

Abstract
Since the approval of sipuleucel-T for men with metastatic castrate resistant prostate cancer in 2010, great strides in the development of anti-cancer immunotherapies have been made. Current drug development in this area has focused primarily on antigen specific [i.e. cancer vaccines and antibody based therapies)] or checkpoint inhibitor therapies, with the checkpoint inhibitors perhaps gaining the most attention as of late. Indeed, drugs blocking the inhibitory signal generated by the engagement of cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1) found on T-cells has emerged as potent means to combat the immunosuppressive milieu. The anti-CTLA-4 monoclonal antibody ipilimumab has already been approved in advanced melanoma and two phase III trials evaluating ipilimumab in men with metastatic castrate-resistant prostate cancer are underway. A phase III trial evaluating ProstVac-VF, a poxvirus-based therapeutic prostate cancer vaccine, is also underway. While there has been reason for encouragement over the past few years, many questions regarding the use of immunotherapies remain. Namely it is unclear what stage of disease is most likely to benefit from these approaches, how best to incorporate said treatments with each other and into our current treatment regimens and which therapy is most appropriate for which disease. Herein we review some of the recent advances in immunotherapy as related to the treatment of prostate cancer and outline some of the challenges that lie ahead.
Introduction
Prostate cancer is the most common non-cutaneous malignancy afflicting men in the United States. In 2013 it is expected that approximately 240,000 American men will be diagnosed with prostate cancer and nearly 30,000 of those individuals will die as result of the disease [1]. In the 1940s Charles Huggins discovered that prostate cancer would regress in response to androgen ablation, and since then targeting of the androgen/androgen receptor (AR) signaling axis has remained the cornerstone of advanced prostate cancer treatment [2,3]. In spite of the initially high success rates of androgen deprivation therapies, these drugs are far from curative, and ultimately the disease will progress to a clinical state known as castration-resistant prostate cancer (CRPC) [4].
In 2004, docetaxel was the first agent shown to prolong life for men with CRPC [5,6]. It garnered Food and Drug Administration (FDA) approval in 2005, and was followed in suit by sipuleucel-T in 2010. [7]. An autologous antigen presenting cell (APC) based immunotherapy, sipuleucel-T remains the only cancer vaccine approved for the treatment of any malignancy. Subsequently cabazitaxel, abiraterone, enzalutamide and radium-223 have also been shown to prolong life for men with advanced prostate cancer, with all subsequently gaining approval for men with CRPC [8-12]. With this recent explosion in therapies approved for the treatment of advanced prostate cancer, the question remains for how to best incorporate immune based treatment approaches with these newer agents.
The majority of work has gone into developing immune-based therapies that are either antigen (Ag) specific [i.e. cancer vaccines and antibody (Ab) based therapies)] or monoclonal antibodies that function as immune checkpoint inhibitors. It should be noted, however, that while not typically thought of as immunotherapies, a complex dynamic exists between androgen-directed therapies, cytotoxics and radiotherapies and their effect on the immune system; with all therapeutic approaches generating some degree of anti-cancer immune response [13-15]. If these primarily immune-based therapies are to truly make an impact in the treatment of prostate cancer they will likely need to be combined with some of the aforementioned ‘traditional’ therapies. Determining the sequence and most rational combinations of treatments will be a crucial next step in fully harnessing an anti-prostate cancer immune response.
Overview of the immune system
On a fundamental level, one major role for the immune system is to recognize and destroy cells displaying foreign antigens, regardless of whether they belong to infectious elements or tumors [16,17]. A variety of cell types comprise the immune system; they can broadly be categorized as either contributing to the host's innate or adaptive immunity. Cells of the innate immune system include neutrophils, macrophages and natural killer (NK) cells. An individual's innate immunity acts as the first line of defense against foreign antigens, and functions through the opsonization, phagocytosis, and release of protein mediators such as cytokines, chemokines and proteolytic enzymes. An innate immune response can in turn lead to the activation of the acquired immune system – which is in turn made up of T and B lymphocytes, responsible for cellular and humoral immune responses respectively. In general, the adaptive immune system is the predominate means through which cancer directed immunity occurs.
