

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease associated with aging that is characterized by the histopathological pattern of usual interstitial pneumonia. Although an understanding of the pathogenesis of IPF is incomplete, recent advances delineating specific clinical and pathologic features of IPF have led to better definition of the molecular pathways that are pathologically activated in the disease. In this review we highlight several of these advances, with a focus on genetic predisposition to IPF and how genetic changes, which occur primarily in epithelial cells, lead to activation of profibrotic pathways in epithelial cells. We then discuss the pathologic changes within IPF fibroblasts and the extracellular matrix, and we conclude with a summary of how these profibrotic pathways may be interrelated.
Keywords: pulmonary fibrosis, epithelial-to-mesenchymal transition, unfolded protein response, alveolar type II cell, fibrocytes, myofibroblast, epigenetics
Clinical Features of Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is defined as a chronic fibrosing interstitial lung disease of unknown etiology that occurs in adults and is characterized by the histopathological pattern of usual interstitial pneumonia (1). It is the most common of the idiopathic interstitial pneumonias and one of the most common forms of interstitial lung disease overall, with an estimated prevalence of 50 per 100,000 (2). Importantly, the prevalence of IPF rises dramatically with age; it is virtually absent in patients younger than 50 but is present in an estimated 0.2% of those older than 75 (2).
Among the various forms of interstitial lung disease, IPF has received the most attention because of its uniquely poor prognosis and its unresponsiveness to traditional therapies. Median survival for IPF is just 3 years; progressive breathlessness and eventual respiratory failure are the most common natural history (3). Many patients with IPF experience acute episodes of respiratory worsening that are associated with a high risk of death (4). In approximately half of these acute events, no cause can be found; these idiopathic acute worsenings are termed acute exacerbations (5). It has been hypothesized that acute exacerbations represent intrinsic acceleration of the underlying disease process in IPF, perhaps triggered by an occult stressor such as viral infection, microaspiration, or ambient pollution (6, 7).
There are no widely accepted therapies for IPF, although the novel antifibrotic agent pirfenidone has been approved for use in some countries (8). Notably, traditional therapy for interstitial lung disease using prednisone and immunomodulatory agents such as azathioprine is ineffective. In fact, a recent randomized controlled trial of combination therapy with prednisone, azathioprine, and the antioxidant acetylcysteine was stopped prematurely for futility and safety concerns in the active therapy arm (9). Because of this lack of efficacy, drug development has focused on targeting fibrogenesis and fibroproliferation, and an increasing number of targeted therapies are in early-phase clinical trials. It seems likely that the therapeutic landscape for IPF will change dramatically in the next decade.
Comprehensive management of the patient with IPF involves establishing an accurate diagnosis through careful multidisciplinary review; management of common comorbidities such as gastroesophageal reflux (GER) disease, obstructive sleep apnea, and depression; vaccination against influenza and pneumococcal infection; structured exercise and education through formal pulmonary rehabilitation classes; and, in progressive or advanced disease, assessment of appropriateness for lung transplantation. In the absence of effective therapies, all patients with IPF should be evaluated for clinical trials of novel drug agents. In patients who do not qualify for a clinical trial, empiric therapy with acetylcysteine and/or proton pump inhibition should be considered (1).
Pathologic Features of Idiopathic Pulmonary Fibrosis
The gross appearance of the lungs in IPF shows a characteristic fibrosis that is distributed along the inferior portions of the lobes with subpleural accentuation. The pleural surface has a bosselated or cobblestone appearance, and on cut section, these regions correspond to areas of airspace enlargement and fibrotic retraction. This pattern of fibrosis has been termed gross honeycombing. The typical microscopic appearance of IPF has been termed usual interstitial pneumonia and is one of spatial (or geographic) and temporal heterogeneity. The spatial heterogeneity of this lesion results in a patchy fibrotic reaction with fibrosis that is prominent in the peripheral portion of the secondary pulmonary lobule and spares the central portion of the lobule. This distribution of fibrosis is also reflected in the temporal heterogeneity of the lesion. As one moves from the peripheral to the central portion of the pulmonary lobule, the lung injury pattern changes from chronic to acute to absent. The regions in the immediate subpleural parenchyma often show advanced scarring and microscopic honeycombing characterized by irregular airspaces lined by bronchiolar epithelium and surrounded by dense fibrosis. The central portion of the pulmonary lobule often shows delicate alveolar septa without significant inflammation or fibrosis (Figure 1). A characteristic finding of usual interstitial pneumonia is the presence of fibroblast foci at the interface between the fibrotic and less involved regions of the lobule (1). These fibroblast foci manifest as proliferations of spindled fibroblasts, often arranged parallel to the alveolar surface, within a lightly basophilic myxoid background (Figure 2). The epithelial surface above the fibroblast focus often has a cuboidal appearance similar to that seen in in reactive type II pneumocyte hyperplasia.
Figure 1.
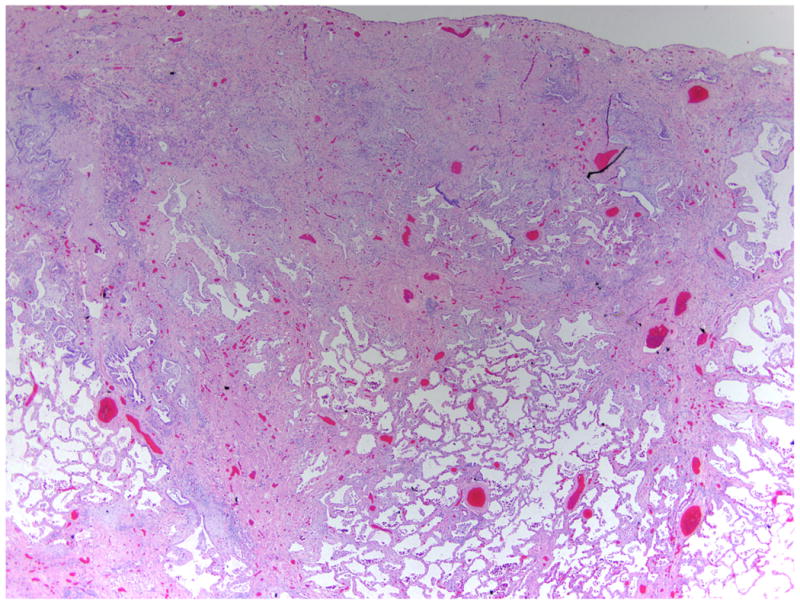
Pathologic changes in usual interstitial pneumonia. This low-power image of usual interstitial pneumonia shows the typical subpleural accentuation of dense fibrosis with irregular airspaces and central delicate alveolar septa. Stained with hematoxylin and eosin; magnification of 20×.
Figure 2.
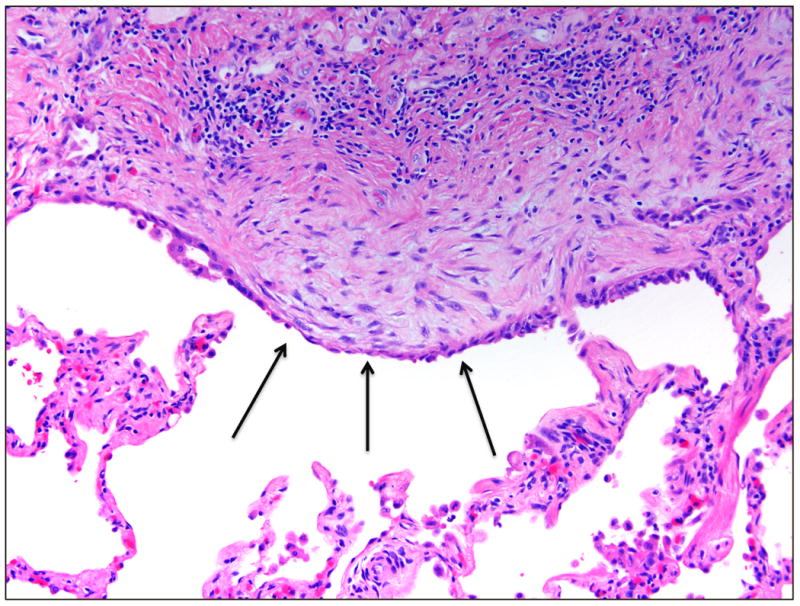
Fibroblast foci. The fibroblast focus shows a proliferation of spindled fibroblasts within a myxoid matrix, producing a bulge of tissue into the airspace. The overlying epithelium often shows plump reactive-appearing cells (arrows). Stained with hematoxylin and eosin; magnification of 200×.
