

Abstract
Lung fibrosis is the final result of a large number of disorders and is usually considered an irreversible process. However, some evidence suggests that fibrosis could eventually be reversible. In this study we aimed to document the time-related reversibility of bleomycin-induced lung fibrosis and to examine the gene expression profile associated with its initial progression and subsequent resolution. C57BL/6 mice were instilled with a single dose of bleomycin and euthanized at 1, 4, 8, 12, and 16 wk. Control animals received an equal volume of saline. Lung fibrosis was examined by morphology and hydroxyproline content and the transcriptional signature by gene microarray analysis. Our results showed that bleomycin-injured mice developed prominent inflammation at 1 wk, followed by fibrosis that peaked at 2 mo. Then fibrosis resolved until lungs displayed almost normal architecture at 4 mo. Genomewide transcriptional profiling revealed 533 significantly changed genes. Self-organizing maps analysis of these genes identified four clusters based on the temporal pattern of gene expression. Clusters 1 and 2 contained genes upregulated during the inflammatory and fibrotic response and were enriched for extracellular matrix-related genes including several collagens, matrix metalloproteinases, and TIMP-1. Cluster 3 identified upregulated genes during the fibrotic response, and cluster 4 contained genes decreased during inflammation and fibrosis that increased during resolution. Most enriched pathways included genes involved in cell cycle and in regulation of transcription. Our findings corroborate the reversibility of bleomycin-induced lung fibrosis and reveal transcriptional signatures that characterize the progression and resolution.
Keywords: pulmonary fibrosis, fibrosis resolution, MMPS, NFκB2
lung fibrosis represents the final common pathway of a variety of lung injuries, and it is characterized, independently of the etiology, by the expansion of the fibroblast/myofibroblast population and by the abnormal accumulation of extracellular matrix (ECM) replacing normal functional parenchyma (18). In general, there is no effective therapy, and currently the only helpful available treatment for advanced lung fibrosis is transplantation.
However, a growing body of evidence suggests that tissue fibrosis may be a potentially reversible process, providing new insights in the understanding of the mechanisms of remodeling as well as in the achievement of novel therapeutic approaches. In other tissues, evidence for regression has been observed usually after the causative agent is removed or if animals are treated effectively. For example, resolution of experimental kidney fibrosis has been obtained by blocking or antagonizing the action of the renin-angiotensin system, and fibrosis following steatohepatitis may regress when diet is controlled (2, 16). Furthermore, antiviral treatment for infection with either hepatitis B or C virus supports that viral eradication is associated with at least partial regression of liver fibrosis in humans (11). Spontaneous resolution of liver fibrosis has been also demonstrated in a model of CCl4-induced liver injury that has been associated with the presence of macrophage-derived MMP13 (4, 6).
The notion that lung fibrosis may resolve spontaneously in experimental models is suggested by some observations in bleomycin-treated animals (13, 15). However, the molecular mechanisms likely implicated in lung fibrosis resolution in this model are unknown.
In the present study we aimed 1) to document the time-related reversibility of bleomycin-induced lung fibrosis and 2) to determine the gene expression patterns associated with the initial progression and the subsequent regression of the fibrotic response by microarray in this bleomycin mouse model of lung fibrosis.
METHODS
Bleomycin treatment.
Mice (C57BL/6) age 8 to 10 wk were instilled intratracheally with 0.10 U/10 g of bleomycin (Blenoxane, Bristol-Myers Squibb) in 50 μl sterile saline. Control animals received an equal volume of sterile saline (1, 19). Mice were housed in specific pathogen-free conditions and provided with food and water ad libitum. All studies and procedures were approved by the Science and Ethics Committee at the National Institute of Respiratory Diseases and were performed according to the protocols and guidelines of the institutional animal care and use committee. Mice were euthanized at 1, 4, 8, 12, and 16 wk after bleomycin or saline treatment.
Morphology.
Lungs were lavaged with saline solution through the main pulmonary artery, and the right lung was removed and fixed by inflation with 4% paraformaldehyde in PBS at continuous pressure of 25 cmH2O and embedded in paraffin. Sections were stained with hematoxylin-eosin and Masson's trichrome stain. Lung fibrosis was quantified by using a semiquantitative score, as previously described (9).
Hydroxyproline assay.
To quantify collagen concentration, left lungs were hydrolyzed in 6 N of HCl for 24 h at 110°C, and hydroxyproline was quantified as described elsewhere (26). Each sample was tested in triplicate. Data are expressed as micrograms of hydroxyproline per left lung.
