

Abstract
Because doxorubicin (DOX)-containing chemotherapy causes left ventricular (LV) dysfunction and remodeling that can progress to heart failure, strategies to alleviate DOX cardiotoxicity are necessary to improve health outcomes of patients surviving cancer. Although clinical evidence suggests that aerobic exercise training (ET) can prevent cardiotoxicity in patients undergoing DOX chemotherapy, the physiological mechanisms involved have not been extensively studied, nor is it known whether compounds [such as resveratrol (RESV)] have similar beneficial effects. With the use of a murine model of chronic DOX exposure, this study compared the efficacy of modest ET to RESV treatment on exercise performance, LV remodeling, and oxidative stress resistance. Mice were divided into four groups that received saline, DOX (8 mg/kg ip, one time per week), DOX + RESV (4 g/kg diet, ad libitum), and DOX + ET (45 min of treadmill exercise, 5 days/wk) for 8 wk. LV function and morphology were evaluated by in vivo echocardiography. DOX caused adverse LV remodeling that was partially attenuated by modest ET and completely prevented by RESV. These effects were paralleled by improvements in exercise performance. The cardioprotective properties of ET and RESV were associated with reduced levels of atrial natriuretic peptide and the lipid peroxidation by-product, 4-hydroxy-2-nonenal. In addition, ET and RESV increased the expression of cardiac sarcoplasmic/endoplasmic reticulum calcium-ATPase 2a, superoxide dismutase, mitochondrial electron transport chain complexes, and mitofusin-1 and -2 in mice administered DOX. Compared with modest ET, RESV more effectively prevented DOX-induced LV remodeling and was associated with the reduction of DOX-induced oxidative stress. Our findings have important implications for protecting patients against DOX-associated cardiac injury.
Keywords: cardiotoxicity, exercise, oxidative stress, resveratrol
survival of cancers has improved with advancement in screening and therapy over the last four decades (6). Anthracycline-containing chemotherapies [e.g., doxorubicin (DOX)] have broad-spectrum anti-tumor activity, making DOX central for the treatment of several types of cancer, such as breast, ovarian, lymphoma, and multiple myelomas (50). However, DOX may trigger dose-dependent, cumulative, and progressive left ventricular (LV) remodeling (e.g., cavity dilation and decreased global systolic function) that may progress to heart failure (33, 34, 42, 48). The incidence of heart failure in modern adjuvant trials with DOX-containing regimens is 0–2.1% (46), with corresponding rates of asymptomatic LV systolic dysfunction between ∼30 and 50% (28, 33). The onset of cardiotoxicity is clinically important because chemotherapy is withheld when LV ejection fraction (LVEF) falls below the lower limit of normal (i.e., 50% or >10% absolute decrease from baseline) (15). Because of the anti-cancer benefits of DOX, strategies to alleviate its cardiotoxic side effects are necessary to improve the quality of life for these patients.
While research has suggested that the development of DOX-induced cardiotoxicity is multifactorial in nature (37, 54), the majority of studies support the involvement of increased oxidative stress due to increased levels of reactive oxygen species (ROS) as contributing to DOX-induced cardiotoxicity (39). Mitochondria are a major source of ROS, and data indicate that DOX causes disruption of the electron transport chain (3) that results in impaired mitochondrial function and increased ROS production. Therefore, strategies aimed at reducing ROS and improving mitochondrial function could prevent and/or halt the progression of DOX-induced cardiotoxicity and significantly improve the long-term survival of chemotherapy patients. Interestingly, aerobic exercise training (ET) augments mitochondrial function, in part by reducing ROS levels and increasing mitochondrial biogenesis (reviewed in Ref. 1). Moreover, long-term ET reduces ROS levels in muscle (1), suggesting that ET may assist with lessening DOX-induced cardiotoxicity.
Consistent with the beneficial effects associated with exercise in DOX-treated patients, clinical evidence suggests that ET may prevent LV dysfunction and improve exercise performance in patients undergoing chemotherapy (22, 23, 53). Despite this benefit, the improvement in aerobic capacity in patients undergoing adjuvant therapy is small (23), a finding due in part to low adherence with programs of exercise (19). As such, pharmaceutical agents or nutritional supplements that possess similar biological benefits as ET should provide significant clinical value. Of importance, the naturally occurring polyphenol resveratrol (RESV) has recently been shown to both mimic (26) and augment ET (11) via improving LV function and increasing skeletal muscle mitochondrial biogenesis and fatty acid oxidation. Similar to ET, RESV also reduces ROS production in the hearts of several animal models (9, 44, 45). Based on these findings, we performed the first study to directly compare the effects of modest ET to RESV on DOX-induced cardiotoxicity, exercise performance, and markers of mitochondrial function and oxidative stress resistance.
Unlike several recent studies that have used supraclinical dosages of DOX to investigate mechanisms of DOX-induced cardiotoxicity (24, 43, 53, 55), we used a clinically relevant chronic DOX dose regime in our mouse model to mimic chemotherapy of breast cancer patients. Herein, we show that DOX induced a significant decline in LVEF, whereas modest ET attenuated and RESV completely prevented this. Both modest ET and RESV improved exercise tolerance in the setting of DOX-induced cardiotoxicity. These favorable adaptations were associated with reduced levels of lipid peroxidation and enhanced antioxidant defenses in both ET and RESV mice. Furthermore, in mice undergoing DOX treatment, modest ET and RESV preserved the expression of mitochondrial electron transport chain complexes and stimulated the expression of mitochondrial fusion proteins mitofusin-1 and -2, which may be necessary for a healthy mitochondrial environment (2). As such, modest ET and/or administration of RESV to patients receiving DOX-based therapies may provide significant benefit by reducing the risk for chemotherapy-induced LV dysfunction.
MATERIALS AND METHODS
Materials.
Most antibodies used in this study were purchased from Cell Signaling Technology or Santa Cruz Biotechnology. The mitofusin-1 antibody was from the University of California-Davis/NIH NeuroMab Facility, and the mitofusin-2 antibody was from Sigma. Most other reagents and chemicals were purchased from Sigma. RESV was purchased from Lalilab (Durham, NC).
Animal care.
All animals and procedures were conducted in accordance with the University of Alberta Animal Policy and Welfare Committee. Eight-week-old female C57BL6 mice were purchased from Charles River Laboratories (Pointe-Claire, QC). Before randomization, all mice were lightly exercised on the treadmill (10 m/min) for 30 min/day for 5 days to acclimatize them to the treadmill so as not to bias the final endurance test. Mice were under a 12:12-h light-dark cycle (0600–1800) and had free access to drinking water and food. Following acclimatization, at 10 wk of age, the mice were randomly assigned to one of four groups (n = 9–11/group): 1) sedentary and treated with saline (CON), 2) sedentary and treated with doxorubicin (DOX), 3) aerobic training together with doxorubicin treatment (DOX + ET), and 4) sedentary and treated with doxorubicin and resveratrol (DOX + RESV). Mice were permitted ad libitum consumption of either an AIN93G standard chow diet or an AIN93G standard chow diet that was supplemented with 4 g RESV/kg of diet (Dyets, Bethlehem, PA), a dosage that is equivalent to ∼320 mg RESV·kg−1·day−1. The dosage of RESV was consistent with previous studies (11, 13). The CON group received weekly intraperitoneal injection of 0.9% saline. DOX (Adriamycin hydrochloride; Sigma Aldrich Canada, Oakville, ON) was administered via weekly intraperitoneal injections of 8 mg·kg−1·wk−1 for a total of 4 wk. The sedentary animals were handled daily. Aerobic ET was performed between 0900 and 1200 and consisted of progressive treadmill running up to 18 m/min at 0% grade for 45 min 5 days/wk for 8 wk on a motorized treadmill (EXER 3/6; Columbus Instruments, Columbus, OH) as previously described (11). ET began at 10 m/min, 0% grade, for 10 min before the first DOX injection and was systematically increased until the desired exercise intensity was achieved. Forty-eight hours after the final exercise session, all mice were killed. Organs of interest were excised and snap-frozen in liquid nitrogen and stored at −80°C for biochemical analysis.
