

Abstract
The design of polyvalent molecules, presenting multiple copies of a specific ligand, represents a promising strategy to inhibit pathogens and toxins. The ability to control independently the valency and the spacing between ligands would be valuable for elucidating structure-activity relationships and for designing potent polyvalent molecules. To that end, we designed monodisperse polypeptide-based polyvalent inhibitors of anthrax toxin in which multiple copies of an inhibitory toxin-binding peptide were separated by flexible peptide linkers. By tuning the valency and linker length, we designed polyvalent inhibitors that were over four orders of magnitude more potent than the corresponding monovalent ligands. This strategy for the rational design of monodisperse polyvalent molecules may not only be broadly applicable for the inhibition of toxins and pathogens, but also for controlling the nanoscale organization of cellular receptors to regulate signaling and stem cell fate.
Keywords: anthrax toxin, multivalency in, periodic polypeptides in, protein engineering in, structure-activity relationships
Synthetic polyvalent molecules are currently being developed for applications ranging from pathogen inhibition to drug delivery, the control of cellular signaling, and the inhibition of bacterial toxins.[1–8] Anthrax lethal toxin, which is responsible for the symptoms and mortality of anthrax,[9] is composed of two proteins: lethal factor (LF), an enzyme responsible for host cell death; and protective antigen (PA), which binds to cell-surface receptors and facilitates toxin internalization. Upon binding the host cell, PA is proteolytically processed into a 63 kDa fragment, PA63, which assembles into heptamers ((PA63)7) on the surface of the host cell. The binding of the toxic enzyme, LF, to the receptor-bound (PA63)7 results in the formation of a complex which is internalized by the host cell (Figure S1). This complex formation is one of the key steps in the intoxication pathway (Figure S1B),[10,11] and therapeutics have been designed to inhibit these interactions.[3] In previous work, phage display was used to identify a 12-mer peptide (HTSTYWWLDGAP) that binds to (PA63)7.[10] Polyvalent inhibitors have been created by attaching multiple copies of this peptide to a variety of scaffolds such as polymers,[12] liposomes,[13] and β-cyclodextrin.[14]
Structure-based design is a particularly effective strategy to design polyvalent inhibitors of anthrax lethal toxin and other targets. [11–16] Ligand spacing and valency are key parameters that influence the efficacy of polyvalent molecules (Scheme 1A).[12–14,17] The ligand spacing is simply the distance separating adjacent ligands on a polyvalent molecule.[5,13,14] Matching the ligand spacing on the polyvalent molecule to the distance between ligand binding sites on the target molecule has been shown to be an effective strategy to design potent polyvalent ligands.[16,18] For a linear polymer or polypeptide, ligand spacing can be manipulated by choosing a linker of appropriate length to separate adjacent ligands on a polyvalent molecule. Optimizing the valency is another effective strategy to enhance the binding of a synthetic polyvalent molecule to its target.[3,5,12,13,14] For instance, Gujraty et al. designed polyvalent inhibitors of anthrax toxin using polymeric scaffolds of controlled molecular weight and reported an initial sharp increase in potency with increasing valency, followed by a plateau where potency was independent of valency.[12] Matching the valency of the polyvalent molecule to the number of binding sites on the target molecule may also be an effective strategy.[5,12,14]
Scheme 1. Controlling the activity of polyvalent molecules.
