro. Infect Immun 56: 3173–3179, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Krishnan S, Prasadarao NV. Outer membrane protein A and OprF: versatile roles in Gram-negative bacterial infections. FEBS J 279: 919–931, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kurahashi K, Sawa T, Ota M, Kajikawa O, Hong K, Martin TR, Wiener-Kronish JP. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 296: L198–L209, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lai PS, Thompson BT. Why activated protein C was not successful in severe sepsis and septic shock: are we still tilting at windmills? Curr Infect Dis Rep 15: 407–412, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lavoie EG, Wangdi T, Kazmierczak BI. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect 13: 1133–1145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Le Berre R, Nguyen S, Nowak E, Kipnis E, Pierre M, Quenee L, Ader F, Lancel S, Courcol R, Guery BP, Faure K. Relative contribution of three main virulence factors in Pseudomonas aeruginosa pneumonia. Crit Care Med 39: 2113–2120, 2011 [DOI] [PubMed] [Google Scholar]
- 80. Lee DJ, Cox D, Li J, Greenberg S. Rac1 and Cdc42 are required for phagocytosis, but not NF-kappaB-dependent gene expression, in macrophages challenged with Pseudomonas aeruginosa. J Biol Chem 275: 141–146, 2000 [DOI] [PubMed] [Google Scholar]
- 81. Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol 175: 7512–7518, 2005 [DOI] [PubMed] [Google Scholar]
- 82. Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, Kim KC. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 280: L181–L187, 2001 [DOI] [PubMed] [Google Scholar]
- 83. Lillehoj EP, Kim BT, Kim KC. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am J Physiol Lung Cell Mol Physiol 282: L751–L756, 2002 [DOI] [PubMed] [Google Scholar]
- 84. Lovewell RR, Collins RM, Acker JL, O'Toole GA, Wargo MJ, Berwin B. Step-wise loss of bacterial flagellar torsion confers progressive phagocytic evasion. PLoS Pathog 7: e1002253, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Luzar MA, Thomassen MJ, Montie TC. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun 50: 577–582, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2: 1051–1060, 2000 [DOI] [PubMed] [Google Scholar]
- 87. Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15: 194–222, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory? Am J Physiol Cell Physiol 291: C218–C230, 2006 [DOI] [PubMed] [Google Scholar]
- 89. Mahenthiralingam E, Speert DP. Nonopsonic phagocytosis of Pseudomonas aeruginosa by macrophages and polymorphonuclear leukocytes requires the presence of the bacterial flagellum. Infect Immun 63: 4519–4523, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maille E, Trinh NT, Prive A, Bilodeau C, Bissonnette E, Grandvaux N, Brochiero E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-α after injury. Am J Physiol Lung Cell Mol Physiol 301: L945–L955, 2011 [DOI] [PubMed] [Google Scholar]
- 91. Mariencheck WI, Savov J, Dong Q, Tino MJ, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of a live, mucoid strain of P. aeruginosa. Am J Physiol Lung Cell Mol Physiol 277: L777–L786, 1999 [DOI] [PubMed] [Google Scholar]
- 92. Martin DW, Schurr MJ, Mudd MH, Govan JR, Holloway BW, Deretic V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci USA 90: 8377–8381, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JI, Jensen P, Johnsen AH, Givskov M, Ohman DE, Molin S, Hoiby N, Kharazmi A. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145: 1349–1357, 1999 [DOI] [PubMed] [Google Scholar]
- 94. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA 105: 2562–2567, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107: 3076–3080, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Migneault F, Boncoeur E, Morneau F, Pascariu M, Dagenais A, Berthiaume Y. Cycloheximide and lipopolysaccharide downregulate αENaC mRNA via different mechanisms in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 305: L747–L755, 2013 [DOI] [PubMed] [Google Scholar]
- 97. Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R, Kazmierczak BI. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J Immunol 186: 7080–7088, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, Schlesinger LS, Wozniak DJ. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14: 95–106, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Moraes TJ, Martin R, Plumb JD, Vachon E, Cameron CM, Danesh A, Kelvin DJ, Ruf W, Downey GP. Role of PAR2 in murine pulmonary pseudomonal infection. Am J Physiol Lung Cell Mol Physiol 294: L368–L377, 2008 [DOI] [PubMed] [Google Scholar]
- 100. Moreau-Marquis S, Bomberger JM, Anderson GG, Swiatecka-Urban A, Ye S, O'Toole GA, Stanton BA. The DeltaF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. Am J Physiol Lung Cell Mol Physiol 295: L25–L37, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Moreau-Marquis S, Stanton BA, O'Toole GA. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther 21: 595–599, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Muramatsu S, Tamada T, Nara M, Murakami K, Kikuchi T, Kanehira M, Maruyama Y, Ebina M, Nukiwa T, Ichinose M. Flagellin/TLR5 signaling potentiates airway serous secretion from swine tracheal submucosal glands. Am J Physiol Lung Cell Mol Physiol 305: L819–L830, 2013 [DOI] [PubMed] [Google Scholar]