Test of exercise capacity.
Exercise tests were performed by a technician blinded to the identity of the mice. A motorized treadmill (EXER 3/6; Columbus Instruments) equipped with electrical stimulation (0.25 mA, 1 Hz for 200 ms in length) was used according to the following protocol: 10 m/min for 1 min, 11 m/min for 1 min, 12 m/min for 1 min, 13 m/min for 2 min, 15 m/min for 5 min, 17 m/min for 5 min, and 20 m/min until fatigue. The test was terminated when animals were unwilling to run despite >30 total seconds at the beginning of the treadmill and were unmotivated to run despite repeated prodding by an air puff. Exercise capacity is defined as the total amount of time to reach fatigue and the distance covered.
In vivo LV function and morphology.
Transthoracic echocardiography was performed by an experienced and blinded research animal echocardiographer, using the 30-MHz transducer (RMV-716; Visual Sonics, Toronto, ON) as previously described (12). Mice were mildly anesthetized (sedated with 3% isofluorane and 1.0 l/min oxygen and maintained at 1–1.5% isofluorane and 1 l/min oxygen). Rodent paws were taped to ECG metal strips on a mouse-handling platform (P/N 11437; Visual Sonics) to obtain ECG tracings simultaneously with the cardiac images. Two-dimensional M-mode recordings were obtained by transthoracic echocardiography short-axis views at the level of the papillary muscles, and standard calculations using this view were made. Ventricular dimensions were obtained from M-mode measurements of at least three to six cardiac cycles, and percent ejection fraction and percent fractional shortening were determined. Doppler tissue imaging from the apical four-chamber view was used to assess mitral valve annular velocities, E′ and A′. In addition, pulse wave Doppler of the mitral E and A wave velocities were taken from the four-chamber view.
Blood pressure measurements.
Noninvasive systolic blood pressure measurements were made in conscious restrained mice using a tail-cuff system with a temperature and sound-controlled environment (IITC Life Science) as previously described (52). These noninvasive systolic blood pressure measurements have also been validated by radiotelemetry (52). After 3 days of training in the same restrainer used for the blood pressure measurements, each mouse was assessed between 0900 and 1200, a minimum of four times per session.
Analysis of mouse tissues.
Mice were sacrificed with an intraperitoneal injection of euthanyl (0.2 ml/kg body wt) at 18 wk of age. Tissues were homogenized, and the protein concentration was assayed using Bradford protein reagent. Protein (15–20 μg) was used for SDS-PAGE and transferred to nitrocellulose. Membranes were immunoblotted with antibodies and visualized using the Perkin-Elmer enhanced chemiluminescence Western blotting detection system. Citrate synthase activity was measured in freshly made soleus muscle homogenates (1:20 dilution) using oxaloacetate (0.1 mM final concentration) as the substrate.
Quantification of HNE-protein adduct.
HNE-protein adducts were quantified from plasma and frozen mouse heart samples using the commercially available ELISA (Cell Biolabs, San Diego, CA) according to instructions of the manufacturer.
Statistical analysis.
Results are presented as means ± SE. Endpoints were analyzed using a one-way ANOVA, and Bonferroni multiple-comparison's tests were performed post hoc to compare differences between experimental groups. A probability value of <0.05 was considered significant.
RESULTS
DOX caused pathological LV remodeling in mice.
To mimic DOX-induced cardiotoxicity observed in breast cancer patients, we established a murine model of chronic DOX exposure in which female mice were subjected to weekly DOX (8 mg·kg−1·wk−1) or placebo (CON) (0.9% saline) for 4 wk. This DOX dose and schedule has been reported to cause progressive LV remodeling in mice (30). Furthermore, because the incidence of heart failure in modern adjuvant trials with DOX-containing regimens is <2.1% (46), we chose this mouse model that would reduce LV systolic function and not cause congestive heart failure as characterized by pulmonary edema. That said, because LV dysfunction decreases life expectancy in humans (49), this is a clinically important phenotype induced by DOX exposure.
In this model, DOX prevented body weight gain (P < 0.05; Table 1) and reduced heart weight and heart weight-to-tibia length (HW/TL) ratios (P < 0.05; Fig. 1C). Atrophy does not appear to be involved in this observation since cardiomyocyte cross-sectional area was not reduced in the hearts of the DOX-treated mice (data not shown). Furthermore, these differences cannot be attributed to altered food intake since the amount of food consumed was similar across the groups of mice (data not shown). Although we do report a very minor thinning of the LV in DOX-treated mice (P < 0.05; Fig. 1D), we currently have no evidence to explain why this occurs. Furthermore, significant differences in TL were not observed (Table 1), suggesting that growth of the mouse was not impaired.
Table 1.