RNA extraction and preparation of labeled cRNA.
RNA was prepared from right lungs by using TRIzol reagent (Life Technologies, Grand Island, New York, NY) following the manufacturers' instructions. An additional cleanup of total RNA was carried out with the RNeasy Mini kit (Qiagen). Purity of the samples and efficiency of the extractions were verified by spectrophotometry (NanoDrop; Wilmington, DE) and bioanalysis (Agilent; Palo Alto, CA). Total RNA was used as template for double-stranded cDNA synthesis. cDNA was then purified with a QIAquick purification kit (Qiagen, Valencia, CA) and used to obtain biotin-labeled cRNA by an in vitro transcription reaction. Biotin-labeled cRNA was recovered by use of the RNeasy kit.
Microarray analysis.
Biotin-labeled cRNA from each mouse lung was fragmented and hybridized with an individual CodeLink Mouse Uniset 20K BioArray (Amersham Biosciences/GE Healthcare). After hybridization, arrays were stained with Cy5-streptavidin and washed before scanning. The arrays were scanned with a GenePix 4000B microarray scanner (Axon Instruments, Union City, CA). Images of each array were analyzed using CodeLink Expression Analysis software (Amersham BioSciences). Raw intensity values for each spot were corrected for background by subtracting median local background intensity determined at the default setting in CodeLink Expression Analysis software as suggested by the manufacturer (27).
Microarray analyses were performed with GeneSpring GX software (Agilent, Santa Clara, CA) or BRB array tools (21, 22). Temporally differentially expressed genes were identified by one-way ANOVA [P < 0.05, false discovery rate (FDR) < 5%]. Hierarchical clustering and self-organizing maps (SOM) were performed with GeneSpring. For clustering and SOM, all sample values were compared with the geometric mean of the saline controls at the initial time point. This was performed by log base 2 transformation of the data and subtraction of the geometric mean of the saline control mice expression level. Microarray data were submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov./geo/) accession number GSE42301.
Quantitative real-time RT-PCR.
Total lung RNA was extracted from mouse lungs with TRIzol reagent (Invitrogen Life Technologies, Grand Island, NY); 1 μg of RNA was treated with 1 unit of DNase and reversed transcribed into cDNA (Advantage RT-for-PCR Kit; Clontech, Palo Alto, CA) according to the manufacturer's instructions. Quantitative real-time PCR amplification was performed with specific FAM dye-labeled TaqMan probes for MMP-8, TIMP-1, NFκB2, and 18S rRNA (PE Applied Biosystems) and i-Cycler iQ Detection System (Bio-Rad, Hercules, CA). PCR was performed under the following conditions: 95°C for 10 min; 40 cycles at 95°C for 15 s, and 60°C for 1 min. Results were expressed as the ratio of the target gene normalized to 18S rRNA.
Immunohistochemistry.
Mouse lung sections were incubated with rabbit polyclonal anti NFκB2 (Abcam ab. 31409 Cambridge, UK) as previously reported. A secondary biotinylated anti-immunoglobulin followed by horseradish peroxidase-conjugated streptavidin (BioGenex, San Ramon, CA) was used according to the manufacturer. 3-Amino-9-ethylcarbazole (BioGenex) was used as substrate (1).
Statistical analysis.
Statistical differences between groups were determined by one-way ANOVA followed by Tukey's test for quantitative PCR and hydroxyproline measurement. Fibrosis score was evaluated by the nonparametric Kruskal-Wallis test followed by nonparametric Mann-Whitney U-test. Results are expressed as means ± SD. P value < 0.05 was considered statistically significant.
RESULTS
Bleomycin injury induced a spontaneously reversible lung fibrotic reaction.