- 103. Murphy TF. Pseudomonas aeruginosa in adults with chronic obstructive pulmonary disease. Curr Opin Pulm Med 15: 138–142, 2009 [DOI] [PubMed] [Google Scholar]
- 104. Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr 19: 83–88, 2007 [DOI] [PubMed] [Google Scholar]
- 105. O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30: 295–304, 1998 [DOI] [PubMed] [Google Scholar]
- 106. Ofek I, Goldhar J, Keisari Y, Sharon N. Nonopsonic phagocytosis of microorganisms. Annu Rev Microbiol 49: 239–276, 1995 [DOI] [PubMed] [Google Scholar]
- 107. Ohman DE, Chakrabarty AM. Genetic mapping of chromosomal determinants for the production of the exopolysaccharide alginate in a Pseudomonas aeruginosa cystic fibrosis isolate. Infect Immun 33: 142–148, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Oliver AM, Weir DM. The effect of Pseudomonas alginate on rat alveolar macrophage phagocytosis and bacterial opsonization. Clin Exp Immunol 59: 190–196, 1985 [PMC free article] [PubMed] [Google Scholar]
- 109. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol 83: 1345–1353, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Park YS, Lillehoj EP, Kato K, Park CS, Kim KC. PPARγ inhibits airway epithelial cell inflammatory response through a MUC1-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 302: L679–L687, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81: 2043–2052, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol 21: 10–16, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB, Jr, Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487: 109–113, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pier GB, Grout M, Zaidi TS. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci USA 94: 12088–12093, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pier GB, Meluleni G, Goldberg JB. Clearance of Pseudomonas aeruginosa from the murine gastrointestinal tract is effectively mediated by O-antigen-specific circulating antibodies. Infect Immun 63: 2818–2825, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pier GB, Takeda S, Grout M, Markham RB. Immune complexes from immunized mice and infected cystic fibrosis patients mediate murine and human T cell killing of hybridomas producing protective, opsonic antibody to Pseudomonas aeruginosa. J Clin Invest 91: 1079–1087, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Plotkowski MC, Costa AO, Morandi V, Barbosa HS, Nader HB, de Bentzmann S, Puchelle E. Role of heparan sulphate proteoglycans as potential receptors for nonpiliated Pseudomonas aeruginosa adherence to non-polarised airway epithelial cells. J Med Microbiol 50: 183–190, 2001 [DOI] [PubMed] [Google Scholar]
- 118. Pollard AJ, Currie A, Rosenberger CM, Heale JP, Finlay BB, Speert DP. Differential post-transcriptional activation of human phagocytes by different Pseudomonas aeruginosa isolates. Cell Microbiol 6: 639–650, 2004 [DOI] [PubMed] [Google Scholar]
- 119. Portnoy DA, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun 60: 1263–1267, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268: 1899–1902, 1995 [DOI] [PubMed] [Google Scholar]
- 121. Rajan S, Saiman L. Pulmonary infections in patients with cystic fibrosis. Semin Respir Infect 17: 47–56, 2002 [DOI] [PubMed] [Google Scholar]
- 122. Reszka KJ, Xiong Y, Sallans L, Pasula R, Olakanmi O, Hassett DJ, Britigan BE. Inactivation of the potent Pseudomonas aeruginosa cytotoxin pyocyanin by airway peroxidases and nitrite. Am J Physiol Lung Cell Mol Physiol 302: L1044–L1056, 2012 [DOI] [PubMed] [Google Scholar]
- 123. Robriquet L, Collet F, Tournoys A, Prangere T, Neviere R, Fourrier F, Guery BP. Intravenous administration of activated protein C in Pseudomonas-induced lung injury: impact on lung fluid balance and the inflammatory response. Respir Res 7: 41, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rogers CS, Abraham WM, Brogden KA, Engelhardt JF, Fisher JT, McCray PB, Jr, McLennan G, Meyerholz DK, Namati E, Ostedgaard LS, Prather RS, Sabater JR, Stoltz DA, Zabner J, Welsh MJ. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 295: L240–L263, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rollag H, Hovig T. Phagocytosis of non-opsonized Escherichia coli by mouse peritoneal macrophages. An electron microscopic study. Zentralbl Bakteriol Mikrobiol Hyg A 257: 93–107, 1984 [PubMed] [Google Scholar]
- 126. Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2: 11, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ryder C, Byrd M, Wozniak DJ. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr Opin Microbiol 10: 644–648, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171: 1209–1223, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Schulert GS, Feltman H, Rabin SD, Martin CG, Battle SE, Rello J, Hauser AR. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J Infect Dis 188: 1695–1706, 2003 [DOI] [PubMed] [Google Scholar]