Physical characteristics and LV morphology and function in mice
| CON |
DOX |
DOX + ET |
DOX + RESV |
|||||
|---|---|---|---|---|---|---|---|---|
| Baseline | 8 Weeks | Baseline | 8 Weeks | Baseline | 8 Weeks | Baseline | 8 Weeks | |
| Body weight, g | 19.9 ± 0.3 | 23.5 ± 0.5† | 19.6 ± 0.3 | 19.1 ± 0.3* | 19.4 ± 0.3 | 18.6 ± 0.6* | 19.6 ± 0.3 | 18.6 ± 0.3* |
| Systolic BP, mmHg | 115.3 ± 3.1 | 115.1 ± 1.8 | 121.9 ± 2.7 | 93.9 ± 3.7†* | 118.1 ± 2.8 | 102.1 ± 5.0† | 117.8 ± 2.7 | 111.9 ± 7.4 |
| Heart weight, mg | ND | 104.3 ± 1.7 | ND | 93.2 ± 4.6 | ND | 98.6 ± 3.9 | ND | 88.3 ± 2.4* |
| Tibia length, mm | ND | 19.3 ± 0.1 | ND | 19.4 ± 0.1 | ND | 18.9 ± 0.2 | ND | 19.2 ± 0.2 |
| HR, beats/min | 434 ± 15 | 466 ± 17 | 452 ± 12 | 418 ± 17† | 448 ± 15 | 438 ± 18 | 453 ± 19 | 443 ± 20 |
| CO, ml/min | 16.2 ± 0.8 | 16.0 ± 1.0 | 18.9 ± 1.8 | 12.3 ± 1.1* | 17.6 ± 1.4 | 12.5 ± 0.7†* | 15.1 ± 1.3 | 15.8 ± 1.6§ |
| SV, μl | 36.6 ± 1.3 | 32.8 ± 1.3† | 34.2 ± 1.1 | 27.4 ± 0.9†* | 35.4 ± 1.6 | 31.3 ± 2.4† | 36.1 ± 1.1 | 33.1 ± 1.9 |
| E′, mm/s | 30.7 ± 1.2 | 29.3 ± 2.3 | 32.2 ± 2.3 | 20.1 ± 1.8†* | 28.3 ± 1.1 | 22.8 ± 1.6 | 26.0 ± 1.0 | 30.4 ± 1.6§ |
| A′, mm/s | 24.2 ± 0.8 | 22.1 ± 2.1 | 23.0 ± 1.5 | 20.7 ± 1.6 | 22.5 ± 1.1 | 22.4 ± 1.8 | 24.0 ± 2.1 | 22.5 ± 0.9 |
| S′, mm/s | 21.3 ± 1.1 | 20.2 ± 1.4 | 20.7 ± 1.6 | 16.8 ± 0.9 | 19.5 ± 1.0 | 21.4 ± 2.3 | 20.9 ± 1.6 | 19.8 ± 0.8 |
| E/E′ | 21.3 ± 1.1 | 20.2 ± 2.1 | 20.8 ± 1.6 | 28.1 ± 2.4 | 24.0 ± 1.5 | 27.7 ± 3.0 | 29.4 ± 1.7 | 21.8 ± 1.1 |
| E/A | 2.1 ± 0.1 | 1.9 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.2 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.0 ± 0.1 | 2.2 ± 0.1 |
| LVPWs, mm | 0.89 ± 0.03 | 1.05 ± 0.03† | 0.97 ± 0.03 | 0.93 ± 0.04* | 0.94 ± 0.06 | 1.02 ± 0.03 | 0.87 ± 0.03 | 1.09 ± 0.07§ |
| LVPWd, mm | 0.62 ± 0.01 | 0.67 ± 0.01† | 0.64 ± 0.01 | 0.66 ± 0.03 | 0.64 ± 0.02 | 0.70 ± 0.02 | 0.63 ± 0.02 | 0.73 ± 0.03 |
| IVSs, mm | 0.89 ± 0.03 | 1.08 ± 0.03† | 0.97 ± 0.03 | 0.94 ± 0.03* | 0.93 ± 0.06 | 1.03 ± 0.02 | 0.88 ± 0.03 | 1.11 ± 0.04§ |
| IVSd, mm | 0.63 ± 0.01 | 0.69 ± 0.01† | 0.66 ± 0.00 | 0.69 ± 0.03 | 0.65 ± 0.02 | 0.68 ± 0.02 | 0.65 ± 0.03 | 0.73 ± 0.02 |
| LVIDs, mm | 2.70 ± 0.05 | 2.33 ± 0.11† | 2.51 ± 0.06 | 2.85 ± 0.10†* | 2.67 ± 0.11 | 2.62 ± 0.06§ | 2.74 ± 0.06 | 2.62 ± 0.06§ |
| LVIDd, mm | 3.53 ± 0.09 | 3.53 ± 0.09† | 3.66 ± 0.05 | 3.73 ± 0.09 | 3.82 ± 0.10 | 3.65 ± 0.09 | 3.86 ± 0.05 | 3.49 ± 0.11§ |
| FS, % | 29.7 ± 1.3 | 34.2 ± 1.6† | 31.6 ± 1.1 | 23.8 ± 1.0†* | 30.2 ± 1.4 | 28.0 ± 0.7* | 28.9 ± 0.9 | 35.4 ± 2.1§ |
Data presented as means ± SE. CON, no treatment; DOX, doxorubicin only; DOX + ET, doxorubicin plus exercise training; DOX + RESV, doxorubicin plus resveratrol; CO, cardiac output; FS, fractional shortening; HR, heart rate; IVS, intraventricular septum; LV, left ventricle; LVID, LV internal dimension; LVPW, LV posterior wall; SV, stroke volume; ND, not determined.
P < 0.05 for difference within groups from baseline to 8 wk.
P < 0.05, value for difference vs. CON group at 8 wk.
P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups at 8 wk.
Fig. 1.
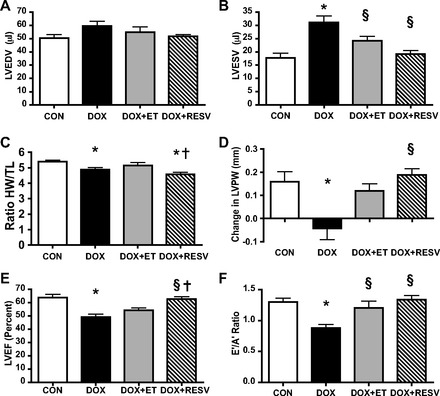
Exercise training (ET) and resveratrol (RESV) both attenuate doxorubicin (DOX)-induced cardiotoxicity. Echocardiography analysis of sedentary saline-injected controls (CON), DOX, DOX + ET, and DOX + RESV mice; left-ventricular (LV) end-diastolic volume (LVEDV) (A); LV end-systolic volume (LVESV) (B); ratio of heart weight-to-tibia length (HW/TL) (C); change in LV posterior wall thickness (LVPW) (D); LV ejection fraction (LVEF) (E); and the E′/A′ ratio (F). Values are means ± SE. (n = 9–10). *P < 0.05, value for difference vs. CON group; §P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups; †P < 0.05, value for DOX + ET vs. DOX + RESV groups.
Both the systolic LV internal dimension (LVIDs) and LV end-systolic volume (LVESV, P < 0.05; Table 1 and Fig. 1, C and D) were significantly increased in the hearts of the DOX-treated mice, although the diastolic LV internal dimension (LVIDd) and LV end-diastolic volume (LVED) were not significantly affected by DOX. These findings together with the markedly reduced LVEF (P < 0.05; Fig. 1E) indicate that DOX impaired cardiac contractility in the mice. In addition, DOX reduced systolic blood pressure (P < 0.05; Table 1) and heart rate (P < 0.05; Table 1), likely because of insufficient cardiac output (P < 0.05; Table 1) necessary to maintain systolic blood pressure. Because the echocardiographic measures were performed on anesthetized mice, the absolute values of the heart rate and fractional shortening were lower than would be expected in conscious mice. Nevertheless, all groups were treated similarly, and the comparisons across groups as well as to the baseline state are appropriate. That said, it is uncertain whether these results can be directly extrapolated to the conscious state in the absence of anesthesia.
DOX-induced LV remodeling is partially attenuated by ET in mice.
To mimic the modest level of exercise that would be expected from a patient undergoing chemotherapy and to characterize the efficacy of this ET during concurrent DOX treatment, mice performed 45 min of forced treadmill ET (i.e., a combination of electrical stimulation and an air puff to encourage the mice to run) 5 days/wk for a total of 8 wk, at a speed of 18 m/min, throughout the DOX treatment. The addition of modest ET to the DOX regimen did not alter the reduction of body weight in the mice, although ET did partially attenuate the reduced systolic blood pressure (Table 1). In addition, ET prevented several features of DOX-induced cardiotoxicity, including reduced LVESV (P < 0.05; Fig. 1B) and LVIDs (P < 0.05; Table 1). Because our ET protocol was modest, it did not produce a significant endurance ET effect, and thus the LVIDd and LVED were not significantly affected. DOX + ET also attenuated the DOX-induced reduction of systolic LV posterior wall (LVPWs, P < 0.05; Fig. 1D), although DOX + ET did not prevent the reduced HW/TL ratio (Fig. 1C). Nevertheless, DOX + ET partially attenuated the DOX-induced alterations in LV systolic (LVEF; Fig. 1E) and diastolic (E′/A′; P < 0.05; Fig. 1F) function.