Within the adaptive immune system, T-cells generate a diverse repertoire of T-cell receptors (TCR) through somatic gene rearrangement occurring in the thymus. T-lymphocyte progenitors migrate from the bone marrow compartment to the thymus where they undergo T-cell receptor gene rearrangements, generating on the order of 1015 unique TCR. In the process of generating different TCR, a small fragment of episomal DNA is cleaved, producing a so called T-cell receptor rearrangement excision circle (TREC). These TREC can be detected in the circulation, and quantification of TREC serves as a sort of surrogate readout for a patient's production of new T cells. In contrast to B-cells, which produce secreted antibodies that recognize antigens in their natural state, T-cells recognize small peptide antigens bound within the groove of major histocompatibility (MHC) molecules. Given that TCR are specific for proteins in complex with MHC molecules, it is necessary for T-cells to be able to recognize MHC-foreign antigen complexes without being auto-reactive. In order to achieve this balance, T-cells undergo positive and negative selection while in the thymus. Only those T-cells able to interact with MHC complexes on an individual's thymic epithelial cells (i.e. positive selection) and not displaying high affinity for MHC complexes found on dendritic cells and macrophages (i.e. negative selection) will escape apoptosis and survive to migrate out of the thymus. Upon entering puberty, the thymus begins to involute and subsequent T-cells are generated predominately via peripheral expansion, with a small component of new diversity generated through poorly understood mechanisms. Peripherally expanded T-cells are clones of their predecessors, maintaining identical TCR to their forbearers.
After exiting the thymus and prior to being exposed to an antigen specific to their TCR, T-cells are considered naïve. In order to become activated, a T-cell requires two discrete signals. The first occurs when T cells recognize a cognate antigen-MHC complex on the surface of an antigen presenting cell (APC). Full T cell activation, however, also requires a second signal mediated by the appropriate signaling from co-stimulatory molecules on APC [18]. Co-stimulatory signaling typically occurs via the interaction between B7 proteins (CD80 and CD86) on APC and the CD28 family of proteins on T-cells [i.e. CD28,) [19]. In addition to these co-stimulatory interactions, there are a series of co-inhibitory interactions as well; i.e. the interaction between CTLA-4 and PD-1 and their respective ligands results in inhibitory signals which effectively turn off T cell function. The molecules that mediate these negative interactions have collectively been termed immune “checkpoints”, and have been blocked clinically using highly specific monoclonal antibodies. Additional inhibitory signals can be generated by additional immune cells such as myeloid derived suppressor cells (MDSCs), T-regulatory cells (CD4+CD25+ T-cells) (Tregs) and tumor-associated macrophages.
In addition to its role as an immune checkpoint, CTLA-4 has more recently been found to negatively mediate T-cell function through a number of additional mechanisms [20,21]. For one, it has been found that CTLA-4 is critical to Treg function, with mice harboring CTLA-4 deficient Tregs developing fatal T-cell mediated autoimmune disease [22]. It has also been found in humans that an anti-CTLA-4 antibody given in conjunction with the therapeutic tumor vaccine GVAX leads to an increased effector T-cells to Treg ratio – implying that drugs targeting CTLA-4 may work in part through the down-regulation of Tregs [23]. In preclinical models, relevant data show that anti-CTLA-4 antibodies specifically deplete tumor-associated regulatory T cells [24,25].
Once activated, a specific T-cell undergoes expansion and generates effector T-cells (i.e. CD4+ T helper cells and CD8+ cytotoxic T-lymphocytes). The two effector T-cells, CD4+ cells and CD8+ cells, function mainly as cytokine producers and cytotoxic killer cells respectively [18]. CD4+ T-cells can be further subclassified as either type 1 (Th1) or type 2 (Th2) helper cells. The Th1 cells secrete interleukin-2 (IL2) and interfreron-γ whereas Th2 cells secrete IL-4, 5, 10 and 13, leading to predominant cellular and humoral immune responses respectively. CD8+ cells ultimately go on to differentiate into cytotoxic T-lymphocytes (CTL); eliminating cells that display foreign antigens (e.g. virally infected cells) displayed by class I MHC molecules. CD8+ cells cause cell death typically through one of two mechanisms: 1) the insertion of perforins into the target-cell membranes which allows for the subsequent passage of granzymes into the target cell, or 2) by binding the target cell's Fas molecule, leading to activation of caspases within the target cell [17]. In addition to their immediate effector functions, both CD4 and CD8 T cells give rise to long-lasting memory T cells, which may persist throughout an individual's entire lifespan.