Usual interstitial pneumonia secondary to idiopathic pulmonary fibrosis is often identified by a paucity of inflammation and a lack of uniform involvement of the lung tissue. Other diseases with similar features often manifest as fibrosis secondary to alveolitis and may yield additional histologic findings that aid in their identification. In autoimmune connective tissue diseases, the lung tissue may have some features of usual interstitial pneumonia, such as peripheral fibrosis and honeycombing; however, there is often additional centrilobular fibrosis in which the less involved lung tissue shows a lacy fibrosis or chronic inflammation (10). In addition, these cases often show prominent lymphoid aggregates or evidence of pleuritis (11). In hypersensitivity pneumonia, the lung may also have peripheral fibrosis with honeycombing; however, many cases also show prominent peribronchiolar fibrosis and may show scattered, poorly formed granulomas (12, 13).
Acute exacerbation of usual interstitial pneumonia shows a pattern of acute lung injury (diffuse alveolar damage) superimposed upon the typical findings of pulmonary fibrosis (Figure 3). These changes are most pronounced in the regions of the lungs that lack significant chronic fibrosis. The fibrotic regions often appear unchanged at the periphery of the lung. The central portions of the lobule show histologic changes typical of diffuse alveolar damage, including diffuse alveolar septal thickening by edema and type II pneumocyte hyperplasia as well as accumulation of airspace fibrin and edema. Other clues of acute exacerbation include nonspecific changes such as hyaline membranes, distal airway squamous metaplasia, and thrombi within small pulmonary arteries.
Figure 3.
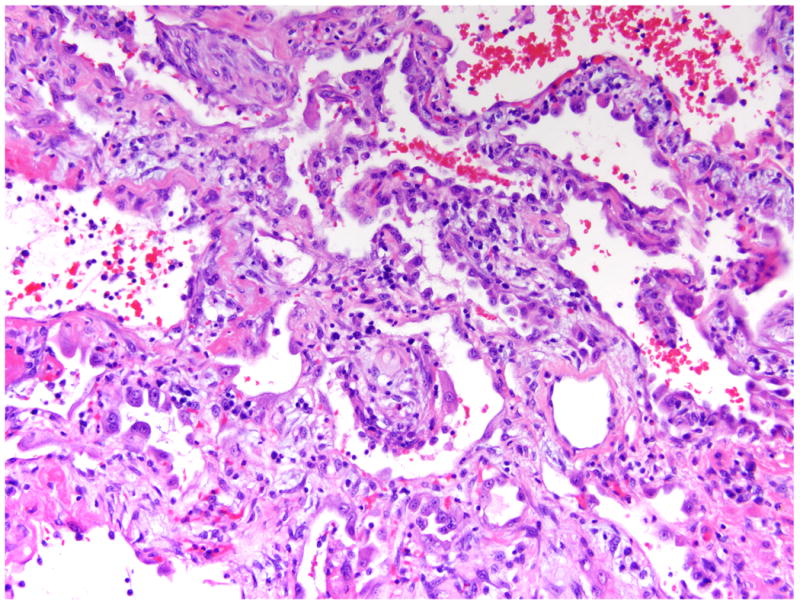
Pathologic changes in acute exacerbation of idiopathic pulmonary fibrosis. In acute exacerbation of usual interstitial pneumonia, the central lung tissue shows histologic changes of diffuse alveolar damage including edematous alveolar septa, type II pneumocyte hyperplasia, and hyaline membrane formation. Stained with hematoxylin and eosin; magnification of 200×.
Pathobiology of Idiopathic Pulmonary Fibrosis
In the following review of the pathobiology of IPF we focus on data obtained from patients with the disease. We do so because there is no animal model that reliably recapitulates the pathologic changes in idiopathic pulmonary fibrosis (14, 15). The bleomycin model of lung injury is commonly used to study molecular pathways of pulmonary fibrosis. However, pathologic changes seen in this model do not recapitulate those seen in idiopathic pulmonary fibrosis. In the bleomycin model, the fibrosis evolves over months, whereas in idiopathic pulmonary fibrosis, the fibrosis evolves over years. In the bleomycin model, the fibrosis is patchy and tends to be airway centric. In idiopathic pulmonary fibrosis, the fibrosis has subpleural accentuation, and the fibroblasts are arranged into a highly organized reticulum (14, 15). These changes are not found in the bleomycin model (15). Therefore, although the bleomycin lung fibrosis model can be used to identify mediators that have potential to cause lung fibrosis, the findings in mice need confirmation in translational studies in fibrotic lung diseases in humans. Where mentioned, data from animal models are used to emphasize specific profibrotic molecular mechanisms and pathways that have been more clearly defined using animal models.
Epithelium
Genetics
Early reports of cases of pulmonary fibrosis occurring in families suggested a genetic predisposition to some forms of the disease (16–18). These cases were followed by reports that pulmonary fibrosis developed in a subset of patients with defined clinical syndromes such as Hermansky-Pudlak syndrome and dyskeratosis congenita (19–21). Having established that pulmonary fibrosis occurs in patients with defined clinical syndromes enabled investigators to collect cohorts of individuals with uniform disease phenotypes and analyze them for the genetic mutations associated with these diseases. This analysis led to the association of mutations in the HPS gene in patients with Hermansky-Pudlak syndrome (22, 23) and mutations in the DKC, TERT, and TERC genes in patients with dyskeratosis congenita (24). These genetic mutations were the first to be linked to syndromes complicated by lung fibrosis and empowered subsequent genetic analysis of pedigrees of families with other forms of lung fibrosis.
In 2001, Nogee et al. (25) described a family with interstitial lung disease. Using a candidate-gene approach, they identified a heterozygous mutation in the surfactant protein C (SPC)-encoding gene that causes skipping of exon 4 and deletion of the terminal 37 amino acids (SPCΔexon4). This report indicates that mutations in SPC, a major protein manufactured and secreted by alveolar type II cells, could lead to interstitial lung diseases. Shortly thereafter, Thomas et al. (26) reported a Q188L missense mutation of the C-terminal portion of SPC in a large kindred of patients with familial pulmonary fibrosis (FPF). Subsequent reports have identified an additional six mutations in the SPC gene that are associated with pulmonary fibrosis (27). Most of these mutations occur in the BRICHOS domain (28), which serves a chaperone function for SPC that prevents the aggregation of SPC while aiding its insertion into membranes (28). Although these studies provide strong evidence for linkage of SPC mutations to specific familial cases of pulmonary fibrosis, these mutations reportedly occur in only 1% of sporadic cases of pulmonary fibrosis (Figure 4) (29, 30).
Figure 4.

Estimated frequencies of genetic mutations predisposing to idiopathic pulmonary fibrosis (IPF). This pie chart illustrates the estimated prevalence of mutations in surfactant protein C (SPC), surfactant protein A (SPA), mucin 5B (MUC5B), and telomerase (TERT, TERC) that predispose patients to develop sporadic IPF. All percentages are estimates derived from reports in specific patient cohorts.
Whole-genome linkage analysis on a large kindred of individuals with pulmonary fibrosis was performed using a single-nucleotide polymorphism (SNP) panel of more than 6,000 markers (31). The analysis identified chromosome 10q22 as the affected region. The genes encoding surfactant proteins A1, A2, and D, which reside in this region of the chromosome, were identified as candidate genes. Sequencing these genes identified a missense mutation predicting a G231V missense mutation in surfactant protein A2 (31). Subsequent sequencing of the surfactant protein A genes in other FPF kindreds identified a second missense mutation (F198V). Both mutations predict instability of the translated protein, and the mutated proteins are abnormally retained in the endoplasmic reticulum (ER) and fail to form higher-order oligomers.
The observation that patients with dyskeratosis congenita, a disease of premature aging, develop pulmonary fibrosis provided the first clue that mutations in age-related genes may predispose to pulmonary fibrosis (20, 32). This association led to the sequencing of TERT and TERC as candidate genes in 73 probands of FPF kindreds (33). Mutations in TERT or TERC were identified in 6 of the 73 probands, establishing that these mutations are associated with cases pulmonary fibrosis independently of other features of dyskeratosis congenita. Similarly, a whole-genome linkage scan of two large FPF kindreds displayed linkage to chromosome 5p15 with a logarithm of odds (LOD) score of 2.8. Sequencing the TERT gene, which resides in this region of the chromosome, revealed a missense R865H mutation and a frameshift V747fs mutation in the probands of the two kindreds (34). Sequencing additional families identified four additional TERT mutations and one TERC mutation in affected probands. Similarly, mutations in TERT or TERC have been identified in individuals with sporadic IPF (35, 36). Interestingly, although mutations in TERT or TERC are identified in a small minority of sporadic IPF patients (Figure 4), a significantly higher proportion of affected patients have short telomeres when measured in either their peripheral blood (36) or alveolar epithelial cells (AECs) (35). Collectively, these studies show that mutations in the aging-related genes TERT and TERC are associated with up to 15% of FPF cases and rare sporadic IPF cases. The questions of why patients with IPF have short telomeres despite having no TERT or TERC mutation and how these mutations or shortened telomeres contribute to the development of lung fibrosis have yet to be answered.