DOX-induced LV remodeling is prevented by RESV in mice.
To determine whether RESV provided similar benefits as modest ET in DOX-treated mice, we also treated mice with RESV for the entire duration of the DOX treatment (DOX + RESV). Even though DOX + RESV did not prevent the reduction of body weight compared with the DOX mice, DOX + RESV attenuated the DOX-induced reduction of systolic blood pressure (Table 1). Remarkably, RESV completely prevented the increase of DOX-induced LVESV and LVIDs (P < 0.05; Fig. 1B and Table 1) in the mice. Interestingly, hearts from DOX + RESV-treated mice had a reduced HW/TL ratio compared with CON mice (P < 0.05; Fig. 1C). DOX + RESV prevented the reduced LVPWs (P < 0.05; Fig. 1D and Table 1) and systolic intraventricular septum (P < 0.05; Table 1) observed in the hearts of DOX-treated mice, and these values were similar to the CON group. Importantly, RESV markedly improved LVEF in DOX-treated mice (P < 0.05; Fig. 1E). In addition, the hearts of the DOX + RESV also had a higher E′ velocity and an improved ratio of E′/A′ (P < 0.05; Fig. 1F and Table 1) compared with the DOX mice, although RESV did not significantly affect other measures of diastolic function such as E/E′ and mitral E/A (Table 1). Taken together, these data suggest that RESV improves systolic function (i.e., increases LVEF) in hearts of DOX-treated mice to an extent that exceeds the benefits provided by modest ET (P < 0.05; Fig. 1E).
DOX-induced impairment in exercise capacity is prevented by modest ET and RESV supplementation in mice.
Because echocardiography revealed that modest ET partially attenuated the DOX-induced decline in cardiac function under resting conditions, we next determined whether ET also improved coordinated cardiovascular endurance. To do this, we subjected mice to a forced test of exercise capacity on a motorized treadmill to assess time to fatigue and total distance, as described previously (11). Citrate synthase activity was significantly increased in the soleus muscle tissue of DOX + ET and DOX + RESV mice (P < 0.05; Fig. 2A), indicating that these interventions increased skeletal muscle oxidative capacity. Surprisingly, even with the impairment in LV function with DOX administration, neither time to fatigue (Fig. 2C) nor total distance to fatigue (Fig. 2B) was lower in DOX-treated mice compared with the CON group. However, modest ET during DOX treatment (DOX + ET) resulted in significant improvements in exercise performance compared with DOX or CON groups (P < 0.05; Fig. 2, B and C), suggesting that modest ET could improve exercise performance even in the presence of DOX. Although chronic ET is widely established as one of the most effective interventions to augment aerobic capacity in mammals, the effects of modest ET on exercise capacity in mice undergoing DOX treatment have not been shown previously. Together, our findings implicate modest ET as a method to prevent DOX-induced damage to the cardiac and skeletal muscle and potentially the whole oxygen cascade involving the integration of heart, blood, vasculature, and skeletal muscle.
Fig. 2.
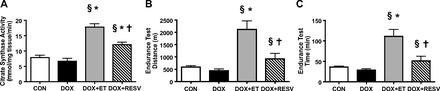
ET and RESV improve exercise performance during concurrent DOX treatment. A: citrate synthase activity; B: distance to fatigue during an endurance test; C: time to fatigue during an endurance test. Values are means ± SE (n = 9–10). *P < 0.05, value for difference vs. CON group; §P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups; †P < 0.05, value for DOX + ET vs. DOX + RESV groups.
Although modest ET during DOX treatment preserved LVEF in mice (P < 0.05; Fig. 1E) and improved exercise performance, adherence to an equivalent program of ET by patients undergoing chemotherapy may be difficult and may not be of sufficient intensity to observe benefit (19). Therefore, because RESV mimics many of the benefits of ET, we investigated whether the addition of RESV to the diets of mice undergoing treatment with DOX was sufficient to improve exercise performance. Interestingly, DOX + RESV significantly increased time to fatigue (P < 0.05; Fig. 2C) and distance to fatigue (P < 0.05; Fig. 2B) compared with DOX or CON groups. Collectively, these data demonstrate that modest ET and/or RESV can significantly improve exercise performance even in the presence of DOX.
Reduced expression of molecular markers of cardiac dysfunction and oxidative stress in DOX-treated mice following ET and RESV.
The cardiac levels of atrial natriuretic peptide (ANP), a gene that is expressed in response to increased afterload or cardiac injury, was increased by DOX compared with the CON group (P < 0.05; Fig. 3A). Notably, the cardiac expression of ANP was reduced in both the DOX + ET and DOX + RESV groups compared with the DOX mice (P < 0.05; Fig. 3A). Consistent with the reduction of the expression of the calcium transporter, sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA) 2a, being associated with reduced contractility in cardiomyopathies (32), we observed that SERCA2a expression was significantly lower in DOX relative to CON (P < 0.05; Fig. 3B). However, DOX + ET and DOX + RESV partially prevented the decline of SERCA2a expression (Fig. 3B). Collectively, these data suggest that ET and RESV supplementation could attenuate alterations of gene expression and possibly impaired calcium handling associated with DOX-induced cardiotoxicity.
Fig. 3.
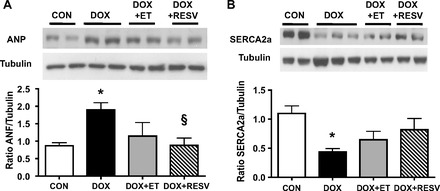
ET and RESV attenuate DOX-induced changes in molecular markers of heart failure and mitochondrial dysfunction. Cardiac levels of atrial natriuretic peptide (ANP) protein were quantified by densitometry and normalized against tubulin (A); cardiac sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA)-2a protein was quantified by densitometry and normalized against tubulin (B). Values are means ± SE (n = 9–10). *P < 0.05, value for difference vs. CON group; §P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups.
DOX-induced ROS is a central cause of cardiac injury associated with DOX-mediated cardiotoxicity (39). Consistent with this notion, concentrations of 4-hydroxy-2-nonenal (HNE), an established molecular biomarker of ROS activity (27), were significantly elevated in both the circulation (P < 0.05; Fig. 4A) as well as the hearts (P < 0.05; Fig. 4B) of DOX mice compared with CON mice. Although chronic ET reduces ROS levels (1), it was not known whether these findings extend to mice undergoing modest ET in the presence of DOX. However, our results indicated that both serum and cardiac levels of HNE were significantly lower in the DOX + ET group relative to DOX and in the case of HNE levels in the heart, were comparable to levels in the CON group (P < 0.05; Fig. 4, A and B). Consistent with the antioxidant properties associated with RESV, serum levels of HNE were also significantly lower in the DOX + RESV group relative to DOX (P < 0.05; Fig. 4A). Likewise, HNE levels were markedly lower in the hearts of DOX + RESV mice relative to DOX and were similar to the CON group (P < 0.05; Fig. 4B).
Fig. 4.