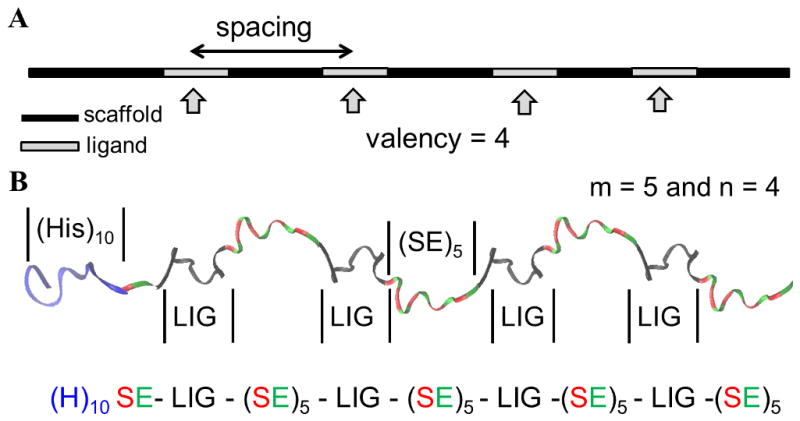
(A) Cartoon showing the key parameters that influence the efficacy of polyvalent molecules – valency and spacing. The valency of the polyvalent molecule depicted in the figure is 4. (B) Cartoon showing a representative image of (H)10-SE[LIG-(SE)5]4 polypeptide. Color scheme: black, 12-mer peptide ligand (HTSTYWWLDGAP); blue, decahistidine tag; red, serine; green, glutamic acid.
For polyvalent conjugates, the number of ligand molecules attached to the scaffold can usually be adjusted by varying the relative molar ratio of ligands to scaffold during a conjugation reaction. Bioconjugation coupling strategies such as amine-activated ester coupling or sulfhydryl-maleimide coupling have been the preferred strategies used to attach multiple copies of ligand molecules to scaffolds.[12,13] However, limited control over the specificity of bioconjugation reactions may result in final products with inconsistency in the number and position of ligand attachment. As a result, it is difficult to control the final ligand density and ligand spacing precisely and independently. In the case of polymeric scaffolds, the polydispersity of the scaffold may further increase the variability of the polyvalent inhibitor product, which can further complicate characterization efforts. Insufficient control over ligand spacing and valency and scaffold polydispersity are therefore important limitations of some of the typical approaches for designing polyvalent inhibitors.
To address these limitations, we describe an approach to design polyvalent inhibitors of anthrax toxin that are monodisperse and allow for exquisite control over ligand spacing and valency. The ability to make monodisperse biomacromolecular scaffolds via protein engineering approaches has long been appreciated.[19–23] Kiick and co-workers have used a combination of biosynthetic and chemical approaches to design well-defined polyvalent molecules based on polypeptide scaffolds with different architectures.[24,25] Here we designed polypeptides in which multiple instances of the inhibitory peptide ligand (LIG - HTSTYWWLDGAP) were incorporated within the polypeptide sequence in a serial fashion, separated by flexible peptide linker sequences. This biosynthetic approach therefore resulted in polypeptides that were monodisperse and displayed a predefined number of inhibitory peptide instances at defined spacings without the need for a chemical conjugation step.
Specifically, we designed monodisperse polypeptides with the sequence (H)10-SE[LIG–(SE)m]n, where the decahistidine tag aids in the purification of the polypeptides, ‘m’ represents the number of sequential repeats of serine and glutamic acid and therefore controls the ligand spacing, and ‘n’ represents the number of LIG repeats, i.e., the valency. The polar amino acids serine (S) and glutamic acid (E) were included in the linker sequences to enhance the solubility of the polypeptides. Scheme 1B is a representation of one such polypeptide sequence for m = 5 and n = 4.