In 2011, a large genome-wide association study reported the linkage of a SNP in 82 families with FPF (37). The strongest linkage occurred in chromosome 11: marker D11S1318, with a LOD score of 3.3. Finer mapping of this region located a minor allele of the SNP rs35705950, located 3 kb upstream of the MUC5B gene, that was present in 34% of individuals with FPF. Further analysis found this SNP in 38% of individuals in a cohort of sporadic IPF patients and 9% of healthy controls. The rs35705950 allele was associated with lung expression of MUC5B that was 37.4 times higher than in unaffected subjects. Independent analysis confirmed association of the rs35705950 SNP with sporadic IPF in two additional cohorts (38): The SNP was present in 34% of patients with IPF and only 11% of healthy controls. Although these data provide strong evidence for a linkage of MUC5B polymorphisms to IPF, the molecular consequences of the SNP and how increased MUC5B protein expression may contribute to development of lung fibrosis require further explanation.
Collectively, analysis for genetic mutations and polymorphisms in patients with FPF or sporadic IPF has yielded significant advances in understanding the genetic predisposition to development of lung fibrosis. A common theme to all the mutations identified to date is that they affect genes that are either expressed in lung epithelial cells (e.g., SPA, SPC, MUC5B) or lead to identifiable molecular changes in AECs (TERT, TERC). For example, telomeres are short in the AECs of nearly all patients with sporadic IPF (35). As discussed below, this knowledge has centered the understanding of the pathology of IPF around the role that the epithelial cell, and its dysfunction, plays in the development of lung fibrosis.
Endoplasmic reticulum stress
The ER is the cellular compartment where secreted and membrane proteins are made, folded, and matured for packaging and trafficking via the Golgi complex. ER stress occurs when there is an imbalance between cellular demand for protein synthesis and the ER's capacity to synthesize, process, and package the requisite proteins (39). In response to this stress, the cell activates a cellular response termed the unfolded protein response (UPR) that leads to activation of biochemical pathways designed to match the protein production capacity of the ER. If the UPR cannot match the demand, a terminal UPR is activated and the cell sacrifices itself through apoptotic pathways (39, 40). Mammalian cells contain three sensors of unfolded proteins—IRE1α (inositol-requiring enzyme 1α), PERK (protein kinase RNA-like ER kinase), and ATF6 (activating transcription factor 6)—which, when activated, initiate the UPR. Of these sensors, IRE1α is a key mediator of the apoptotic switch (41, 42). When the ER is under stress, IRE1α senses the unfolded proteins associated with such stress and undergoes trans-autophosphorylation. This phosphorylation signals the activation of proapoptotic pathways (41). In addition, IRE1α has an RNase that, when active, splices XBP1 (X-box-binding protein 1) mRNA to produce the transcription factor XBP1s, whose targets enhance ER protein-folding capacity (43). Thus, two markers of ER stress and the UPR are phosphorylation of IRE1α and splicing of XBP1.
Studies have reported that markers of UPR activation are elevated in alveolar type II cells of patients with IPF (44–46). AECs in IPF lungs express increased levels of p50ATF6 (the processed form of ATF6) (45, 47), CHOP [CCAAT/enhancer-binding protein (C/EBP) homologous protein], BiP (binding immunoglobulin protein), and IRE1α (44–46). In addition, type II cells isolated from IPF lungs demonstrated increased levels of active phosphorylated of IRE1α, XBP1s.
Many possible causes of ER stress have been identified in patients with IPF. Herpesvirus protein expression colocalizes with markers of the UPR in AECs from patients with IPF (46), suggesting that ectopic production of viral proteins may activate the UPR. As mentioned above, mutations in SPC and SPA have been identified in patients with lung fibrosis. When the SPC mutants SPCΔexon4 or L188Q are expressed in lung epithelial cells, they activate mediators of the UPR, as evidenced by increased levels of BiP, EDEM (ER degradation–enhancing α-mannosidase-like protein), p-eIF2α (phosphorylated - eukaryotic initiation factor 2 alpha), and XBP1s (48-50). Similarly, expression of the G231V or F198S mutant of SPA in epithelial cells leads to increased levels of BiP and XBP1s (51). These potential causes of UPR activation in IPF lungs cannot be the only ones, because surfactant mutations account for no more than 5% of cases of familial or sporadic IPF (Figure 4), and herpesviruses have been identified in patients with sporadic IPF (46, 52). Therefore, if UPR activation is a central pathway promoting development of IPF, other triggers of UPR activation need to be identified.
TGF-β activation
Levels of active transforming growth factor β (TGF-β) are increased in the lungs of patients with IPF. All three isoforms (TGF-β1, TGF-β2, and TGF-β3) of inactive TGF-β are synthesized and secreted bound to the latency-associated peptide (LAP) (53). Under normal conditions, TGF-β is bound in its inactive form to LAP, which is disulfide-linked to latent TGF-β-binding proteins, cross-linked to the extracellular matrix. During the development of lung fibrosis, AECs express increased levels of the integrin αvβ6 (54), which can bind to the arginine-glycine-aspartate (RGD) sequence of LAP (55). Binding of αvβ6 is restricted to the TGF-β1 and TGF-β3 isoforms because the TGF-β2 isoform lacks an RGD sequence. Activation of epithelial cells expressing αvβ6 through binding of mediators such as thrombin (55b) and the phospholipids sphingosine 1-phosphate (56) and lysophosphatidic acid (57) to their receptors induces actin/myosin-mediated contraction of the epithelial cell. This contraction pulls on the latent TGF-β tethered to the epithelial cell via binding of LAP to the integrin αvβ6. This retraction induces TGF-β activation and signaling in localized, spatially restricted lung regions because the active TGF-β does not appear to be released into a soluble form. Whether αvβ6-mediated TGF-β activation contributes to IPF has yet to be proved, but possible profibrotic processes associated with TGF-β activation include inhibition of AEC proliferation, differentiation of fibroblasts to myofibroblasts (59), and activation of programming that promotes mesenchymal transition of epithelial cells (60).
Epithelial-to-mesenchymal transition
Epithelial-to-mesenchymal transition (EMT) is the process by which epithelial cells acquire molecular and cell physiologic features commonly associated with mesenchymal cells following activation by specific growth factors, of which TGF-β is the prototype (60, 61). This molecular reprogramming, which causes epithelial cells to express mesenchymal cell–associated genes, occurs in three biologic contexts: development, cancer, and fibrosis (62). Many molecular pathways for these three subtypes of EMT overlap regardless of the biologic context. For example, growth factor activation of epithelial cells triggers upregulation of a specific set of proteins (e.g., slug, FSP1, αSMA) and downregulation of others (e.g., E-cadherin, syndecan 1) (62). These changes in protein expression cause epithelial cells to lose their polarity and tight junctions and to become more mobile (62).
Normal tissue homeostasis does not require EMT (62, 63); rather, EMT programming is activated during conditions of tissue injury and remodeling (62, 63). The evidence that alveolar cells exhibit EMT in IPF patients is based on immunohistochemical studies colocalizing epithelial cell– and mesenchymal cell-associated proteins within IPF lungs (60, 64, 65). In one report, the alveolar type II cell markers TTF-1 (thyroid transcription factor 1) and prosurfactant protein B colocalized with αSMA (α smooth muscle actin) in the majority of AECs in IPF lungs (65). Similarly, immunostaining colocalized the alveolar type II cell marker prosurfactant protein C and the mesenchymal proteins N-cadherin, αSMA, and calponin 1 in IPF lungs (60, 64). These proteins did not colocalize in normal lungs. mRNA analysis confirms that epithelial cells express mesenchymal proteins in IPF lungs. Epithelial cells isolated by laser capture express higher levels of mRNA for the mesenchymal proteins type I collagen, αSMA, and calponin 1 (64). This finding was confirmed in AECs isolated from IPF lungs, which also demonstrated elevated mRNA levels for type I collagen, αSMA, and calponin 1 as well as the transcription factor SLUG (64), which has been shown to promote EMT.