B-cells are somewhat analogous to T-cells in that the B-cell receptor (BCR), composed of an immunoglobulin light (IgL) and heavy chain (IgH), also undergoes an immune editing process [26]. Whereas T-cells undergo positive and negative selection in the thymus, B-cell immune editing occurs predominately in the bone marrow (i.e. central tolerance). In the case where an auto-antigen activates BCR signaling, that immature B-cell's developmental progression is blocked. Continued BCR editing can, however, rescue that cell and allow for its continued maturation. Those B-cells lacking autoimmunity ultimately go on to migrate from the bone marrow and into circulation. In the case where a circulating B-cell encounters a BCR stimulating auto-antigen, apoptosis of that clone serves to prevent autoimmunity (i.e. peripheral tolerance), with BCR editing generally not observed in the periphery. At the present time, the precise role of B cells in the clinical response to cancer vaccines or immune checkpoint blockade is not completely clear, but recent data show that the PAP-directed vaccine sipulueucel-T does generate a significant antibody response in patients [27].
The Concept of Immunoediting
Conceptually, the role of the immune system plays in controlling cancer can be thought of in three sequential stages: elimination, equilibrium and escape [28] (figure 1). This so-called immunoediting hypothesis has been born out of a number of observations in immune competent and immune deficient mouse tumor models [28,29]. Immunoediting holds that early in oncogenesis, tumors may be recognized by the immune system in a clinically beneficial manner. Thus, early tumors appear to be “eliminated” through a combination of innate and adaptive responses, leading to the destruction of tumor cells. In instances where this elimination process is incomplete, a malignant clone(s) that was not able to be destroyed by the initial immune response can enter an equilibrium phase. This phase can potentially persist for the lifetime of the host, and is thought to rely exclusively on the adaptive immune system. A number of different mechanisms may lead to immune escape, with the cancer ultimately growing into clinically apparent malignancies. These mechanisms of immune escape include: 1) tumor antigen loss, 2) resistance to immune effector cells and 3) emergence of an immunosuppressive microenvironment. Prostate cancer in the clinic has certainly “escaped” immune recognition.
Figure 1.
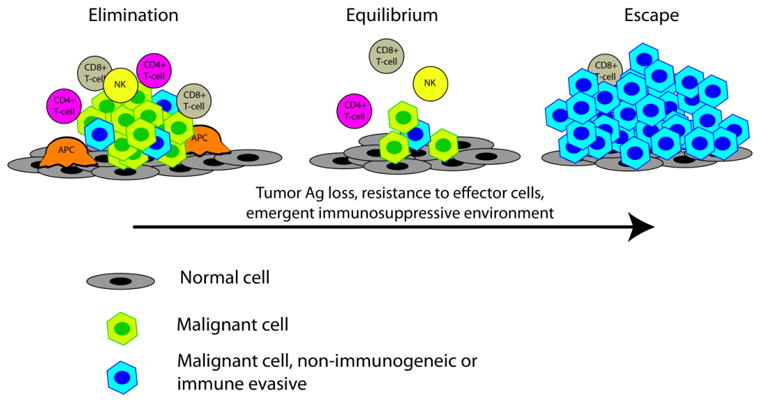
Immune editing hypothesis. Abbreviations: APC, antigen presenting cell; NK, natural killer cell.