Thermodynamic models of polyvalent interactions have suggested that the highest avidity of multivalent binding is obtained when the root mean square (RMS) end-to-end length of the linker separating adjacent ligands on a polyvalent molecule matches the separation between the ligand binding sites on the target molecule.[26,27] Therefore we expected that the greatest binding avidity could be obtained if the inter-ligand spacing in the polypeptide inhibitors matched the distance between adjacent binding sites on (PA63)7. Our previous work concluded that the 6-mer peptide sequence “TYWWLD” in the 12-mer peptide “HTSTYWWLDGAP” was both necessary and sufficient for ligand binding to PA63.[28] In addition, previous mutagenesis studies investigating the interactions between a library of PA mutants and phage presenting the sequence HYTYWWLD had found that the residues P184, L187, K197 and R200 on PA63 (indicated by black dots in Scheme 2A and represented by a black rectangular sector symbol in Scheme 2B and 2C) were important for the binding of the peptide ligand to (PA63)7.[14] From the crystal structure of (PA63)7,[29] we calculated that the distances between these four amino acid residues on adjacent PA63 monomers of (PA63)7 were in the 25–35 Å range (Table S1 and Scheme 2A). Assuming that the binding of the divalent (H)10-SE[LIG(SE)m]2 polypeptides to two adjacent PA63 monomers of (PA63)7 (Scheme 2C) occurs primarily via the interactions between the 6-mer “TYWWLD” sequence and the predicted ligand binding site on PA63, the sequence “GAP-(SE)m-HTS” serves as the linker separating adjacent ligands on the polypeptide. Based on the thermodynamic models, enhanced divalent binding could be expected if the root mean square (RMS) end-to-end distance of the “GAP-(SE)m-HTS” polypeptide sequence was also in the range of 25–35 Å. Linkers with RMS end-to-end distances in this range might be obtained by choosing appropriate values of ‘m’.
Scheme 2. Cartoon representing the binding of the polypeptide-based inhibitors to adjacent sites on (PA63)7.
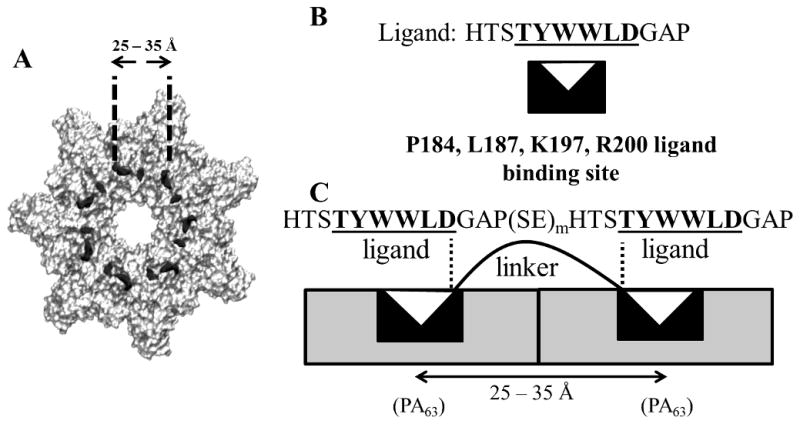
(A) Structure of (PA63)7 with black dots on individual PA63 monomers representing the predicted binding sites (P184, L187, K197, and R200) for the 12-mer ligand (B) Cartoon showing the interaction of the 6-mer “TYWWLD” (white triangle) with its binding site on PA63 (black sector symbol). (C) Cartoon representing interactions between a divalent polypeptide and binding sites on adjacent PA63 monomers of (PA63)7.
We performed replica exchange molecular dynamics (REMD) simulations to guide our choice of optimal values for ‘m’. Conformational sampling studies were performed using implicit solvent REMD simulations to predict the range of conformations that “GAP–(SE)m–HTS” linkers can attain for different values of ‘m’. As shown in Table S2, these studies suggested that GAP-(SE)m-HTS linkers for which the values of ‘m’ were in the range of 5 to 7 would have root mean-squared (RMS) end-to-end distances in the desired range of 25–35 Å. Therefore, we hypothesized that values of ‘m’ from 5 to 7 would enhance the polyvalent binding of the polypeptides to (PA63)7 and correspondingly increase the inhibitory efficacy.