Although evidence indicates that epithelial cells acquire mesenchymal features in IPF lungs, the contribution of these mesenchymal changes to the fibrotic process remain unresolved. One ongoing controversy is whether epithelial cells acquire sufficient mesenchymal characteristics that they can be classified as fibroblasts (61, 66). In humans, the only datum supporting this possibility is the finding that fibroblasts isolated from IPF patients express the epithelial cell protein keratin 18 (67). However, this finding does not prove that fibroblasts are derived from epithelial cells, because it is possible that fibroblasts express this epithelial cell–associated protein in isolation or are undergoing a mesenchymal-to-epithelial transition (68). Although it is difficult to resolve whether fibroblasts are derived from epithelial cells in human studies, elegant lineage tracing studies have shown that the transition of alveolar type II cells accounts for 30–40% of cells expressing the mesenchymal proteins vimentin or S100A4 in models of lung fibrosis (60, 69). Similar lineage tracing studies have reported that myofibroblasts, in contrast, are rarely derived from epithelial cells in the bleomycin lung fibrosis model (66, 69). Whether AECs ever express a sufficient number of mesenchymal markers to identify them as fibroblasts in IPF lungs remains unresolved. Resolution of this issue is complicated by the lack of definitive, cell-specific markers for fibroblasts. Although it may be important to understand whether epithelial cells become fibroblasts in IPF patients, what is more important is how activation of EMT programming, or production of mesenchymal proteins by epithelial cells, contributes to the fibrotic process. This may be determined through additional studies using genetically modified mice that lack either mesenchymal proteins or signaling mediators of EMT in epithelial cells in models of lung fibrosis.
Relationships between the genetic predisposition to idiopathic pulmonary fibrosis, epithelial-to-mesenchymal transition, endoplasmic reticulum stress, AND TGF-β
The UPR, TGF-β, and EMT are activated in patients with IPF (Figure 5), but how this activation occurs remains largely undefined. Genetic predisposition may explain the activation of these processes in some patients. As outlined above, surfactant mutations lead to activation of the UPR. Although there is strong evidence for activation of the molecular pathways of the UPR in the AECs of IPF patients, whether this activation is sufficient to lead to development of lung fibrosis remains unresolved. For example, ectopic expression of the L188Q mutated form of SPC in the type II cells of mice for 6 months did not lead to fibrosis despite activation of the UPR (49). However, these mice demonstrated increased apoptosis and exaggerated lung fibrosis in response to bleomycin (49). These data show that activation of the UPR for 6 months is not sufficient to promote development of lung fibrosis in mice, but that such UPR activation alters the epithelium to a phenotype that, when subjected to a second external stimulus, is more prone to activation of profibrotic pathways. Whether chronic UPR activation in the alveolar epithelium over many years—which may be the situation in humans—is sufficient to cause lung fibrosis needs further study.
Figure 5.

Profibrotic attributes of epithelial cells in idiopathic pulmonary fibrosis lungs. Genetic mutations in epithelial cell–associated proteins predispose to the development of lung fibrosis by leading to the development of short telomeres or endoplasmic reticulum (ER) stress. ER stress may be profibrotic by causing apoptosis or release of TGF-β. Epithelial cells release profibrotic mediators such as Wnts, PDGF, and TGF-β, which activate fibroblasts, or CXCL12 and CCL2, which recruit fibrocytes to the lung. Epithelial cells produce the integrin αvβ6, which activates TGF-P and may thereby cause epithelial cells to undergo epithelial-to-mesenchymal transition.
Mutations of the SPA protein lead to UPR activation in epithelial cells (51, 70). Patients heterozygous for the G231V SPA mutation have higher levels of TGF-β in their bronchoalveolar lavage fluid than healthy control subjects do (70). These observations suggest a link between activation of the UPR and production of TGF-β. In support of this relationship, epithelial cells heterologously expressing the G231V or F198S SPA mutations express higher levels of TGF-β and of the TGF-β-binding proteins LTBP-1 and LTBP-4, which are required for TGF-β secretion (70). This increased TGF-β secretion is at least in part dependent on activation of the UPR, because siRNA gene silencing of IRE1α, PERK, or ATF6 significantly decreases mutant SPA–mediated TGF-β secretion. This UPR-mediated TGF-β secretion appears limited to specific genetic mutations, because not all the examined SPA or SPC mutations mediate TGF-β secretion. Overall, these results suggest that, in some circumstances, activation of the UPR may contribute to development of pulmonary fibrosis by enhancing TGF-β secretion.
A relationship between activation of the UPR and EMT has also been reported (50, 71). When AECs are exposed to the ER stress inducers thapsigargin, tunicamycin, or the SPCΔexon4 mutation, the cells increase expression of the ER chaperone BiP and XBP1s, change morphology to a shape more consistent with mesenchymal cells, decrease expression of E-cadherin and zonula occludens 1, and increase expression of αSMA (71). This ER stress–induced EMT depends on Src kinase, because inhibition of Src using PP2 [4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4]pyrimidine] blocks the ER stress–mediated EMT (71). Similarly, stable expression of the L188Q mutated form of SPC leads to decreased expression of E-cadherin and zonula occludens 1, increased expression of S1004A and αSMA, and a morphologic change of the epithelial cells to a mesenchymal shape (50). The TGF-β inhibitor SB431542 or siRNA knockdown of the TGF-β signaling molecule Smad2 blocks EMT changes. Further, these changes depend on UPR activation, because siRNA knockdown of IRE1 blocks Smad2/3 phosphorylation (50). Collectively, these reports suggest that ER stress and activation of the UPR could contribute to fibrotic remodeling in the lungs, in part via TGF-β activation and EMT-mediated pathways.
External stressors in idiopathic pulmonary fibrosis
The sections above outline genetic mutations predisposing to the development of lung fibrosis and how these mutations may lead to profibrotic molecular changes originating within epithelial cells. In these examples, the primary pathologic change is a genetic mutation that causes the epithelial cells to become intrinsically abnormal. These genetic abnormalities lead to activation of molecular pathways such as the UPR, TGF-β secretion, and EMT, and ultimately to lung fibrosis (Figure 5). Although in theory these mutations could lead directly to lung fibrosis, there are no data to support this possibility. For example, the telomerase-deficient mice have not been reported to develop lung fibrosis (72, 73). Similarly, when the L188Q mutated form of SPC is overexpressed in mouse alveolar type II cells, the mice do not develop lung fibrosis despite activation of the UPR and increased apoptosis of alveolar type II cells (49). Why the genetic mutations do not lead directly to the development of lung fibrosis in mouse models remains unresolved. One possibility is that mice, because of their short life span, are not exposed to the effects of the mutations long enough to cause fibrosis, whereas humans are exposed to the effects of the mutations for decades before lung fibrosis develops. An alternative explanation for the lack of confirmation that genetic mutations lead directly to fibrosis is that exposure to additional environmental insults in genetically predisposed individuals is required for development of lung fibrosis. For example, aged mice develop more severe lung fibrosis after exposure to bleomycin or murine gammaherpesvirus infection (74–76). In the case of gammaherpesvirus infection, the predisposition to more severe fibrosis in aged mice is associated with greater activation of the UPR and increased apoptosis (76). Similarly, mice expressing the L188Q mutated form of SPC develop lung fibrosis after administration of bleomycin in doses that do not cause fibrosis in control animals (49). Collectively, these observations suggest that genetic mutations may not be sufficient to cause lung fibrosis; rather, a second event—that is, an environmental exposure—may act in concert with a genetically predisposed epithelium.
What such key external exposures are in IPF patients remain to be established. One possibility is that typical environmental exposures such as pollution, dust, or smoke may contribute to development of lung fibrosis in genetically predisposed individuals. Evidence for this possibility comes from epidemiologic studies reporting that smoke and dust exposure are found more often in patients with IPF than in healthy controls (77). An exposure that has received considerable attention is GER (and associated microaspiration). Patients with IPF are more likely to have GER than are control subjects (78, 79). Treatment of GER with either proton pump inhibitors or surgical fundoplication is associated with slower disease progression and better survival (80, 81), suggesting that GER and microaspiration may contribute to disease pathogenesis and/or exacerbation in genetically vulnerable individuals. What environmental exposures activate profibrotic molecular programming in the epithelium of genetically predisposed individuals and whether an exposure exceeding routine everyday exposures of the lung need further study.