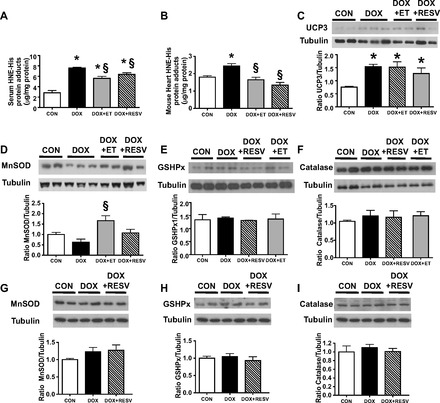
ET and RESV attenuate DOX-induced lipid peroxidation by-product formation. Levels of 4-hydroxy-2-nonenal (HNE)-His protein adducts were quantified in the serum (A) and the heart (B); cardiac levels of uncoupling protein 3 (UCP3) (C); manganese superoxide dismutase (MnSOD) protein (D); glutathione-S-peroxidase (GSHPx1) protein (E); catalase (F) and soleus muscle levels of MnSOD (G); and GSHPx1 (H) and catalase proteins (I) were quantified by densitometry and normalized against tubulin. Values are means ± SE (n = 9–10). *P < 0.05, value for difference vs. CON group; §P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups.
To further investigate the mechanisms responsible for the reduced HNE levels in the hearts of DOX-treated mice undergoing concurrent ET or RESV treatment regimens, we measured the expression of uncoupling protein 3 (UCP3) in the hearts, since UCP3 could be a potential source of ROS. Interestingly, compared with CON, DOX increased UCP3 protein expression (P < 0.05; Fig. 4C), although neither ET nor RESV significantly affected UCP3 expression compared with DOX-treated mice (Fig. 4C). On the other hand, cardiac manganese superoxide dismutase (MnSOD) protein expression was reduced in the DOX group compared with CON (P < 0.05; Fig. 4D). MnSOD expression was approximately threefold higher in the DOX + ET group compared with DOX (P < 0.05; Fig. 4D). However, in DOX + RESV mice, MnSOD expression was maintained at CON levels (Fig. 4D). Interestingly, the expression of other antioxidant defense enzymes such as glutathione-S-peroxidase (GSHPx1) and catalase were not altered in the hearts from all groups of mice (Fig. 4, E and F). Furthermore, we did not observe changes in the expression of MnSOD, GSPHx, or catalase in soleus muscle tissues of the mice in this study (Fig. 4, G-I). Collectively, these data indicate that modulation of oxidative stress and MnSOD expression in the heart may be key factors underlying protection against DOX-induced cardiac injury and that ET and RESV appear to have overlapping although not identical mechanisms of action with respect to modifying these factors.
Although both exercise and RESV have been reported to activate the AMP-activated protein kinase (AMPK) in the heart, we did not observe changes in AMPK phosphorylation at its activation site (Thr172) (Fig. 5A). In addition, the expression of the silent information regulator of transcription (SIRT)-1 was not significantly changed across all the groups of mouse hearts (Fig. 5B). Because impaired mitochondrial function has been suggested to be a crucial factor in the pathogenesis of DOX-induced cardiotoxicity, we measured the expression of the electron transport chain complexes in cardiac tissue from all groups of mice. While DOX treatment caused a significant reduction in the cardiac protein levels of complexes I and II (P < 0.05; Fig. 5C), both the DOX + ET and the DOX + RESV groups had similar levels of expression of the electron chain complexes as the CON group. This was likely not due to alterations in mitochondrial numbers since changes in the mRNA expression of genes encoded by the mitochondrial genome were not observed (Fig. 5, D and E). Because a major adaptation to exercise is the improvement in mitochondrial fusion, we also measured the levels of the mitochondrial fusion proteins mitofusin-1 and -2. Whereas DOX did not alter cardiac levels of mitofusin-1 and -2, the addition of ET or RESV to the DOX treatment regimen was sufficient to increase the expression of both mitofusin-1 and -2 (Fig. 5, F and G), suggesting that these adaptations may contribute to improved mitochondrial function and ultimately contractility in the hearts of mice undergoing DOX treatment.
Fig. 5.
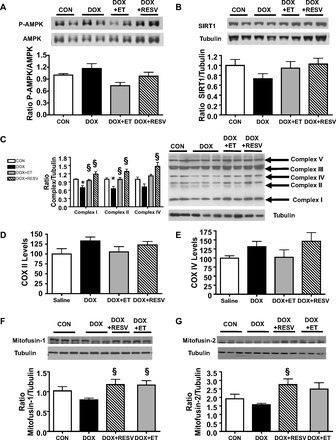
ET and RESV increase levels of mitochondrial complexes and fusion proteins in DOX-treated mice. Cardiac levels of phosphorylated (P) AMP-activated protein kinase (AMPK) at T172 were quantified by densitometry and normalized against total AMPK (A); silent information regulator of transcription-1 (SIRT1) protein was quantified by densitometry and normalized against tubulin (B); mitochondrial electron transport chain complexes in the mouse heart (C); cytochrome c oxidase (COX)-II mRNA levels (D); COX-IV mRNA levels (E); mitofusin-1 protein was quantified by densitometry and normalized against tubulin (F); mitofusin-2 protein was quantified by densitometry and normalized against tubulin (G). Values are means ± SE (n = 9–10). *P < 0.05, value for difference vs. CON group; §P < 0.05, value for DOX vs. DOX + ET or DOX + RESV groups.
DISCUSSION
DOX-containing chemotherapies are cardiotoxic and can cause LV dysfunction and progressive LV remodeling, which may ultimately progress to heart failure (33, 34, 42, 48). Clinically, the consequence of impaired LV function in chemotherapy patients is an inability to perform the activities of daily living (7). Because many patients are living longer after the diagnosis and treatment of their cancer, strategies to alleviate the cardiotoxic side effects of DOX are of extreme clinical importance.
This study is the first to directly compare the efficacy of modest ET with RESV supplementation for the protection against DOX-induced cardiotoxicity. While both modest ET and RESV prevented DOX-induced LV dysfunction, the results of this study demonstrate for the first time that RESV improves cardiac contractility and LVEF during DOX treatment beyond those achieved by modest ET alone. Consistent with previous rodent studies (5, 21, 29, 30, 54), we show that chronic DOX treatment significantly decreased global LV systolic and diastolic function (e.g., decreased LVEF and E′/A′ ratio, respectively). Several mechanisms have been proposed to explain the cardiotoxic side effects of DOX, including cardiomyocyte apoptosis, myofibrillar damage, impaired calcium handling, impaired mitochondrial activities, and increased generation of ROS (37). In this study, we observed that DOX elevated HNE and inhibited SERCA2a expression and that these changes were associated with cardiac dysfunction. Given that ROS can cause alterations in calcium homeostasis and DOX stimulates calcium release and inhibits sarcoplasmic reticulum calcium reuptake (32), our data suggest that these two major factors could contribute to systolic and diastolic dysfunction in our chronic DOX model of LV dysfunction. Therefore, modest ET or RESV may act to reduce DOX-induced cardiotoxicity by targeting several of these cellular processes and pathways.