Next, we performed experiments to test the validity of the simulation predictions and to study the effect of ligand spacing (‘m’) and valency (‘n’) on inhibitory potency. To characterize the effect of ligand spacing, we determined the ability of divalent polypeptides ((H)10-SE[LIG(SE)m]2) with values of m ranging from 3 to 13 to inhibit anthrax lethal toxin in a cytotoxicity assay. The procedures used for the generation of plasmids encoding the polypeptide sequences (Figure S2) and for polypeptide expression and purification have been described in detail in the Supporting Information. Half maximal inhibitory potency (IC50) values for the polypeptide inhibitors were determined using the dose response curves for individual polypeptides and expressed on a per-peptide basis (Figure 1). As shown in Figure 1A, all (H)10-SE[LIG(SE)m]2 polypeptides (m = 3 to 13) exhibited submicromolar IC50 values; polypeptides with values of m ranging from 5 to 13 exhibited the lowest IC50 values (ca. 60–90 nM). These results are consistent with the results from our REMD simulations which had predicted that polypeptides with values of m ranging from 5 to 7 would exhibit enhanced polyvalent binding. Based on these results we selected ‘m = 5’ as the value for ‘m’ going forward because it represented the minimum linker length required for a divalent polypeptide to exhibit maximum inhibition of anthrax lethal toxin.
Figure 1.
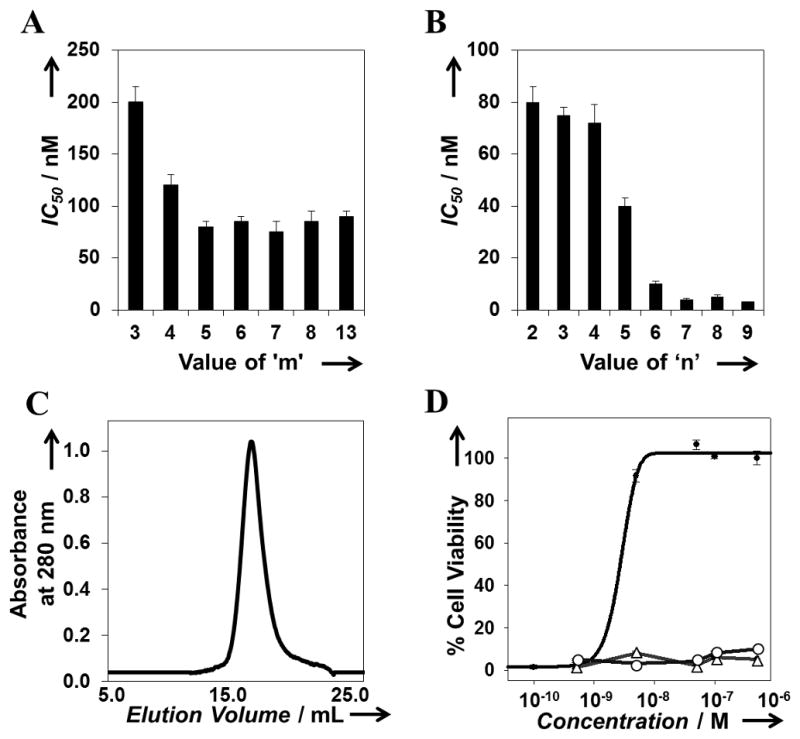
Influence of ligand spacing and valency on inhibitory potency of polypeptide inhibitors. (A) Influence of ligand spacing on the inhibitory potency of polypeptide inhibitors of the form (H)10-SE[LIG(SE)m]2. Estimates of the corresponding RMS linker lengths can be found in Table 2. (B) Influence of valency on the inhibitory potency of polypeptide inhibitors of the form (H)10-SE[LIG(SE)5]n. (C) SEC elution profile of (H)10-SE[LIG(SE)5]7 polypeptide inhibitors. (D) Comparison of dose response curves for (H)10-SE[LIG(SE)5]7 (line with filled circles), Ac-HTSTYWWLDGAPK-Am inhibitory peptide (line with open triangles), and negative control (H)10-SE[CTRL(SE)5]7 polypeptide (line with open circles).