Mesenchyme
Given that the epithelium is abnormal in IPF, the relationship between epithelial cell dysfunction and deposition of excess collagen within remodeled lung tissue needs explanation. The simple view—that subjecting epithelial cells to ER stress, leading to TGF-β activation and transition of epithelial cells to collagen-secreting mesenchymal cells, is a major source of the pathologic deposition of collagen—is likely inadequate. Limitations of this view are noted above. More likely, the development of lung remodeling and fibrosis depends on an interaction between (a) an epithelium that is dysregulated by the underlying genetic mutations, UPR activation, and/or EMT and (b) fibroblasts, the professional collagen-secreting cells of the lung. Whether this relationship is simply stimulation of fibroblasts by TGF-β that is activated by the epithelial cell integrin αvβ6 or whether there are more complex interactions between the epithelium and mesenchyme is the subject of ongoing research. The following section highlights recent advances in understanding the relevant interactions between epithelial and mesenchymal cells.
Fibrocytes
Fibrocytes are bone marrow-derived mesenchymal cells that can be identified in the circulation or in tissues by their coexpression of the hematopoietic precursor markers CD34 or CD45 and the mesenchymal cell protein type I collagen (82). Fibrocytes are proposed to contribute to development of lung fibrosis by directly producing extracellular matrix proteins such as type I and type III collagen, by differentiating into fibroblasts or myofibroblasts, or by producing cytokines that induce collagen deposition (83, 84). Fibrocytes have been found both in circulation (85–87) and in the lung parenchyma (85) of patients with IPF. An increase in the percentage of circulating fibrocytes correlates with disease exacerbations in IPF patients (87). These elevated fibrocyte levels return to baseline after the exacerbation resolves. Furthermore, IPF patients whose fibrocytes represent >5% of their total circulating blood leukocytes have a worse prognosis than do those whose fibrocytes represent <5% of total circulating blood leukocytes. These epidemiologic studies suggest that fibrocytes are significant contributors to lung fibrosis.
The alveolar epithelium may play a role in recruiting fibrocytes to IPF lungs. Fibrocytes express the chemokine receptors CXCR4 (84, 85) and CCR2 (88). The alveolar epithelium in IPF lung expresses CXCL12, a ligand for CXCR4 (85, 86). Furthermore, circulating levels of CXCL12 are increased in the plasma of IPF patients (85, 86). Similarly, the alveolar epithelium of IPF patients expresses CCL2 (89), the ligand for CCR2. These observations suggest that circulating fibrocytes may be recruited to IPF lungs through a CXCR4/CXCL12 or CCR2/CCL2 axis and that expansion of fibrocytes in the lungs may contribute to IPF.
Fibroblasts
Epithelial cell dysfunction and aberrant epithelial-mesenchymal signaling lead to the activation of fibroblasts and the deposition and remodeling of matrix. This chronic activation appears to lead to profibrotic, pathologic changes in IPF fibroblasts (90– 94). The myofibroblast is the classic pathologic fibroblast phenotype described in IPF lungs (59). Several mediators, including TGF-β, can elicit the differentiation of fibroblasts to myofibroblasts. Compared with resident lung fibroblasts, myofibroblasts secrete excessive amounts of matrix, including type I collagen (95). This excess matrix deposition may lead to pathologic lung fibrosis and remodeling. The myofibroblast phenotype may not be durable and appears to require ongoing activation by myofibroblast differentiating factors (e.g., TGF-β), because the phenotype can be reversed by withdrawal of a differentiating stimulus such as myoD (myogenic differentiation 1) or by stimulation with factors such as FGF (fibroblast growth factor) (96, 97). Additional phenotypic changes of fibroblasts that could contribute to the development of lung fibrosis include resistance to apoptosis and acquisition of an invasive phenotype (Figure 6) (90, 98). These phenotypic changes appear durable; they persist after the fibroblasts are removed from patients.
Figure 6.

Mechanisms by which mesenchymal cells or matrix contribute to lung fibrosis. Idiopathic pulmonary fibrosis fibroblasts acquire pathologic, profibrotic traits such as resistance to apoptosis, production of excess matrix, and an invasive phenotype. The remodeled matrix is abnormally stiff, and the composition of the extracellular matrix is extensively altered compared with that in normal lungs. In turn, these pathologic changes within the matrix may further promote profibrotic traits within fibroblasts.
During normal wound healing, unneeded fibroblasts are removed through the activation of apoptotic pathways (99). This clearance mechanism for fibroblasts limits ongoing matrix deposition and fibrosis. Unlike normal fibroblasts, IPF fibroblasts resist apoptosis when exposed to FAS ligand (91) and have greater proliferative capacity when grown on polymerized collagen (94, 100). Molecular changes of IPF fibroblasts that potentially protect them from apoptosis include SPARC (101) or survivin production (102), increased STAT-3 signaling (103), and lower levels of the prostaglandin E2 generator cyclooxygenase 2 (COX-2) (104) and of the transcriptional activator of apoptosis-inducing proteins forkhead box 03a (FoxO3a) (100). IPF fibroblasts interact with polymerized collagen via the α2β1 integrin (94). The levels of this integrin are abnormally low in IPF fibroblasts, resulting in pathologic activation of the PI3K (phosphoinositide 3-kinase)/Akt pathway due to inappropriately low PTEN (phosphatase and tensin homolog) function (94). This leads to high levels of FoxO3a, which enable IPF fibroblasts to escape the negative regulation of proliferation normally exerted by a type I collagen–rich environment (100). Activation of these prosurvival pathways in IPF fibroblasts may lead to their retention in IPF lungs, allowing the fibroblasts to deposit collagen in an ongoing manner, which ultimately leads to pathologic levels of lung remodeling and fibrosis.
It has been commented that IPF has features similar to those of cancer. Most notable is the relentless progression of IPF, which is similar to that of cancer, and the appearance of foci of fibroblasts, which are reminiscent of tumorlets (105). Although the foci of fibroblasts are not monoclonal and are more accurately described as reticula of fibroblasts (105), their anatomic appearance suggests the possibility that fibroblasts isolated from IPF patients may be able to invade the extracellular matrix much like metastatic cancer cells do (90, 93, 98). Significantly, IPF fibroblasts invade artificial basement membranes more readily than control fibroblasts do (90, 93, 98). The mechanisms for this enhanced invasion are poorly understood; however, the invasive properties correlate with levels of αSMA expression. Furthermore, the invasive phenotype of lung fibroblasts can be induced by the α5β1 integrin and negatively regulated by α4β1 integrin signaling (98). Additional clues to regulation of the invasive phenotype of IPF fibroblasts come from studies reporting that overexpressing hyaluronan synthase 2 (HAS2) in myofibroblasts results in a more severe fibrotic phenotype following bleomycin-induced lung injury (90). The development of this fibrosis depends on the hyaluronan receptor CD44, because the invasive phenotype and degree of fibrosis are diminished in CD44-deficient mice and in those treated with a CD44-blocking antibody. Similarly, siRNA knockdown of HAS2 or treatment with CD44 blocking antibodies limits the invasive capacity of IPF fibroblasts (90). These studies demonstrate that IPF fibroblasts have an enhanced invasive phenotype and that this invasive phenotype is regulated by a range of mediators, including CD44 and HAS2.
Epithelial-mesenchymal cross talk
In regions of normal or IPF lung, epithelial cells and fibroblasts are in close apposition. The best evidence of this association is fibroblast foci, which have a layer of epithelium overlying the fibroblasts (Figure 2). At these regional sites, epithelial cells may signal directly to fibroblasts by releasing soluble mediators that can act in trans to activate adjacent fibroblasts. One example is the release of TGF-β by epithelial cells (106) and its activation by the epithelial integrin αvβ6, which could signal to fibroblasts, converting them to αSMA-expressing myofibroblasts. Another cytokine IPF AECs produce in increased amounts is PDGF (platelet-derived growth factor) (107), which can contribute to lung fibrosis by promoting fibroblast proliferation (108). This possibility is currently being tested in a clinical trial examining whether a tyrosine kinase inhibitor that targets a several receptors, including PDGF receptor, slows the progression of IPF (109).