Aerobic ET and not strength training has been well documented to attenuate pathological LV remodeling and improve exercise tolerance in patients with heart failure (18, 41, 51). Aerobic ET also provides a safe cardioprotective intervention for DOX-induced cardiotoxicity without reducing the antitumor efficacy of DOX (22). Several studies have demonstrated that ET before acute DOX administration improves LV systolic and diastolic function in rodents (5, 21, 53). However, the molecular mechanisms underlying the effects of ET in DOX-induced cardiac dysfunction are incompletely understood. What is known is that ET protects the heart against ROS by enhancing the antioxidant pathways (1) and improves calcium handling (25). ET has also been shown to mitigate DOX-induced ROS production (24). In agreement with these effects of ET, we show that modest ET reduced DOX-induced production of HNE, likely via increased MnSOD expression. Consistent with the finding that ET increased the expression of the cardiac calcium transporter SERCA2a (25), we showed that modest ET also increased SERCA2a expression in DOX-treated mice. Thus, we suggest that modest ET-induced improvements in these pathways may play a major role in the cardioprotection afforded by ET against DOX-induced cardiotoxicity.
RESV mimics the improved cardiovascular functions and exercise performance associated with ET through targeting similar molecular pathways (11). RESV has been reported to stimulate SIRT1 and AMPK signaling pathways (10). While some recent studies suggest that DOX inhibits SIRT1 and AMPK in the hearts of male Wistar rats (16, 55) and cultured cells (4), we did not detect changes in AMPK phosphorylation and SIRT1 expression in the hearts of DOX-treated female mice. These differences could be attributed to the differences in species/models used in these different studies as well as the acute supraclinical dosages of DOX used in previous studies (4, 16, 55) compared with the chronic clinically relevant DOX treatment regimen used in this study. Although it is beyond the scope of the current study to test all of the possible mechanisms responsible for the cardioprotective effects of RESV, it has been shown that RESV regulates nuclear factor-kB and the levels of inflammatory cytokines (36, 38). We speculate that RESV could also prevent DOX-induced cardiotoxicity through blocking the inflammatory pathways that are induced in heart failure, although further experimentation is required to confirm this.
Although AMPK and SIRT1 do not appear to be involved in the protective effects of RESV, we did observe that RESV attenuated DOX-induced production of ROS by-products (i.e., HNE). We speculate that several mechanisms could be responsible for this observation. For example, RESV may increase the expression of enzymes involved in oxidative stress resistance. In agreement with this, we observed that RESV attenuated inhibition of MnSOD expression by DOX. These results are in line with previous findings that showed that the ability of RESV to prevent DOX-induced cardiac dysfunction in rats involved increased antioxidant defenses (8, 43). DOX also increased cardiac UCP3 expression, suggesting that UCP3 could be a potential source of ROS. However, neither DOX + ET nor DOX + RESV attenuated the DOX-induced increase in cardiac UCP3 expression (Fig. 4C); therefore, changes in UCP3 expression did not explain the reduction of HNE levels by ET and RESV.
In addition to inducing oxidative stress resistance, ET and RESV improve mitochondrial function in healthy rodents (1, 11, 26). In agreement with this, we observed that ET and RESV attenuated the reduction of electron transport chain complexes I and II in the DOX-treated mice. Whereas these interventions did not increase mitochondrial numbers, ET and RESV did markedly increase the expression of mitofusin-1 and -2, which are required for the formation of a dynamic mitochondrial network (2). Although all of the potential effects on the mitochondria have likely not been identified herein, this is the first study to report increased mitofusin-1 and -2 in the hearts of DOX + ET and DOX + RESV mice. Interestingly, it was recently reported that the prevention of heart failure by RESV in Dahl salt-sensitive rats involved increased expression of mitofusin-1 and -2 (35). In addition, the induction of mitofusin-2 expression was associated with the attenuation of DOX-induced LV dysfunction by cyclosporin A. These findings suggest that this potential mechanism should be explored with further experiments to determine whether mitofusin-1 and -2 are required to mediate the positive effects of ET and RESV in the hearts of DOX-treated mice and determine whether these mitochondrial changes translate into improved mitochondrial respiration by cardiac myocytes.
We showed that RESV attenuated the inhibition of SERCA2a expression by DOX, suggesting that RESV also prevented impaired calcium handling, thereby improving myocardial contractility. Whereas these observations are consistent with beneficial reductions in ROS and the rescue of MnSOD and SERCA2a expression in the hearts of modest ET and RESV mice, they do not explain how RESV improves the percent LVEF and fractional shortening to a greater extent than modest ET in mice treated with DOX. Given that RESV is a molecule with pleiotropic effects (10), further work is required to identify additional molecular pathways in the heart that are targeted by RESV and protect against DOX-induced cardiotoxicity. It is possible that other organ systems could contribute to the reduction in ROS by-products, although we did not observe changes in the expression of MnSOD, GSPHx, or catalase in soleus muscle tissues of the mice in this study. That said, we did not directly measure ROS production in the heart so we cannot completely rule out changes in ROS production from being involved in how RESV provides significant benefits for LVEF beyond modest ET in mice treated with DOX.
Currently, a diagnosis of DOX-induced cardiotoxicity results in lowering the dosage or discontinuation of DOX at the expense of lowering the antineoplastic activity associated with the administration of higher dosages (34). Despite this strategy, some patients develop LV dysfunction even after chemotherapy has been completed (20). Therefore, the need to protect the heart from cardiotoxicity while maximizing the anti-cancer benefits of therapeutic agents is essential for the long-term health and survival of patients. While randomized controlled trials demonstrate that supervised ET is a safe and tolerated adjunct during chemotherapy (7, 22), adherence to even a modest exercise program may be difficult for most patients (19). Indeed, a large heart failure trial found a nonsignificant reduction in rehospitalizations and mortality in patients randomized to ET, which may be attributed to insufficient exercise intensity to significantly improve clinical outcomes (31).
Based on what we know about ET in patients undergoing chemotherapy, our results are particularly exciting because they suggest that RESV could be used to prevent the cardiotoxic effects of DOX in patients that are unable to perform exercise while undergoing chemotherapy. At present, the only small molecule approved for the prevention of anthracycline-induced cardiotoxicity during chemotherapy is dexrazoxane (40, 47). However, dexrazoxane is limited in its clinical use since some studies suggested it interfered with anthracycline activity and diminished oncologic efficacy (40, 47).
While the present study employed a cancer-free mouse model of chronic DOX treatment to avoid the confounding variables involved with malignancy, it is reasonable to assume that the cardioprotective effects of modest ET and RESV will extend to rodents with tumors. Indeed, RESV is a particularly appealing molecule since several studies suggest that RESV is an anti-cancer agent on its own (see Ref. 17 for review). Furthermore, it was recently reported that RESV inhibits the growth of DOX-resistant B16 melanoma cells and sensitizes a variety of cancer cells to DOX-mediated apoptosis (14). Nevertheless, it will be important to evaluate the antineoplastic effects of DOX in mouse models of cancer undergoing concurrent ET and/or RESV treatment in future experiments.
In conclusion, our findings have important clinical implications for the prevention and/or treatment of DOX-induced cardiotoxicity in the oncology setting.