To evaluate the putative long-term resolution of the fibrotic changes induced by bleomycin, lung collagen content and histopathology were assessed at 1, 4, 8, 12, and 16 wk after 0.1 U/10 g bleomycin instillation. Intratracheal treatment with this dose of bleomycin resulted in 36% mortality during the first week and 50% through the following 8 wk without additional mortality after this time point. Representative photomicrographs of the lung histology are shown in Fig. 1, A–F. Lungs of saline-treated animals were normal. After 1 wk of bleomycin treatment, tissue sections showed a prominent inflammatory response, characterized by neutrophils, lymphocytes, and macrophages infiltrating the alveolar spaces and the interstitium. At 4 wk, multifocal areas of interstitial fibrosis and epithelial bronchiolization near to fibrotic areas were observed. Fibrosis was more extensive after 8 wk postbleomycin and was characterized by intra-alveolar and septal fibrosis with dense collagen accumulation and loss of pulmonary architecture. At 12 wk after bleomycin the extent of the lesions markedly decreased and at 16 wk the lungs showed minimal lesions. Semiquantitative analysis of the histopathological changes corroborated these observations (Fig. 1G). In accordance with the results of the morphological scoring, collagen accumulation examined by hydroxyproline content was markedly increased in the lungs of mice at 4 and 8 wk after bleomycin treatment. As illustrated in Fig. 1H, lung hydroxyproline increased at 4 wk postbleomycin (90.1 ± 5.5 vs. 50.1 ± 4.3 μg/left lung in saline controls), reaching a peak at 8 wk (187.2 ± 22.0; P < 0.001), which was followed by a significant reduction at 12 and 16 wk (92.2 ± 23.0 and 70.9 ± 13.0, respectively; P < 0.05 compared with 8 wk). Hydroxyproline levels at 16 wk showed no statistical difference with saline control levels.
Fig. 1.
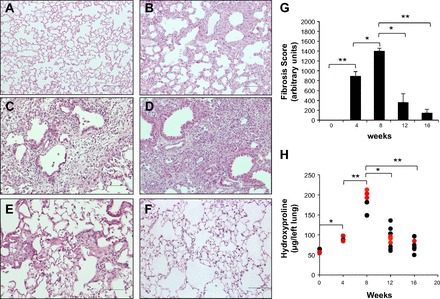
Histological assessment and collagen content in lung tissue during the time course of bleomycin-induced lung fibrosis. A–F: representative light micrographs in lung tissue sections of saline- and bleomycin-instilled mice stained with hematoxylin-eosin. A: saline-instilled mouse. B–F: bleomycin-instilled mice at 1, 4, 8, 12, and 16 wk, respectively. Scale bars indicate 50 μM. Histopathology shows dense areas of lung fibrosis at 4 wk (C) with a more severe and diffused fibrotic reaction at 8 wk (D). At 12 wk (E) there was a focal and markedly less-pronounced fibrosis and at 16 wk (F) most areas of the lung parenchyma were preserved. G: fibrosis score. Data are expressed as means ± SD (n = 6–9 mice in each time point). H: total lung collagen levels were measured by hydroxyproline content (n = 6–9 mice in each time point). Red points represent mice selected for microarray analysis. *P < 0.05; **P < 0.001.
Microarray analysis identified 533 differentially expressed genes in the time course of bleomycin-induced lung injury.
To better understand the mechanisms involved in epithelial injury, inflammation, and fibroproliferation during the time course of bleomycin-induced lung injury, and to identify candidate genes that might be involved in lung repair and reversibility after fibrosis, we performed microarray-based analysis of global transcription on whole lung mRNA from saline control and bleomycin-treated mice euthanized at 1, 4, 8, 12, and 16 wk postinstillation. Three representative animals of each group were selected for microarray analysis (red points from Fig. 1H). Using one-way ANOVA, we identified 533 genes that were differentially expressed (P < 0.05, FDR <5%; Fig. 2A). Consistent with the histopathological fibrosis score and total lung collagen content, several genes that encode proteins that constitute ECM, such as fibrillar collagens (Col1a1, Col1a2, Col5a1, Col5a2, Col5a3), and genes that play a role in ECM remodeling, such as matrix metalloproteinases (Mmp8, Mmp12, Mmp14, Mmp15, Mmp19), and specific inhibitors, such as Timp1, were significantly increased during the fibrotic response and decreased during fibrosis resolution (Fig. 2B).
Fig. 2.
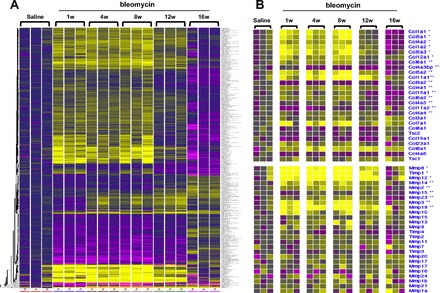
Gene expression infograms for time course of bleomycin-induced lung fibrosis. A: gene expression infogram for all 533 differentially expressed genes. B: gene expression infogram for most informative genes that code for extracellular matrix (ECM) components or play a significant role in ECM metabolism. Every row represents a gene and every column a mouse lung sample. Increased genes are shown in progressively brighter shades of yellow, and decreased genes are shown in progressively darker shades of purple. Genes shown in gray are not different between the groups. The baseline per gene was defined as the geometric mean of the expression of the gene in the saline controls mice at the initial time point.