Additional soluble mediators produced by epithelial cells that may signal adjacent fibroblasts are Wnt proteins. Wnts are secreted glycoproteins that play key roles in lung development, cancer, and fibrogenesis. Lipid modification of Wnts makes them relatively insoluble, which suggests that they may be deposited in the extracellular matrix and act locally after release (110). In humans, 19 Wnt glycoproteins and 10 receptors for Wnts have been identified (111). Microarray screens revealed that Wnts, including Wnt2 and Wnt5a, and the Wnt receptor Fzd7 are overexpressed in IPF lungs. A more focused analysis of Wnt expression in IPF alveolar epithelium revealed that Wnt1 and Wnt3a are produced by IPF alveolar epithelium (112). Further, Wnt3a stimulates type I collagen production by fibroblasts. The downstream canonical Wnt signaling molecule β-catenin has been localized in nuclei of fibroblast foci in IPF lungs, suggesting that IPF fibroblasts may respond to Wnt activation (113, 114). Thus, Wnts produced by epithelial cells are candidate mediators of profibrotic signaling between epithelial cells and the mesenchyme in IPF.
Propagation of Lung Fibrosis
A key issue to resolve to understand the pathobiology of IPF is why the disease has a relentlessly progressive clinical course. A common explanation is that IPF progresses due to repetitive, often microscopic injury to the alveolar epithelium (115). The term injury implies an external force that causes harm to the lung. There is little clinical or histopathologic evidence to support this claim. In contrast, histopathologic analysis of IPF tissues commonly demonstrates normal lung epithelium adjacent to remodeled lung (Figure 1). The possibility that the epithelium is preferentially injured at the deepest recesses of the lung is hard to confirm. In contrast, the injury hypothesis is supported by histopathologic findings in patients suffering an acute exacerbation of IPF (Figure 3). In these cases, patients have the pathologic hallmarks of lung injury—hyaline membranes, alveolar filling with proteinaceous material, and organizing pneumonia—diffusely throughout their lungs (116). Because the pathologic changes of an acute exacerbation of IPF are diffuse and occur in a minority of patients with IPF (5, 117), other explanations for the relentless progression of IPF should be considered in patients who do not exhibit such acute exacerbations.
Alternative explanations for IPF progression outlined in the preceding sections include the genetic mutations associated with IPF and the phenotypic changes identified in IPF fibroblasts. Genetic mutations are durable, irreversible, changes that could contribute to disease progression via persistent activation of the UPR or secretion of TGF-β. However, the surfactant mutations are the only genetic mutations with defined molecular pathways that can explain their contribution to lung fibrosis. The absence of an understanding of molecular mechanisms leading to pulmonary fibrosis in patients with MUC5B, TERT, or TERC mutations or in individuals with no identifiable mutation means that nongenetic explanations for the relentless progression of IPF must be considered. Another possible explanation is that the durable phenotypic changes of IPF fibroblasts lead to IPF progression. However, these changes have yet to be proved relevant in in vivo model systems.
Role of the extracellular matrix
The extracellular matrix is extensively modified in IPF lungs. One possible explanation for IPF progression is that the pathologically remodeled extracellular matrix includes molecules that signal mesenchymal cells to release additional extracellular matrix. If a critical threshold of profibrotic matrix is deposed, there may be a feed-forward, autoamplified loop of matrix deposition and remodeling. A recent study used proteomic analysis identified more than 40 proteins that are over- or underrepresented in decellularized IPF matrix (118). Many of these proteins are known to activate signaling pathways on the cells that engage them. Further, decellularized IPF matrix is significantly stiffer than normal controls (118). When fibroblasts are cultured on stiff matrices or the stiffer IPF matrix, they adopt an activated myofibroblast phenotype (118– 120) that includes increased αSMA and decreased COX-2 expression, which are features of IPF fibroblasts. Thus, it is possible that a change in the composition or stiffness of the extracellular matrix can contribute to the propagation of lung fibrosis. How these changes in extracellular matrix proteins relate to epithelial cells, and whether they are sufficient to explain the phenotypes of IPF fibroblasts, requires further study.
Epigenetics
Regulation of gene expression by epigenetic influences has recently been recognized as an important mechanism by which environmental or other stresses can induce lasting changes in the phenotype of a cell, tissue, or organism (121). Epigenetic changes include alterations in DNA methylation, histone modification, or microRNA expression (121, 122). Exposure to environmental stresses such as tobacco smoke (123, 124), air pollution (125), and aging (126) can lead to epigenetic DNA changes. In a study of epigenetic changes in IPF lung, microRNA microarray analysis revealed that levels of 18 microRNAs, including let-7d, were decreased in IPF lungs (127). The let-7d microRNA localized to the alveolar epithelium, and the decreased let-7d levels in epithelial cells were associated with increased expression of the mesenchymal proteins N-cadherin, vimentin, and αSMA. Similarly, genome-wide methylation analysis identified a large number of differentially methylated genes in IPF lungs (128, 129). Changes in DNA methylation corresponded to mRNA expression for numerous genes, some of which have known roles in IPF, such as regulation of apoptosis and biosynthetic cellular processes (128–130). These findings suggest that DNA methylation and/or other epigenetic changes are important in the pathogenesis of IPF and that their enduring influences on gene expression could in part explain the relentless progression of the illness.
Subclassification of Patients with Idiopathic Pulmonary Fibrosis
IPF has specific clinical, radiologic and histopathologic features that are required for diagnosis. Within the clinical entity of IPF, subgroups of IPF patients with distinct cellular or molecular phenotypes predicting differences in rates of progression are beginning to be distinguished. For example, histopathology can be used to sub-categorize patients with IPF. IPF patients with greater numbers of fibroblastic foci have more rapid progression of IPF (131) and patients with greater numbers of lung mast cells have slower progression of IPF (132). Similarly, genetics have been used to sub-categorize IPF patients. IPF patients with the TT or GT MUC5B genotype have slower rates of progression than IPF patients with the GG MUC5B genotype (133). IPF patients carrying the minor allele rs5743890 in the TOLLIP gene are at increased risk for mortality from IPF (134). Finally, circulating molecular markers have been used to sub-phenotype patients with IPF. Measured markers include MMP7 (135), CCL18 (136), KL-6 (137, 138), and SPD (139), all of which have been reported to predict poor survival when elevated in IPF patients. These studies demonstrate that patients meeting clinical criteria for the diagnosis of IPF may have distinct cellular or molecular subtypes. Whether these subtypes represent distinct diseases, different phases of the same disease or can be used to predict response to therapy requires further research.
Conclusions
IPF is a progressive lung disorder of increasing prevalence that is a major cause of death for individuals suffering from the disease. Recent advances in the mechanistic understanding of IPF pathogenesis permit the consideration of a plausible sequence of events that lead to the development of pulmonary fibrosis. This sequence of events can be subdivided into three pathophysiologic stages (Figure 7). The predisposition stage includes genetic mutations or variations (i.e., SNPs) that predispose individuals to develop lung fibrosis; the chronic epithelial cell turnover during the lifetime of an individual, which leads to shortened telomeres; and environmental exposures. These factors ultimately lead to epithelial cell dysfunction. Not all individuals in this stage will necessarily develop clinically relevant disease; whether they do depends on the degree and duration of exposure to these factors. In initiation stage, molecular mediators of epithelial cell dysfunction such as ER stress; excessive TGF-β activation; and growth factor, chemokine, or Wnt secretion lead to EMT, fibrocyte recruitment, and fibroblast differentiation. These lead to the progression stage, during which pathologic mesenchymal cells release abnormal types and quantities of matrix proteins, which remodel and scar the lung. The pathologically remodeled matrix or epigenetic changes within fibroblasts may lead to a feed-forward loop of mesenchymal cell activation and progressive fibrosis. Some patients with IPF may bypass some of these stages. To develop therapies for IPF, a clearer understanding is needed of the relationships between the specific molecular pathways that are activated during each stage of IPF. This understanding will inform which stage of the disease therapies target as well as whether targeting one stage of the disease is sufficient to slow progression or whether simultaneous targeting of molecular pathways in each stage of disease is required for effective therapy.
Figure 7.

Pathophysiologic stages of idiopathic pulmonary fibrosis (IPF). The pathogenesis of IPF can be delineated into three stages. The initial predisposition stage includes processes such as genetic mutations, environmental exposure (routine or otherwise), and aging that predispose an individual to develop lung fibrosis. The second initiation stage includes defined profibrotic processes, such as TGF-β activation, fibrocyte recruitment, epithelial-to-mesenchymal transition (EMT), and activation of the unfolded protein response (UPR), that, when engaged, accelerate profibrotic processes. The final progression stage includes molecular processes that lead directly to fibrosis, such as pathologic fibroblast differentiation, matrix deposition and remodeling, increased matrix stiffness, and profibrotic epigenetic changes within fibroblasts and epithelial cells. Events in the latter stage may bypass the first two stages, leading to persistent mesenchymal cell activation and matrix remodeling.
Summary Points.