GRANTS
This study was supported by the Canadian Institutes for Health Research (J. R. B. Dyck). V. W. Dolinsky is supported by a research grant from the Heart and Stroke Foundation of Canada. L. W. Jones is supported by research grants from the National Cancer Institute (CA-143254, CA-142566, CA-138634, CA-133895, and CA-164751).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: V.W.D., K.J.R., M.J.H., L.W.J., and J.R.D. conception and design of research; V.W.D., K.J.R., M.M.S., and B.N.Z. performed experiments; V.W.D., K.J.R., M.M.S., and B.N.Z. analyzed data; V.W.D., K.J.R., M.M.S., B.N.Z., M.J.H., M.E.Y., L.W.J., and J.R.D. interpreted results of experiments; V.W.D. and K.J.R. prepared figures; V.W.D., K.J.R., L.W.J., and J.R.D. drafted manuscript; V.W.D., K.J.R., M.M.S., B.N.Z., M.J.H., M.E.Y., L.W.J., and J.R.D. edited and revised manuscript; V.W.D., K.J.R., M.M.S., B.N.Z., M.J.H., M.E.Y., L.W.J., and J.R.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the expert technical assistance of Sarah Aziz, David Fung, Rammy Khadour, Donna Beker, Brandi Sidlick, Jamie Boisvenue, and Carrie-Lynn Soltys of the University of Alberta.
REFERENCES
- 1. Ascensao A, Ferreira R, Magalhaes J. Exercise-induced cardioprotection–biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol 117: 16–30, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Benard G, Karbowski M. Mitochondrial fusion and division: regulation and role in cell viability. Semin Cell Dev Biol 20: 365–374, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berthiaume JM, Wallace KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic Doxorubicin treatment. Cardiovasc Toxicol 7: 178–191, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Chen MB, Wu XY, Gu JH, Guo QT, Shen WX, Lu PH. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem Biophys 60: 311–322, 2011 [DOI] [PubMed] [Google Scholar]
- 5. Chicco AJ, Hydock DS, Schneider CM, Hayward R. Low-intensity exercise training during doxorubicin treatment protects against cardiotoxicity. J Appl Physiol 100: 519–527, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Committee CCSsS. Canadian Cancer Statistics 2009. Toronto, Canada: Canadian Cancer Society, 2009 [Google Scholar]
- 7. Courneya KS, Segal RJ, Mackey JR, Gelmon K, Reid RD, Friedenreich CM, Ladha AB, Proulx C, Vallance JK, Lane K, Yasui Y, McKenzie DC. Effects of aerobic and resistance exercise in breast cancer patients receiving adjuvant chemotherapy: a multicenter randomized controlled trial. J Clin Oncol 25: 4396–4404, 2007 [DOI] [PubMed] [Google Scholar]
- 8. Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med 46: 1589–1597, 2009 [DOI] [PubMed] [Google Scholar]
- 9. Dolinsky VW, Chan AY, Robillard Frayne I, Light PE, Des Rosiers C, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 119: 1643–1652, 2009 [DOI] [PubMed] [Google Scholar]
- 10. Dolinsky VW, Dyck JR. Calorie restriction and resveratrol in cardiovascular health and disease. Biochim Biophys Acta 1812: 1477–1489, 2011 [DOI] [PubMed] [Google Scholar]
- 11. Dolinsky VW, Jones KE, Sidhu RS, Haykowsky M, Czubryt MP, Gordon T, Dyck JR. Improvements in skeletal muscle strength and cardiac function induced by resveratrol contribute to enhanced exercise performance in rats. J Physiol 590: 2783–2799, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dolinsky VW, Morton JS, Oka T, Robillard-Frayne I, Bagdan M, Lopaschuk GD, Des Rosiers C, Walsh K, Davidge ST, Dyck JR. Calorie restriction prevents hypertension and cardiac hypertrophy in the spontaneously hypertensive rat. Hypertension 56: 412–421, 2010 [DOI] [PubMed] [Google Scholar]
- 13. Dolinsky VW, Rueda-Clausen CF, Morton JS, Davidge ST, Dyck JR. Continued postnatal administration of resveratrol prevents diet-induced metabolic syndrome in rat offspring born growth restricted. Diabetes 60: 2274–2284, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duraj J, Bodo J, Sulikova M, Rauko P, Sedlak J. Diverse resveratrol sensitization to apoptosis induced by anticancer drugs in sensitive and resistant leukemia cells. Neoplasma 53: 384–392, 2006 [PubMed] [Google Scholar]
- 15. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol 7: 564–575, 2010 [DOI] [PubMed] [Google Scholar]
- 16. Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chate V, Schnebelen C, Sestili P, Schlattner U, Tokarska-Schlattner M. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc Res 95: 290–299, 2012 [DOI] [PubMed] [Google Scholar]
- 17. Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB. Chemosensitization of tumors by resveratrol. Ann NY Acad Sci 1215: 150–160, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haykowsky MJ, Liang Y, Pechter D, Jones LW, McAlister FA, Clark AM. A meta-analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: the benefit depends on the type of training performed. J Am Coll Cardiol 49: 2329–2336, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Haykowsky MJ, Mackey JR, Thompson RB, Jones LW, Paterson DI. Adjuvant trastuzumab induces ventricular remodeling despite aerobic exercise training. Clin Cancer Res 15: 4963–4967, 2009 [DOI] [PubMed] [Google Scholar]
- 20. Hequet O, Le QH, Moullet I, Pauli E, Salles G, Espinouse D, Dumontet C, Thieblemont C, Arnaud P, Antal D, Bouafia F, Coiffier B. Subclinical late cardiomyopathy after doxorubicin therapy for lymphoma in adults. J Clin Oncol 22: 1864–1871, 2004 [DOI] [PubMed] [Google Scholar]
- 21. Hydock DS, Lien CY, Jensen BT, Schneider CM, Hayward R. Exercise preconditioning provides long-term protection against early chronic doxorubicin cardiotoxicity. Integr Cancer Ther 10: 47–57, 2011 [DOI] [PubMed] [Google Scholar]
- 22. Jones LW, Eves ND, Courneya KS, Chiu BK, Baracos VE, Hanson J, Johnson L, Mackey JR. Effects of exercise training on antitumor efficacy of doxorubicin in MDA-MB-231 breast cancer xenografts. Clin Cancer Res 11: 6695–6698, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Jones LW, Liang Y, Pituskin EN, Battaglini CL, Scott JM, Hornsby WE, Haykowsky M. Effect of exercise training on peak oxygen consumption in patients with cancer: a meta-analysis. Oncologist 16: 112–120, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kavazis AN, Smuder AJ, Min K, Tumer N, Powers SK. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am J Physiol Heart Circ Physiol 299: H1515–H1524, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kemi OJ, Ceci M, Condorelli G, Smith GL, Wisloff U. Myocardial sarcoplasmic reticulum Ca2+ ATPase function is increased by aerobic interval training. Eur J Cardiovasc Prev Rehabil 15: 145–148, 2008 [DOI] [PubMed] [Google Scholar]