Having validated the selected value for ‘m’ (m = 5), we next performed cytotoxicity experiments to determine the effect of valency (i.e., the value of ‘n’) on the inhibitory potency. To that end, we expressed, purified, and assayed polypeptides of the form (H)10-SE[LIG(SE)5]n, where the values of ‘n’ ranged from 2 to 9 and corresponded to the valency of the inhibitor. As shown in Figure 1B, we found that the IC50 values of the polypeptides decreased with an increase in valency. The IC50 values for ‘n = 7, 8, and 9’ were very similar and were in the 2 – 5 nM range. Based on these results we chose the inhibitor with ‘n = 7’ for further characterization and optimization because it represented the minimum number of ligands required by a polypeptide inhibitor with our selected inter-ligand spacing to exhibit the maximum inhibition of anthrax toxin.
Characterization by size exclusion chromatography indicated that the selected polypeptide, (H)10-SE[LIG(SE)5]7, eluted as a single peak suggesting that it was monodisperse (Figure 1C). Secondary structure characterization by far UV-CD spectroscopy indicated that the candidate polypeptide inhibitor was rich in random coil content suggesting that the polypeptide was flexible (data not shown). Figure 1D compares the dose response curves for (H)10-SE[LIG(SE)5]7, the monovalent Ac-HTSTYWWLDGAPK-Am inhibitory peptide, and a control polypeptide (H)10-SE[CTRL(SE)5]7, where the “HTSTYWWLDGAP” inhibitory peptide region was replaced by the sequence “HTSGGGSGGGAP” (CTRL), lacking the PA63-binding “TYWWLD” sequence. Cytotoxicity assays indicated negligible inhibitory activity against anthrax lethal toxin for the monovalent peptide and the polypeptide control (Figure 1D). The IC50 value of the candidate polypeptide (H)10-SE[LIG(SE)5]7, 4 ± 0.5 nM on a per-peptide basis, was more than four orders of magnitude lower than that for the monovalent peptide, which showed negligible inhibitory activity even at concentrations as high as 100 μM.[10]
While an in vivo characterization of the inhibitors is beyond the scope of the current study, we proceeded to characterize and optimize their serum stability, which will be critical for the successful use of polypeptide-based inhibitors in vivo. To test whether the candidate polypeptide was resistant to proteolytic degradation, we performed serum stability studies as described previously.[14] The candidate polypeptide was incubated in 80% mouse serum at 37 °C. Samples were withdrawn at various time intervals and their inhibitory activity was determined using the cytotoxicity assay. As shown in Figure 2A, samples incubated in serum for 24 and 48 h failed to exhibit any inhibitory activity in RAW264.7 cells after exposure to anthrax lethal toxin even at inhibitor doses as high as 1 μM.
Figure 2.
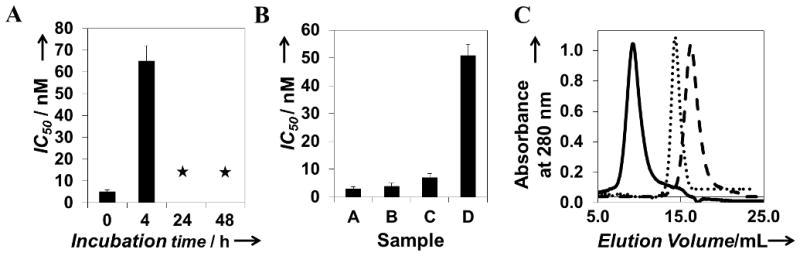
Effect of PEGylation on serum stability, activity, and hydrodynamic radius of (H)10-SE[LIG(SE)5]7. (A) Serum stability of (H)10-SE[LIG(SE)5]7 (B) Effect of PEGylation on inhibitory potency. IC50 values for: A, (H)10-SE[LIG(SE)5]7; B, C(H)9-SE[LIG(SE)5]7; C, PEG20k-C(H)9-SE[LIG(SE)5]7; and D, PEG20k-C(H)9-SE[LIG(SE)5]7 after incubation in 80 % mouse serum for 24 h (C) SEC elution profiles of (H)10-SE[LIG(SE)5]7 (dash line), bovine serum albumin (dotted line), and PEG20k-C(H)9-SE[LIG(SE)5]7 (black solid line). The maximum dose tested in (A) and (B) was 1 μM on a per peptide basis. Asterisks indicate that inhibitory activity was not detected.