SOM clustering of these genes revealed four clusters with distinct gene expression patterns. To approach the mechanistic nature of the genes that differentiate the four clusters, we examined the enrichment of functional annotations in each cluster using the National Institutes of Health DAVID (http://david.abcc.ncifcrf.gov/) (10). The gene expression pattern and functional enrichment of every cluster are shown in Figs. 3 and 4.
Fig. 3.
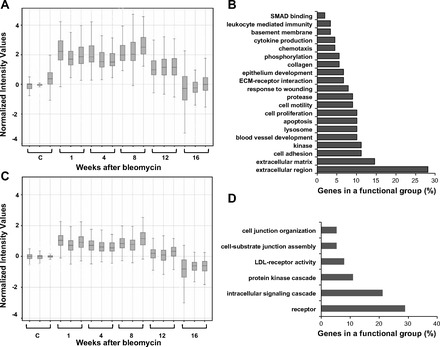
Gene expression patterns of clusters 1 and 2 and enrichment of genes in each cluster with functional categories. Self-organizing maps (SOM) clustering of differentially expressed genes revealed four clusters with distinct gene expression patterns. Time-course profiles of cluster 1 (A) and cluster 2 (C) and enrichment of functional annotations in cluster 1 (B) and cluster 2 (D) are shown. Genes contained within each cluster are listed in Supplementary Tables S1 and S2, respectively.
Fig. 4.
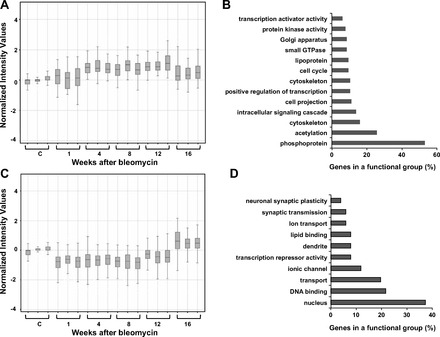
Gene expression patterns of genes in clusters 3 and 4 and enrichment of genes in each cluster with functional categories. Time-course profiles of cluster 3 (A) and cluster 4 (C) and enrichment of functional annotations in cluster 3 (B) and cluster 4 (D) are shown. Genes contained within each cluster are listed in Supplementary Tables S3 and 4, respectively.
Genes in cluster 1 increased (2-fold change) after 1 wk in the inflammatory phase and stayed up during the fibrotic phases until 8 wk postbleomycin, then they decreased at 12 wk and returned to baseline (similar to saline control expression levels) at 16 wk (Fig. 3A). Cluster 1 contained 125 genes grouped in 20 functional categories; among them the most highly represented categories were extracellular region, ECM, cell adhesion, and kinase activity [Fig. 3B, Supplementary Table S1 (Supplemental Material for this article is available online at the Journal website)]. Extracellular region and ECM functional annotations include Col1a1, Col1a2, Col5a1, Col12a1, Fmod, Mmp12, Mmp8, Tnc, Timp1, and Tgfbi genes. These genes are involved in the development and progression of lung fibrosis.
Genes in cluster 2 shared a similar pattern of gene expression with cluster 1 during inflammatory, fibrotic, and resolution phases (Fig. 3C); however, these genes reach lower levels of expression compared with genes in cluster 1. Cluster 2 contained 82 genes grouped in six functional categories (Fig. 3D, Supplementary Table S2). The most representative categories were receptor activity, intracellular signaling cascade, and protein kinase cascade, suggesting that genes involved in signal transduction that influence ECM synthesis, chemotaxis, angiogenesis, proliferation, and apoptosis are upregulated during inflammation and fibrosis.