- 26. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127: 1109–1122, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol 44: 625–633, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meinardi MT, van Veldhuisen DJ, Gietema JA, Dolsma WV, Boomsma F, van den Berg MP, Volkers C, Haaksma J, de Vries EG, Sleijfer DT, van der Graaf WT. Prospective evaluation of early cardiac damage induced by epirubicin-containing adjuvant chemotherapy and locoregional radiotherapy in breast cancer patients. J Clin Oncol 19: 2746–2753, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Migrino RQ, Aggarwal D, Konorev E, Brahmbhatt T, Bright M, Kalyanaraman B. Early detection of doxorubicin cardiomyopathy using two-dimensional strain echocardiography. Ultrasound Med Biol 34: 208–214, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neilan TG, Jassal DS, Scully MF, Chen G, Deflandre C, McAllister H, Kay E, Austin SC, Halpern EF, Harmey JH, Fitzgerald DJ. Iloprost attenuates doxorubicin-induced cardiac injury in a murine model without compromising tumour suppression. Eur Heart J 27: 1251–1256, 2006 [DOI] [PubMed] [Google Scholar]
- 31. O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. J Am Med Assoc 301: 1439–1450, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olson RD, Gambliel HA, Vestal RE, Shadle SE, Charlier HA, Jr, Cusack BJ. Doxorubicin cardiac dysfunction: effects on calcium regulatory proteins, sarcoplasmic reticulum, and triiodothyronine. Cardiovasc Toxicol 5: 269–283, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Perez EA, Suman VJ, Davidson NE, Kaufman PA, Martino S, Dakhil SR, Ingle JN, Rodeheffer RJ, Gersh BJ, Jaffe AS. Effect of doxorubicin plus cyclophosphamide on left ventricular ejection fraction in patients with breast cancer in the North Central Cancer Treatment Group N9831 Intergroup Adjuvant Trial. J Clin Oncol 22: 3700–3704, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Perez EA, Suman VJ, Davidson NE, Sledge GW, Kaufman PA, Hudis CA, Martino S, Gralow JR, Dakhil SR, Ingle JN, Winer EP, Gelmon KA, Gersh BJ, Jaffe AS, Rodeheffer RJ. Cardiac safety analysis of doxorubicin and cyclophosphamide followed by paclitaxel with or without trastuzumab in the North Central Cancer Treatment Group N9831 adjuvant breast cancer trial. J Clin Oncol 26: 1231–1238, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rimbaud S, Ruiz M, Piquereau J, Mateo P, Fortin D, Veksler V, Garnier A, Ventura-Clapier R. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One 6: e26391, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rius C, Abu-Taha M, Hermenegildo C, Piqueras L, Cerda-Nicolas JM, Issekutz AC, Estan L, Cortijo J, Morcillo EJ, Orallo F, Sanz MJ. Trans- but not cis-resveratrol impairs angiotensin-II-mediated vascular inflammation through inhibition of NF-kappaB activation and peroxisome proliferator-activated receptor-gamma upregulation. J Immunol 185: 3718–3727, 2010 [DOI] [PubMed] [Google Scholar]
- 37. Scott JM, Khakoo A, Mackey JR, Haykowsky MJ, Douglas PS, Jones LW. Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer: current evidence and underlying mechanisms. Circulation 124: 642–650, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shadfar S, Couch ME, McKinney KA, Weinstein LJ, Yin X, Rodriguez JE, Guttridge DC, Willis M. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr Cancer 63: 749–762, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 61: 154–171, 2009 [DOI] [PubMed] [Google Scholar]
- 40. Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, Jones SE, Wadler S, Desai A, Vogel C, Speyer J, Mittelman A, Reddy S, Pendergrass K, Velez-Garcia E, Ewer MS, Bianchine JR, Gams RA. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol 15: 1318–1332, 1997 [DOI] [PubMed] [Google Scholar]
- 41. Swank AM, Horton J, Fleg JL, Fonarow GC, Keteyian S, Goldberg L, Wolfel G, Handberg EM, Bensimhon D, Illiou MC, Vest M, Ewald G, Blackburn G, Leifer E, Cooper L, Kraus WE. Modest Increase in Peak VO2 is Related to Better Clinical Outcomes in Chronic Heart Failure Patients: Results from Heart Failure and a Controlled Trial to Investigate Outcomes of Exercise Training (HF-ACTION). Circ Heart Fail 5: 579–585, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan-Chiu E, Yothers G, Romond E, Geyer CE, Jr, Ewer M, Keefe D, Shannon RP, Swain SM, Brown A, Fehrenbacher L, Vogel VG, Seay TE, Rastogi P, Mamounas EP, Wolmark N, Bryant J. Assessment of cardiac dysfunction in a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel, with or without trastuzumab as adjuvant therapy in node-positive, human epidermal growth factor receptor 2-overexpressing breast cancer: NSABP B-31. J Clin Oncol 23: 7811–7819, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Tatlidede E, Sehirli O, Velioglu-Ogunc A, Cetinel S, Yegen BC, Yarat A, Suleymanoglu S, Sener G. Resveratrol treatment protects against doxorubicin-induced cardiotoxicity by alleviating oxidative damage. Free Radic Res 43: 195–205, 2009 [DOI] [PubMed] [Google Scholar]
- 44. Thandapilly SJ, Wojciechowski P, Behbahani J, Louis XL, Yu L, Juric D, Kopilas MA, Anderson HD, Netticadan T. Resveratrol prevents the development of pathological cardiac hypertrophy and contractile dysfunction in the SHR without lowering blood pressure. Am J Hypertens 23: 192–196, 2010 [DOI] [PubMed] [Google Scholar]
- 45. Toklu HZ, Sehirli O, Ersahin M, Suleymanoglu S, Yiginer O, Emekli-Alturfan E, Yarat A, Yegen BC, Sener G. Resveratrol improves cardiovascular function and reduces oxidative organ damage in the renal, cardiovascular and cerebral tissues of two-kidney, one-clip hypertensive rats. J Pharm Pharmacol 62: 1784–1793, 2010 [DOI] [PubMed] [Google Scholar]
- 46. Trudeau M, Charbonneau F, Gelmon K, Laing K, Latreille J, Mackey J, McLeod D, Pritchard K, Provencher L, Verma S. Selection of adjuvant chemotherapy for treatment of node-positive breast cancer. Lancet Oncol 6: 886–898, 2005 [DOI] [PubMed] [Google Scholar]
- 47. van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database of Syst Rev CD003917, 2005 [DOI] [PubMed] [Google Scholar]
- 48. Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 91: 710–717, 1979 [DOI] [PubMed] [Google Scholar]
- 49. Wang TJ, Evans JC, Benjamin EJ, Levy D, LeRoy EC, Vasan RS. Natural history of asymptomatic left ventricular systolic dysfunction in the community. Circulation 108: 977–982, 2003 [DOI] [PubMed] [Google Scholar]
- 50. Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol 19: 670–686, 1992 [PubMed] [Google Scholar]
- 51. Whellan DJ, Nigam A, Arnold M, Starr AZ, Hill J, Fletcher G, Ellis SJ, Cooper L, Onwuanyi A, Chandler B, Keteyian SJ, Ewald G, Kao A, Gheorghiade M. Benefit of exercise therapy for systolic heart failure in relation to disease severity and etiology-findings from the Heart Failure and A Controlled Trial Investigating Outcomes of Exercise Training study. Am Heart J 162: 1003–1010, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Whitesall SE, Hoff JB, Vollmer AP, D'Alecy LG. Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol Heart Circ Physiol 286: H2408–H2415, 2004 [DOI] [PubMed] [Google Scholar]
- 53. Wonders KY, Hydock DS, Schneider CM, Hayward R. Acute exercise protects against doxorubicin cardiotoxicity. Integr Cancer Ther 7: 147–154, 2008 [DOI] [PubMed] [Google Scholar]
- 54. Yi X, Bekeredjian R, DeFilippis NJ, Siddiquee Z, Fernandez E, Shohet RV. Transcriptional analysis of doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol 290: H1098–H1102, 2006 [DOI] [PubMed] [Google Scholar]
- 55. Zhang C, Feng Y, Qu S, Wei X, Zhu H, Luo Q, Liu M, Chen G, Xiao X. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc Res 90: 538–545, 2011 [DOI] [PubMed] [Google Scholar]