We explored PEGylation as an approach to improve the serum stability of the candidate polypeptides, since this strategy has been shown to be effective in several previous reports.[30] To that end, we first used site-directed mutagenesis to replace the N-terminal histidine with a cysteine residue, and expressed and purified the resulting polypeptide (C(H)9-SE[LIG(SE)5]7). As shown in Figure 2B, introduction of a cysteine at the N-terminus did not have a significant effect on the inhibitory potency with C(H)9-SE[LIG(SE)5]7 and (H)10-SE[LIG(SE)5]7 exhibiting IC50 values of 4± 1 nM and 3 ± 0.7 nM respectively. Next, we PEGylated C(H)9-SE[LIG(SE)5]7 using methoxy-PEG-maleimide in a procedure described in the Supplementary Information. 20 kDa PEGs were conjugated to C(H)9-SE[LIG(SE)5]7 to yield “PEG20k-C(H)9-SE[LIG(SE)5]7”. PEGylated polypeptides were separated from non-PEGylated polypeptides and unreacted PEG molecules using size exclusion chromatography. The IC50 value of “PEG20k-C(H)9-SE[LIG(SE)5]7” was 9 ± 1.2 nM (Figure 2B), indicating that PEGylation did not significantly disrupt the activity of the candidate polypeptide.
Next, we tested the effect of PEGylation on the serum stability of the candidate polypeptide. While incubation of (H)10-SE[LIG(SE)5]7 in serum for 24 h resulted in the loss of its ability to inhibit anthrax lethal toxin (Figure 2A), serum-incubated PEG20k-C(H)9-SE[LIG(SE)5]7 successfully neutralized anthrax lethal toxin with an IC50 value of 51 ± 4 nM (Figure 2B). These results suggest that a balance between inhibitory potency and serum stability can be obtained by PEGylating the candidate polypeptides with 20 kDa PEG. Moreover, characterization by SEC indicated that “PEG20k-C(H)9-SE[LIG(SE)5]7” (MW ~ 39 kDa) eluted earlier than bovine serum albumin (BSA) (Figure 2C), indicating that it had a larger hydrodynamic radius than BSA. This result suggests that PEGylation of the polypeptide inhibitors may not only improve their serum stability but also their blood residence time, since proteins with a hydrodynamic radius equal to or greater than that of BSA are expected to have enhanced kidney retention and higher half-life in vivo.[31]
In conclusion, we have demonstrated the structure-based design of potent polyvalent inhibitors of anthrax lethal toxin. This protein engineering approach provides the requisite control over both ligand spacing and valency that is critical for elucidating structure-activity relationships. This strategy for the rational design of monodisperse polyvalent molecules could be broadly applicable not only for the inhibition of toxins and pathogens but also for the control of receptor clustering and cellular signaling in vitro and in vivo.[1,3,6–8]
Supplementary Material
Footnotes
This work was supported by NIH grant U01 AI056546, NIH grant R01 EB015482, and the P.K. Lashmet Chair Fund to R.S.K
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Experimental Details can be found in the Supporting Information.
Contributor Information
Sanket Patke, Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Mohan Boggara, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Ronak Maheshwari, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Sunit K. Srivastava, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA)
Manish Arha, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Marc Douaisi, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Jacob T. Martin, Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180 (USA)
Ian B. Harvey, Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA)
Matthew Brier, Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Tania Rosen, Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180 (USA).
Jeremy Mogridge, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, M5S 1A8 (Canada).