Cluster 3 revealed genes that were upregulated at 4, 8, and 12 wk and returned to base line at 16 wk postbleomycin (Fig. 4A). This cluster contained 185 genes grouped in 13 functional categories including transcription activator activity, intracellular signaling cascade, phosphoprotein, acetylation, and cytoskeleton (Fig. 4B, Supplementary Table S3). Finally, in cluster 4 we identified genes that became downregulated during inflammatory and fibrotic response and returned to normal values or increased during resolution (Fig. 4C). This cluster was composed by 141 genes grouped in 10 functional categories. Most enriched pathways for microarray transcripts that decreased during the inflammatory and fibrotic response and increased during resolution included nucleus, DNA binding, and transport (Fig. 4D, Supplementary Table S4). Nucleus and DNA-binding categories grouped genes involved in cell-cycle such as Cdc14b, Arid3, and G0/G1 switch gene 2, and, in regulation of transcription, genes such as Fkhrl1, Mybl1, T2, Nr2e3, Pax3, Phtf1, Surf 6, Xpa, and Ikbkb.
Transcript levels of two candidate genes that were upregulated in cluster 1 (MMP8 and TIMP1) and one upregulated in cluster 3 (NFκB2) were validated by quantitative real-time RT-PCR. As illustrated in Fig. 5, A–C, the results obtained correlated with the temporal patterns measured by microarray. TIMP-1 and MMP-8 displayed the higher levels at 1 wk, during the inflammatory response, decreasing mildly after 8 wk postbleomycin instillation, reaching almost normal values at 12–16 wk. NFκB2 increased at first week and showed a marked upregulation at 4 and 8 wk with a significant decrease at 12 and 16 wk.
Fig. 5.
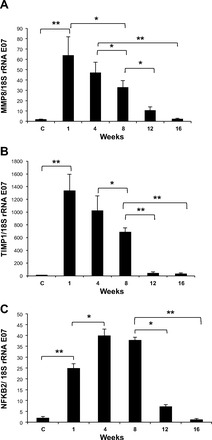
Expression levels of representative genes upregulated in clusters 1 and 3. Lung mRNA was isolated and transcript expression levels of TIMP-1 (A), MMP-8 (B), and NFκB2 (C) were measured by real-time quantitative reverse transcription-polymerase chain reaction. Data are expressed as means ± SD (n = 6 mice in each time point). *P < 0.05; **P < 0.001.
We explored by immunohistochemistry the expression NFκB2 in the lungs of mice instilled with bleomycin. As shown in Fig. 6 strong NFκB2 staining was observed in fibrotic areas mainly at 4 and 8 wk postbleomycin (Fig. 6, C and D) and localized in hyperreactive cuboidal epithelial cells and macrophages. Less immunoreactive NFκB2 was present at 1 and 12 wk (Fig. 6, B and E), mostly in macrophages, whereas it was virtually negative at 16 wk postbleomycin (Fig. 6F).
Fig. 6.
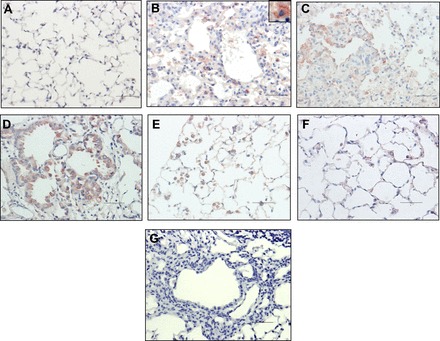
Immunolocalization of NFκB2 in lung tissue during the time course of bleomycin-induced lung fibrosis. Representative photomicrographs of immunohistochemical staining performed with specific antibody against NFκB2 at 1 (B), 4 (C), 8 (D), 12 (E), and 16 (F) wk after bleomycin instillation. Saline-instilled mouse lung is shown in A, and negative control of 8 wk postbleomycin in which the primary antibody was omitted is shown in G. Immunoreactive protein was observed in alveolar epithelial cells (C and D) and macrophages (B inset, E, and F). All sections were counterstained with hematoxylin. Scale bars indicate 50 μM.
DISCUSSION
Lung fibrosis represents the outcome of a repair process characterized by the replacement of the alveolar-capillary structures by permanent scar tissue and, in general, is considered an irreversible process. In other words, pulmonary fibrosis is regarded as the final result of virtually all chronic lung disorders in which the tissue is unable of regeneration and, as consequence, without option to return to the preexisting normal structure. However, a growing body of evidence indicates that “tissue fibrosis” is a highly dynamic pathological state and may eventually be reversible, although key questions remain. Reversibility has been reported mainly in kidney and hepatic fibrosis, primarily in experimental models, although the feasibility to transfer this scientific promise to human fibrosis is presently unclear (2, 4–6, 11, 16, 23). Also, studies in animal models suggest that hypertrophic cardiomyopathy, characterized by cardiomyocyte hypertrophy, myofibrillar disarray, and fibrosis, may be potentially reversible. However, current treatments have not been proved to reverse or even ameliorate cardiac hypertrophy in patients (17). Usually, regression is associated to both the removal of the etiological agent or condition and an effective therapy, whereas research on spontaneous regression is scanty. Regarding lung fibrosis, some evidence suggests that at least bleomycin-induced fibrosis may have a self-limiting response (13, 15).