Epithelial cell dysfunction is a central cause of IPF.
Genetic mutations predispose individuals to develop pulmonary fibrosis.
Pulmonary fibrosis appears to be due to gene-environment interactions.
IPF fibroblasts have distinct phenotypic alterations that contribute to the development of lung fibrosis.
The remodeled matrix may provide a feed-forward loop of profibrotic signaling.
Future Issues.
Establish how MUC5B, TERT or TERC mutations lead to profibrotic molecular changes within the lung.
Define additional genetic mutations that predispose to the development of lung fibrosis.
Define which environmental factors contribute to IPF and how they relate to the molecular pathogenesis of IPF.
Establish novel animal models of pulmonary fibrosis that more reliably recapitulate the human disease.
Define molecular mechanisms that explain how short telomeres and aging lead to pulmonary fibrosis.
Establish molecular links between defined biologic processes that occur in each stage of IPF.
Define the profibrotic molecular pathways of IPF that can be subject to effective therapeutic intervention.
Determine whether IPF consists of a group of disorders that share certain phenotypic features but reflect distinct subgroups of patients, characterized by different pathogenetic molecular mechanisms and clinical phenotypes.
Acknowledgments
This review was funded in part by National Institutes of Health grants HL104422 and HL108794 and by the Nina Ireland Lung Disease Program at UCSF.
Disclosure Statement: Paul Wolters has received grant support from Genentech, Harold Collard has served as a consultant to Biogen, Fibrogen, Genoa, Gilead, Intermune, Medimmune, Mesoblast, and Promedior and received grant support from Boehringer-Ingelheim and Genentech. Kirk Jones has no affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review
Contributor Information
Paul J. Wolters, Email: paul.wolters@ucsf.edu.
Harold R. Collard, Email: hal.collard@ucsf.edu.
Kirk D. Jones, Email: kirk.jones@ucsf.edu.
Literature Cited
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–16. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 3.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:285–92. doi: 10.1513/pats.200601-005TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–63. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 5.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–43. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JS, Song JW, Wolters PJ, Elicker BM, King TE, Jr, et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J. 2012;39:352–58. doi: 10.1183/09031936.00050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:1698–702. doi: 10.1164/rccm.201010-1752OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 377:1760–69. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 9.Idiopath Pulm Fibros Clin Res Netw. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–77. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cipriani NA, Strek M, Noth I, Gordon IO, Charbeneau J, et al. Pathologic quantification of connective tissue disease–associated versus idiopathic usual interstitial pneumonia. Arch Pathol Lab Med. 2012;136:1253–58. doi: 10.5858/arpa.2012-0102-OA. [DOI] [PubMed] [Google Scholar]
- 11.Song JW, Do KH, Kim MY, Jang SJ, Colby TV, Kim DS. Pathologic and radiologic differences between idiopathic and collagen vascular disease–related usual interstitial pneumonia. Chest. 2009;136:23–30. doi: 10.1378/chest.08-2572. [DOI] [PubMed] [Google Scholar]
- 12.Takemura T, Akashi T, Kamiya H, Ikushima S, Ando T, et al. Pathological differentiation of chronic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology. 2012;61:1026–35. doi: 10.1111/j.1365-2559.2012.04322.x. [DOI] [PubMed] [Google Scholar]
- 13.Trahan S, Hanak V, Ryu JH, Myers JL. Role of surgical lung biopsy in separating chronic hypersensitivity pneumonia from usual interstitial pneumonia/idiopathic pulmonary fibrosis: analysis of 31 biopsies from 15 patients. Chest. 2008;134:126–32. doi: 10.1378/chest.08-0033. [DOI] [PubMed] [Google Scholar]
- 14.Baron RM, Choi AJ, Owen CA, Choi AM. Genetically manipulated mouse models of lung disease: potential and pitfalls. Am J Physiol Lung Cell Mol Physiol. 2012;302:L485–97. doi: 10.1152/ajplung.00085.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L152–60. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 16.Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members N Engl J Med. 1986;314:1343–47. doi: 10.1056/NEJM198605223142103. [DOI] [PubMed] [Google Scholar]
- 17.Bonanni PP, Frymoyer JW, Jacox RF. A family study of idiopathic pulmonary fibrosis. A possible dysproteinemic and genetically determined disease Am J Med. 1965;39:411–21. doi: 10.1016/0002-9343(65)90208-1. [DOI] [PubMed] [Google Scholar]
- 18.Mac MJ. Familial pulmonary fibrosis. Dis Chest. 1951;20:426–36. doi: 10.1378/chest.20.4.426. [DOI] [PubMed] [Google Scholar]
- 19.Imokawa S, Sato A, Toyoshima M, Yoshitomi A, Tamura R, et al. Dyskeratosis congenita showing usual interstitial pneumonia. Intern Med. 1994;33:226–30. doi: 10.2169/internalmedicine.33.226. [DOI] [PubMed] [Google Scholar]
- 20.Utz JP, Ryu JH, Myers JL, Michels VV. Usual interstitial pneumonia complicating dyskeratosis congenita. Mayo Clin Proc. 2005;80:817–21. doi: 10.1016/S0025-6196(11)61538-3. [DOI] [PubMed] [Google Scholar]
- 21.Yabe M, Yabe H, Hattori K, Morimoto T, Hinohara T, et al. Fatal interstitial pulmonary disease in a patient with dyskeratosis congenita after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1997;19:389–92. doi: 10.1038/sj.bmt.1700674. [DOI] [PubMed] [Google Scholar]
- 22.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–64. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 23.Oh J, Bailin T, Fukai K, Feng GH, Ho L, et al. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14:300–6. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- 24.Mason PJ, Bessler M. The genetics of dyskeratosis congenita. Cancer Genet. 2011;204:635–45. doi: 10.1016/j.cancergen.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nogee LM, Dunbar AE, 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–79. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 26.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–28. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 27.Ono S, Tanaka T, Ishida M, Kinoshita A, Fukuoka J, et al. Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur Respir J. 2011;38:861–69. doi: 10.1183/09031936.00143610. [DOI] [PubMed] [Google Scholar]
- 28.Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, et al. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci USA. 2012;109:2325–29. doi: 10.1073/pnas.1114740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59:977–80. doi: 10.1136/thx.2004.026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markart P, Ruppert C, Wygrecka M, Schmidt R, Korfei M, et al. Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur Respir J. 2007;29:134–37. doi: 10.1183/09031936.00034406. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Safa WF, Lestringant GG, Frossard PM. X-linked dyskeratosis congenita: restrictive pulmonary disease and a novel mutation. Thorax. 2001;56:891–94. doi: 10.1136/thorax.56.11.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 34.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–57. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–56. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:729–37. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–77. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–86. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 40.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L721–29. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–75. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Upton JP, Wang L, Han D, Wang ES, Huskey NE, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–22. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 44.Cha SI, Ryerson CJ, Lee JS, Kukreja J, Barry SS, et al. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir Res. 2012;13:105. doi: 10.1186/1465-9921-13-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–46. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–26. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 47.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol. 2006;172:395–407. doi: 10.1083/jcb.200508016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108:10562–67. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286:30972–80. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. 2010;285:22103–13. doi: 10.1074/jbc.M110.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kropski JA, Lawson WE, Blackwell TS. Right place, right time: the evolving role of herpesvirus infection as a “second hit” in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L441–44. doi: 10.1152/ajplung.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 54.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, et al. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 55.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, et al. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 55b.Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, et al. Ligation of protease-activated receptor 1 enhances αvβ6 integrin–dependent TGF-β activation and promotes acute lung injury. J Clin Investig. 2006;116:1606–14. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giacomini MM, Travis MA, Kudo M, Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-β through integrin αvβ6–dependent physical force. Exp Cell Res. 2012;318:716–22. doi: 10.1016/j.yexcr.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu MY, Porte J, Knox AJ, Weinreb PH, Maher TM, et al. Lysophosphatidic acid induces αvβ6 integrin–mediated TGF-β activation via the LPA2 receptor and the small G protein Gαq. Am J Pathol. 2009;174:1264–79. doi: 10.2353/ajpath.2009.080160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deleted in proof