Prof. Ravi S. Kane, Email: kaner@rpi.edu, Department of Chemical and Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180 (USA). Center for Biotechnology and Interdisciplinary Studies Rensselaer Polytechnic Institute, Troy, NY 12180 (USA)
References
- 1.Mammen M, Choi SK, Whitesides GM. Angew Chem, Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Krishnamurthy VM, Estroff LA, Whitesides GM. Fragment-based approaches in drug discovery. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2006. pp. 11–53. [Google Scholar]
- 3.Vance D, Shah M, Joshi A, Kane RS. Biotechnol Bioeng. 2008;101:429–434. doi: 10.1002/bit.22056. [DOI] [PubMed] [Google Scholar]
- 4.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 5.Fan EK, Merritt EA, Verlinde CLMJ, Hol WGJ. Curr Opin Struct Biol. 2000;10:680–686. doi: 10.1016/s0959-440x(00)00152-4. [DOI] [PubMed] [Google Scholar]
- 6.Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. J Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 7.Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conway A, Vazin T, Spelke DP, Rode NA, Healy KE, Kane RS, Schaffer DV. Nat Nanotechnol. 2013;8:831–838. doi: 10.1038/nnano.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collier RJ, Young JAT. Annu Rev Cell Dev Bio. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 10.Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Nat Biotechnol. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 11.Nestorovich EM, Bezrukov SM. Chem Rev. 2012;112:6388–6430. doi: 10.1021/cr300141q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gujraty KV, Joshi A, Saraph A, Poon V, Mogridge J, Kane RS. Biomacromolecules. 2006;7:2082–2085. doi: 10.1021/bm060210p. [DOI] [PubMed] [Google Scholar]
- 13.Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Nat Biotechnol. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- 14.Joshi A, Kate S, Poon V, Mondal D, Boggara MB, Saraph A, Martin JT, McAlpine R, Day R, Garcia AE, Mogridge J, Kane RS. Biomacromolecules. 2011;12:791–796. doi: 10.1021/bm101396u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiessling LL, Strong LE, Gestwicki JE. Annu Rep Med Chem. 2000;35:321–330. [Google Scholar]
- 16.Polizzotti BD, Maheshwari R, Vinkenborg J, Kiick KL. Macromolecules. 2007;40:7103–7110. doi: 10.1021/ma070725o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maheshwari R, Levenson EA, Kiick KL. Macromol Biosci. 2010;10:68–81. doi: 10.1002/mabi.200900182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallik S, Plunkett SD, Dhal PK, Johnson RD, Pack D, Shnek D, Arnold FH. New J Chem. 1994;18:299–304. [Google Scholar]
- 19.Maskarinec SA, Tirrell DA. Curr Opin Biotechnol. 2005;16:422–426. doi: 10.1016/j.copbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Boersma YL, Plückthun A. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Davis NE, Karfeld-Sulzer LS, Ding S, Barron AE. Biomacromolecules. 2009;10:1125–1134. doi: 10.1021/bm801348g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Gitlin I, Krishnamurthy VM, Vazquez JA, Costello CE, Whitesides GM. J Am Chem Soc. 2003;125:12392–12393. doi: 10.1021/ja035978l. [DOI] [PubMed] [Google Scholar]
- 23.Amiram M, Quiroz FG, Callahan DJ, Chilkoti A. Nat Mater. 2011;10:141–148. doi: 10.1038/nmat2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Kiick KL. J Am Chem Soc. 2005;127:16392–16393. doi: 10.1021/ja055102+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Top A, Kiick KL. Adv Drug Delivery Rev. 2010;62:1530–1540. doi: 10.1016/j.addr.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gargano JM, Ngo T, Kim JY, Acheson DWK, Lees WJ. J Am Chem Soc. 2001;123:12909–12910. doi: 10.1021/ja016305a. [DOI] [PubMed] [Google Scholar]
- 27.Kane RS. Langmuir. 2010;26:8636–8640. doi: 10.1021/la9047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gujraty K, Sadacharan S, Frost M, Poon V, Kane RS, Mogridge J. Mol Pharmaceutics. 2005;2:367–372. doi: 10.1021/mp050040f. [DOI] [PubMed] [Google Scholar]
- 29.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 30.Pasut G, Veronese FM. Drugs Today. 2009;45:687–695. [PubMed] [Google Scholar]
- 31.Knauf MJ, Bell DP, Hirtzer P, Luo ZP, Young JD, Katre NV. J Biol Chem. 1988;263:15064–15070. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.