- 130. Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 282: L285–L290, 2002 [DOI] [PubMed] [Google Scholar]
- 131. Skerrett SJ, Wilson CB, Liggitt HD, Hajjar AM. Redundant Toll-like receptor signaling in the pulmonary host response to Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 292: L312–L322, 2007 [DOI] [PubMed] [Google Scholar]
- 132. Sonawane A, Jyot J, During R, Ramphal R. Neutrophil elastase, an innate immunity effector molecule, represses flagellin transcription in Pseudomonas aeruginosa. Infect Immun 74: 6682–6689, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Speert DP, Gordon S. Phagocytosis of unopsonized Pseudomonas aeruginosa by murine macrophages is a two-step process requiring glucose. J Clin Invest 90: 1085–1092, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Speert DP, Loh BA, Cabral DA, Salit IE. Nonopsonic phagocytosis of nonmucoid Pseudomonas aeruginosa by human neutrophils and monocyte-derived macrophages is correlated with bacterial piliation and hydrophobicity. Infect Immun 53: 207–212, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Starke JR, Edwards MS, Langston C, Baker CJ. A mouse model of chronic pulmonary infection with Pseudomonas aeruginosa and Pseudomonas cepacia. Pediatr Res 22: 698–702, 1987 [DOI] [PubMed] [Google Scholar]
- 136. Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev 26: 1–43, 2005 [DOI] [PubMed] [Google Scholar]
- 137. Stokes RW, Thorson LM, Speert DP. Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J Immunol 160: 5514–5521, 1998 [PubMed] [Google Scholar]
- 138. Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GAt Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB, Jr, Zabner J, Welsh MJ. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2: 29ra31, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Stotland PK, Radzioch D, Stevenson MM. Mouse models of chronic lung infection with Pseudomonas aeruginosa: models for the study of cystic fibrosis. Pediatr Pulmonol 30: 413–424, 2000 [DOI] [PubMed] [Google Scholar]
- 140. Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity 22: 539–550, 2005 [DOI] [PubMed] [Google Scholar]
- 141. Sukumaran SK, Shimada H, Prasadarao NV. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun 71: 5951–5961, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK, Engelhardt JF. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120: 3149–3160, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sun Y, Karmakar M, Taylor PR, Rietsch A, Pearlman E. ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J Immunol 188: 1884–1895, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204: 3235–3245, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tsai WC, Rodriguez ML, Young KS, Deng JC, Thannickal VJ, Tateda K, Hershenson MB, Standiford TJ. Azithromycin blocks neutrophil recruitment in Pseudomonas endobronchial infection. Am J Respir Crit Care Med 170: 1331–1339, 2004 [DOI] [PubMed] [Google Scholar]
- 146. Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun 68: 4289–4296, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20: 825–852, 2002 [DOI] [PubMed] [Google Scholar]
- 148. van de Weert-van Leeuwen PB, van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, Van der Ent CK, Terheggen-Lagro SW, Arets HG, Beekman JM. Optimal complement-mediated phagocytosis of pseudomonas aeruginosa by monocytes is CFTR-dependent. Am J Respir Cell Mol Biol 49: 463–470, 2013 [DOI] [PubMed] [Google Scholar]
- 149. van Heeckeren AM, Schluchter MD. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim 36: 291–312, 2002 [DOI] [PubMed] [Google Scholar]
- 150. van Heeckeren AM, Tscheikuna J, Walenga RW, Konstan MW, Davis PB, Erokwu B, Haxhiu MA, Ferkol TW. Effect of Pseudomonas infection on weight loss, lung mechanics, and cytokines in mice. Am J Respir Crit Care Med 161: 271–279, 2000 [DOI] [PubMed] [Google Scholar]
- 151. Walters MC, 3rd, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob Agents Chemother 47: 317–323, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wangdi T, Mijares LA, Kazmierczak BI. In vivo discrimination of type 3 secretion system-positive and -negative Pseudomonas aeruginosa via a caspase-1-dependent pathway. Infect Immun 78: 4744–4753, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wei Q, Ma LZ. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int J Mol Sci 14: 20983–21005, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Weichhart T, Saemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis 67, Suppl 3: iii70–iii74, 2008 [DOI] [PubMed] [Google Scholar]
- 155. Zaas DW, Duncan MJ, Li G, Wright JR, Abraham SN. Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. J Biol Chem 280: 4864–4872, 2005 [DOI] [PubMed] [Google Scholar]
- 156. Zaas DW, Swan ZD, Brown BJ, Li G, Randell SH, Degan S, Sunday ME, Wright JR, Abraham SN. Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa. J Biol Chem 284: 9955–9964, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]