In this study we corroborated that lung fibrosis induced by a single intratracheal instillation of bleomycin is a spontaneously reversible process. Following the inflammatory response, evaluated here at 1 wk postinstillation, fibrosis developed, reaching a peak at 8 wk. Progressive lung collagen accumulation was confirmed by histology and fibrosis score as well as by the biochemical increase of hydroxyproline. Then this fibrotic response was followed by a noteworthy reduction at 12 wk with almost total regression at 16 wk.
To better understand the mechanisms underlying progression and regression of the lung fibrotic response, we compared the transcriptional signatures of lung tissues at different times after bleomycin instillation to the initial saline time point. Although some contribution of aging cannot be ruled out after 12–16 wk of aging, we thought that the analysis of the global temporal changes in gene expression during development and resolution of pulmonary fibrosis, comparing to one time point, was an acceptable approach.
SOMs from 533 differentially expressed genes revealed four clusters that grouped the transcripts that increased during the inflammatory and fibrotic response (clusters 1 and 2), those that increased during the fibrotic response (cluster 3), and those that decreased during fibrosis and increased during regression (primarily cluster 4).
As expected, a number of genes implicated in the ECM remodeling were upregulated during the development of fibrosis, including collagens, fibromodulin, and tenascin-C, as well as several matrix metalloproteinases and TIMP-1. Actually, Gene Ontology and functional enrichment analysis revealed that the highly enriched and overrepresented genes belong to this category. We choose to validate two genes from cluster 1 (MMP-8 and TIMP-1) by quantitative real-time PCR, because they have been implicated in lung fibrosis. Thus several studies have demonstrated that increased expression of TIMP-1 plays a critical role in the development of experimental lung fibrosis (1). Moreover, TGF-β1 induces a strong overexpression of TIMP-1 in fibroblasts and lungs of fibrosis-prone mice in contrast to weak induction in fibrosis-resistant mice suggesting that differences in the fibrotic response might be in part due to the increase of this inhibitor (12). On the other hand, MMP-8, an enzyme produced mainly by neutrophils, has been recently implicated in the migration and homing of fibrocytes, one of the likely sources of myofibroblasts, to lung tissue (7). Moreover, MMP-8-deficient mice appear to be resistant to bleomycin-induced lung fibrosis (8). Other ECM genes revealed in cluster 1 are involved in stabilization of fibrillar structures, such as lysyl oxidase-like-2, which catalyzes the formation of cross-links in collagens and elastin proteins.
Lung fibrosis is associated with altered expression of TGFβ receptors, increased activity of TGF-β1 signaling pathways, and upregulation of several TGF-β-regulated genes. In this context, cluster 1 also included several genes involved in TGF-β signaling pathway including the type II TGF-β receptor (TGFβRII). Ligand engagement by this receptor is a major step in TGF-β signaling, and mice with epithelium-specific deletion of TGFβRII are protected from bleomycin-induced pulmonary fibrosis, which is associated with reduced phosphorylation of Smad3 (14).
Several genes involved in the immune and inflammatory response, including two chemokines receptors, Ccr1 and Cxcr7, were also upregulated. Ccr1, a receptor for CCL3/MIP-1α, CCl4/MIP-1β and RANTES, play a crucial role in the recruitment of immune cells to the site of inflammation, and, interestingly, Ccr1-null mice are protected from experimentally induced lung and liver fibrosis (20, 29). Cxcr7 is a chemokine receptor expressed by fibrocytes, but its role in fibrosis has not been explored. Collectively, our findings indicate that most of the cluster 1 genes are implicated in ECM structure, fibroproliferative response, and recruitment and homing of inflammatory cells and fibrocytes, suggesting significant activity in tissue remodeling and reorganization in the lung.
On the other hand, cluster 2, that followed the same time course pattern than cluster 1 included as major categories of significantly upregulated genes those implicated in diverse signaling cascades, including Wnt signaling pathway that may sustain fibrogenesis enhancing the secretion of oxidants, inflammatory cytokines, and growth factors involved in lung fibroblast activation and proliferation. Importantly, the proinflammatory and profibrotic genes from these two clusters significantly decrease after 8-wk postbleomycin instillation, returning to normal values at 16 wk.