- 59.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–21. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 60.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–85. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517–23. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willis BC, Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–34. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 63.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Investig. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marmai C, Sutherland RE, Kim KK, Dolganov GM, Fang X, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L71–78. doi: 10.1152/ajplung.00212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-β1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–32. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–83. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larsson O, Diebold D, Fan D, Peterson M, Nho RS, et al. Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS ONE. 2008;3:e3220. doi: 10.1371/journal.pone.0003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li R, Liang J, Ni S, Zhou T, Qing X, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–65. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maitra M, Cano CA, Garcia CK. Mutant surfactant A2 proteins associated with familial pulmonary fibrosis and lung cancer induce TGF-β1 secretion. Proc Natl Acad Sci USA. 2012;109:21064–69. doi: 10.1073/pnas.1217069110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, et al. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol. 2011;45:498–509. doi: 10.1165/rcmb.2010-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee J, Reddy R, Barsky L, Scholes J, Chen H, et al. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L57–70. doi: 10.1152/ajplung.90411.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–12. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 74.Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L510–18. doi: 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sueblinvong V, Neujahr DC, Mills ST, Roser-Page S, Ritzenthaler JD, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012;344:41–51. doi: 10.1097/MAJ.0b013e318234c132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Torres-Gonzalez E, Bueno M, Tanaka A, Krug LT, Cheng DS, et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol. 2012;46:748–56. doi: 10.1165/rcmb.2011-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–48. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 78.Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27:136–42. doi: 10.1183/09031936.06.00037005. [DOI] [PubMed] [Google Scholar]
- 79.Tobin RW, Pope CE, 2nd, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1804–8. doi: 10.1164/ajrccm.158.6.9804105. [DOI] [PubMed] [Google Scholar]
- 80.Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, et al. Gastroesophageal reflux therapy is associated with longer survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:1390–94. doi: 10.1164/rccm.201101-0138OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raghu G, Yang ST, Spada C, Hayes J, Pellegrini CA. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest. 2006;129:794–800. doi: 10.1378/chest.129.3.794. [DOI] [PubMed] [Google Scholar]
- 82.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 83.Maharaj SS, Baroke E, Gauldie J, Kolb MR. Fibrocytes in chronic lung disease—facts and controversies. Pulm Pharmacol Ther. 2012;25:263–67. doi: 10.1016/j.pupt.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol. 2009;86:1111–18. doi: 10.1189/jlb.0309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andersson-Sjöland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–40. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 86.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–8. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 87.Moeller A, Gilpin SE, Ask K, Cox G, Cook D, et al. Circulating fibrocytes are an indicator for poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–94. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 88.Ekert JE, Murray LA, Das AM, Sheng H, Giles-Komar J, Rycyzyn MA. Chemokine (C-C motif) ligand 2 mediates direct and indirect fibrotic responses in human and murine cultured fibrocytes. Fibrogenes Tissue Repair. 2011;4:23. doi: 10.1186/1755-1536-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Königshoff M, et al. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1–inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:414–25. doi: 10.1164/rccm.200712-1827OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Jiang D, Liang J, Meltzer EB, Gray A, et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208:1459–71. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, et al. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:73–82. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramos C, Montaño M, Garcia-Alvarez J, Ruiz V, Uhal BD, et al. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001;24:591–98. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- 93.Tony DJ, Richards CD, Podor TJ, Gauldie J. Anchorage-independent colony growth of pulmonary fibroblasts derived from fibrotic human lung tissue. J Clin Investig. 1994;93:1525–32. doi: 10.1172/JCI117131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205:1659–72. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229:298–309. doi: 10.1002/path.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hecker L, Jagirdar R, Jin T, Thannickal VJ. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res. 2011;317:1914–21. doi: 10.1016/j.yexcr.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maltseva O, Folger P, Zekaria D, Petridou S, Masur SK. Fibroblast growth factor reversal of the corneal myofibroblast phenotype. Investig Ophthalmol Vis Sci. 2001;42:2490–95. [PubMed] [Google Scholar]
- 98.White ES, Thannickal VJ, Carskadon SL, Dickie EG, Livant DL, et al. Integrin α4β1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med. 2003;168:436–42. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 100.Nho RS, Hergert P, Kahm J, Jessurun J, Henke C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type I collagen matrix. Am J Pathol. 2011;179:2420–30. doi: 10.1016/j.ajpath.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of β-catenin. J Biol Chem. 2010;285:8196–206. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sisson TH, Maher TM, Ajayi IO, King JE, Higgins PD, et al. Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts. Adv Biosci Biotechnol. 2012;3:657–64. doi: 10.4236/abb.2012.326085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moodley YP, Scaffidi AK, Misso NL, Keerthisingam C, McAnulty RJ, et al. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp 130-mediated cell signaling and proliferation. Am J Pathol. 2003;163:345–54. doi: 10.1016/S0002-9440(10)63658-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Investig. 1995;95:1861–68. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med. 2006;174:654–58. doi: 10.1164/rccm.200602-205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu YD, Hua J, Mui A, O'Connor R, Grotendorst G, Khalil N. Release of biologically active TGF-β1 by alveolar epithelial cells results in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:L527–39. doi: 10.1152/ajplung.00298.2002. [DOI] [PubMed] [Google Scholar]
- 107.Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur Respir J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 108.Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183:225–37. doi: 10.1007/s00408-004-2534-z. [DOI] [PubMed] [Google Scholar]
- 109.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 110.Nusse R. Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development. 2003;130:5297–305. doi: 10.1242/dev.00821. [DOI] [PubMed] [Google Scholar]
- 111.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 112.Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, et al. WNT 1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Investig. 2009;119:772–87. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162:1495–502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, et al. Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Investig. 2009;119:213–24. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–51. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 116.Rice AJ, Wells AU, Bouros D, du Bois RM, Hansell DM, et al. Terminal diffuse alveolar damage in relation to interstitial pneumonias. An autopsy study Am J Clin Pathol. 2003;119:709–14. doi: 10.1309/UVAR-MDY8-FE9F-JDKU. [DOI] [PubMed] [Google Scholar]
- 117.Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27:143–50. doi: 10.1183/09031936.06.00114004. [DOI] [PubMed] [Google Scholar]
- 118.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–76. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marinkovic A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently inactivate IPF fibroblasts. Am J Respir Cell Mol Biol. 2012;48:422–30. doi: 10.1165/rcmb.2012-0335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang IV, Schwartz DA. Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med. 2011;183:1295–301. doi: 10.1164/rccm.201010-1579PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rabinovich EI, Selman M, Kaminski N. Epigenomics of idiopathic pulmonary fibrosis: evaluating the first steps. Am J Respir Crit Care Med. 2012;186:473–75. doi: 10.1164/rccm.201208-1350ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Launay JM, Del Pino M, Chironi G, Callebert J, Peoc'h K, et al. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS ONE. 2009;4:e7959. doi: 10.1371/journal.pone.0007959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu F, Killian JK, Yang M, Walker RL, Hong JA, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010;29:3650–64. doi: 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179:572–78. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–29. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE. 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–35. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cisneros J, Hagood J, Checa M, Ortiz-Quintero B, Negreros M, et al. Hypermethylation-mediated silencing of p14ARF in fibroblasts from idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303:L295–303. doi: 10.1152/ajplung.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.King TE, Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA, Jr, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025–32. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- 132.Cha SI, Chang CS, Kim EK, Lee JW, Matthay MA, Golden JA, Elicker BM, Jones K, Collard HR, Wolters PJ. Lung mast cell density defines a subpopulation of patients with idiopathic pulmonary fibrosis. Histopathology. 2012;61:98–106. doi: 10.1111/j.1365-2559.2012.04197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd JE, Gibson KF, Seibold MA, Brown KK, Talbert JL, Markin C, Kossen K, Seiwert SD, Murphy E, Noth I, Schwarz MI, Kaminski N, Schwartz DA. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–9. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski N, Garcia JG. genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Resp Med. 1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, Sciurba F, Dauber J, Selman M, Gochuico BR, Kaminski N. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, Olschewski M, Rottoli P, Muller-Quernheim J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:717–23. doi: 10.1164/rccm.200808-1201OC. [DOI] [PubMed] [Google Scholar]
- 137.Yokoyama A, Kohno N, Hamada H, Sakatani M, Ueda E, Kondo K, Hirasawa Y, Hiwada K. Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1680–4. doi: 10.1164/ajrccm.158.5.9803115. [DOI] [PubMed] [Google Scholar]
- 138.Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M, Bando M, Sugiyama Y, Totani Y, Ishizaki T, Ichiyasu H, Suga M, Hamada H, Kohno N. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. 2006;11:164–8. doi: 10.1111/j.1440-1843.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 139.Greene KE, King TE, Jr, Kuroki Y, Bucher-Bartelson B, Hunninghake GW, Newman LS, Nagae H, Mason RJ. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:439–46. doi: 10.1183/09031936.02.00081102. [DOI] [PubMed] [Google Scholar]