Cluster 3 identified genes that were upregulated during the fibrotic response and returned to control levels at 16 wk postbleomycin. This cluster included genes associated with cytoskeleton and cell adhesion such as Adducin 1, actin bundling protein 1, and Caldesmon 1, and genes involved in proteolysis and cell-cell and cell-matrix interactions like a disintegrin and metalloprotease 8 and 9 (Adam8 and Adam9). There were also several genes related to signaling cascade and inflammatory response such as interferon (alpha and beta) receptor 2 (Ifnar2) and the chemokine Cxcl4.
NFκB2, a member of the NFκB family of transcription factors, was one of the upregulated transcripts in cluster 3 and was also selected for validation by quantitative PCR. NFκB2 plays an important role in inflammation and, actually, recent genetic evidence indicates that negative feedback control mechanisms are necessary to limit the inflammatory effect of sustained activation of NFκB2 signaling (28).
Interestingly, although NFκB2 fulfills a central role in inflammation, we found by real-time PCR and immunohistochemistry that it highly increased during the fibrotic response.
In parallel, a number of genes were downregulated during the fibrotic reaction, increasing during regression (cluster 4), suggesting that their decrease is involved in fibrogenesis. Functional categories in this cluster included nucleus and DNA-binding genes involved in cell-cycle and in transcription regulation. An interesting downregulated gene in this category was G0/G1 switch gene 2 (G0s2), a novel target gene of peroxisome proliferator-activated receptors (PPARs). Interestingly, the reduction of fibroblast proliferation and its differentiation into myofibroblasts by PPARδ is associated with upregulation of this cell cycle inhibitory G0s2 gene (24). Likewise, a zinc finger protein involved in DNA excision repair (Xpa) that is part of the nucleotide excision repair (NER) complex and is involved in the DNA damage recognition was also decreased during the inflammatory/fibrotic reaction. Recently it was demonstrated that downregulation of NER-associated genes (including Xpa) are associated with LPS-induced lung inflammation (9).
In summary, in this study we identify transcriptional signatures that characterize the progression and regression of bleomycin-induced lung inflammation and fibrosis. Enrichment of functional annotations and network analyses identified several areas of biological importance, yielding new insight into mechanisms involved in fibrosis development and resolution. In general terms, the regression of the fibrotic response was caused by the turnoff of the complex ECM remodeling program.
However, some important questions remain. For example, why is this model spontaneously reversible? It is likely that, at least in part, the reversibility observed in this study was caused by the use of a single-dose injury. In this context, it has been recently found that the use of repetitive instillation of bleomycin by laryngeal intubation provokes a marked fibrotic response that persisted 10 wk after the last dose (3). Perhaps, as has been suggested, the term “lung remodeling” should be used to describe the dynamic repair multistep process that occurs in the lung following inflammatory injury, which may be reversible with resolution and return the lung to the preexisting structure (i.e., after a single dose of bleomycin) or progress to the point of irreversible scarring, as was reported with repetitive bleomycin injuries (25).
Supplementary Material
REFERENCES
- 1. Cabrera S, Gaxiola M, Arreola JL, Ramírez R, Jara P, D'Armiento J, Richards T, Selman M, Pardo A. Overexpression of MMP9 in macrophages attenuates pulmonary fibrosis induced by bleomycin. Int J Biochem Cell Biol 39: 2324–2338, 2007. [DOI] [PubMed] [Google Scholar]
- 2. Chatziantoniou C, Dussaule JC. Is kidney injury a reversible process? Curr Opin Nephrol Hypertens 17: 76–81, 2008. [DOI] [PubMed] [Google Scholar]
- 3. Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 299: L442–L452, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duffield J, Forbes S, Constandinou C, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale J. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115: 56–65, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dussaule JC, Guerrot D, Huby AC, Chadjichristos C, Shweke N, Boffa JJ, Chatziantoniou C. The role of cell plasticity in progression and reversal of renal fibrosis. Int J Exp Pathol 92: 151–157, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 178: 5288–5295, 2007. [DOI] [PubMed] [Google Scholar]
- 7. García-de-Alba C, Becerril C, Ruiz V, González Y, Reyes S, García-Alvarez J, Selman M, Pardo A. Expression of matrix metalloproteases by fibrocytes: possible role in migration and homing. Am J Respir Crit Care Med 182: 1144–1152, 2010. [DOI] [PubMed] [Google Scholar]
- 8. García-Prieto E, González-López A, Cabrera S, Astudillo A, Gutiérrez-Fernández A, Fanjul-Fernandez M, Batalla-Solís E, Puente XS, Fueyo A, López-Otín C, Albaiceta GM. Resistance to bleomycin-induced lung fibrosis in MMP-8 deficient mice is mediated by interleukin-10. PLoS One 5: e13242, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Güngör N, Haegens A, Knaapen AM, Godschalk RW, Chiu RK, Wouters EF, van Schooten FJ. Lung inflammation is associated with reduced pulmonary nucleotide excision repair in vivo. Mutagenesis 25: 77–82, 2010. [DOI] [PubMed] [Google Scholar]
- 10. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc 4: 44–57, 2009. [DOI] [PubMed] [Google Scholar]
- 11. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 117: 539–548, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kolb M, Bonniaud P, Galt T, Sime PJ, Kelly MM, Margetts PJ, Gauldie J. Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of “fibrosis-prone” and “fibrosis-resistant” mouse strains. Am J Respir Cell Mol Biol 27: 141–150, 2002. [DOI] [PubMed] [Google Scholar]
- 13. Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 167: 1267–1277, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 121: 277–287, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L152–L160, 2008. [DOI] [PubMed] [Google Scholar]
- 16. Mu YP, Ogawa T, Kawada N. Reversibility of fibrosis, inflammation, and endoplasmic reticulum stress in the liver of rats fed a methionine-choline-deficient diet. Lab Invest 90: 245–256, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Orenes-Piñero E, Hernández-Romero D, Jover E, de la Morena G, Valdés M, Marín F. An insight of novel pharmacological therapies in hypertrophic cardiomyopathy. Med Chem 7: 275–285, 2011. [DOI] [PubMed] [Google Scholar]
- 18. Pardo A, Selman M. Molecular mechanisms of pulmonary fibrosis. Front Biosci 7: d1743–d1761, 2002. [DOI] [PubMed] [Google Scholar]
- 19. Pardo A, Ruiz V, Arreola JL, Ramírez R, Cisneros-Lira J, Gaxiola M, Barrios R, Kala SV, Lieberman MW, Selman M. Bleomycin-induced pulmonary fibrosis is attenuated in gamma glutamyl transpeptidase-deficient mice. Am J Respir Crit Care Med 167: 925–932, 2003. [DOI] [PubMed] [Google Scholar]
- 20. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest 119: 1858–1870, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon R, Korn E, McShane L, Radmacher M, Wright GYZ. Design and Analysis of DNA Microarray Investigations. New York: Springer-Verlag, 2003. [Google Scholar]
- 22. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of Gene Expression Data Using BRB-Array Tools. New York: Springer-Verlag, 2007. [PMC free article] [PubMed] [Google Scholar]
- 23. Snowdon VK, Fallowfield JA. Models and mechanisms of fibrosis resolution. Alcohol Clin Exp Res 35: 794–799, 2011. [DOI] [PubMed] [Google Scholar]
- 24. Teunissen BE, Smeets PJ, Willemsen PH, De Windt LJ, Van der Vusse GJ, Van Bilsen M. Activation of PPARdelta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res 75: 519–529, 2007. [DOI] [PubMed] [Google Scholar]
- 25. Wallace WA, Fitch PM, Simpson AJ, Howie SE. Inflammation-associated remodelling and fibrosis in the lung — a process and an end point. Int J Exp Pathol 88: 103–110, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this amino acid. Arch Biochem Biophys 93: 440–447, 1961. [DOI] [PubMed] [Google Scholar]
- 27. Wu W, Dave N, Tseng GC, Richards T, Xing EP, Kaminski N. Comparison of normalization methods for CodeLink Bioarray data. BMC Bioinformatics 6: 309, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang L, Cui H, Wang Z, Zhang B, Ding J, Liu L, Ding HF. Loss of negative feedback control of nuclear factor-kappaB2 activity in lymphocytes leads to fatal lung inflammation. Am J Pathol 176: 2646–2657, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang X, Walton W, Cook DN, Hua X, Tilley S, Haskell CA, Horuk R, Blackstock AW, Kirby SL. The chemokine, CCL3, and its receptor, CCR1, mediate thoracic radiation-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 45: 127–135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.