In support of this hypothesis, it has been documented in humans that multiple tumor types lead to the induction of tumor-specific T-cells, and in those individuals with increased intra-tumoral infiltration, prolonged survival and/or reduced metastases is often observed [19]. B-cell responses are also likely critical to this immunoediting process. In the case of prostate cancer, it has been shown that a unique antibody “signature” is produced in response to exposure to tumor specific antigens [20,30]. Furthermore, there is some evidence that an enhanced antibody response to tumor antigens, at least in the context of CTLA-4 blockade, may lead to a more robust anti-tumor effect [20].
Androgens and the immune system
Given that the hormonal therapies (HT) (i.e. LHRH agonists/antagonists) have been used to treat prostate cancer for decades, considerable effort has gone into studying their effects. To that end, research has lead to the realization that HTs have several immunomodulatory properties, including: reversing thymic involution and promoting thymopoiesis, augmenting B-cell development, inhibiting tolerance to prostatic antigens and leading to increased prostate immune infiltrates [31,32]. As such, there has been a great deal of interest in using HT and other androgen-directed therapies as a means to augment the host response to cancer vaccines and other immune-based therapies.
While peripheral thymocytes lack AR expression, the AR is expressed on thymic stromal cells, epithelial cells and immature thymocytes; leading to the logical conclusion that androgens exert their effect on T- and B-cells early on in their maturation [33-36]. Congruent with this hypothesis is the observation that the thymus will undergo rapid involution when exposed to rising androgen levels, such as during puberty or when androgens are exogenously administered [37-40]. One proposed mechanism by which this thymic involution occurs in response to androgens is through the androgen stimulated secretion of yet to be determined pro-involution factors by thymic epithelial cells [38,41]. Supporting this hypothesis, experiments have shown that in vitro thymocytes do not undergo apoptosis in response to androgens, whereas in vivo thymocytes do – implying that androgens exert their effect on thymocytes indirectly [42]. Perhaps more pertinent to patients with prostate cancer, the reverse has also been observed, with removal of androgens leading to reversal of thymic regression and subsequent thymopoiesis. This has been seen not only in experimental mouse models, but also in men being treated with HT [40]. The effect of androgens on B-cells has been less well characterized. Analogous to the effect on T-cells, mouse studies have shown that castration can lead to expansion of the immature, pre-B-cell population [43,44]. While other experiments have shown that dihydrotestosterone (DHT) may lead to suppressed B-cell expansion [44]. There is no clear evidence in humans that HT leads to B-cell lymphopoiesis at this time.
Androgens, the immune system and prostate cancer
Given the widespread use of HT in men with prostate cancer, its ability to enhance thymopoiesis and augment immune responses has clear implications relating to immunotherapies. There is some evidence that HT can mitigate immune tolerance to prostate cancer antigens. In a series of mouse studies, it was shown that naïve prostate-specific CD4+ T-cells were mostly ignorant of a model antigen engineered to be expressed in a prostate specific pattern (Pro-HA). When TRAMP mice that develop spontaneous prostate cancer were crossed with mice that express a model antigen in their prostate glands, naïve CD4+ T-cells now recognized the prostate gland. This recognition was found to be tolerogenic; however, leading to abortive proliferation, an absence of effector cytokine production and impaired responsiveness to cancer vaccines. Androgen ablation, however, led to mitigated immune tolerance and the development of effector function in response to vaccination. These experiments imply that the most effective sequence for administering an immunotherapy is likely after androgen ablation [31]. Another set of mouse experiments similarly showed that androgen ablation leads to enhanced antigen specific T-cell proliferation after a CD28 co-stimulatory signal. The same group also reported increased prostate T-cell infiltration in castrated mice – indicating an enhanced restricted T-cell response [45].
In line with the aforementioned mouse studies, Mercader and colleagues showed that HT can lead to a robust restricted T-cell response within prostatectomy samples [32]. When administered prior to a planned prostatectomy, HT led to enhanced T-cell infiltration in both benign and tumor bearing prostate tissue [32]. It was noted that the T-cell infiltrates were predominately CD4+ cells, with CD8+ cells present to a lesser extent. While CD8+ T-cells are generally thought to serve as the dominant effector cell in anti-tumor T-cell responses, CD4+ cells are still thought to be essential in orchestrating a host response to cancer antigens [46-48]. Gannon et al extended these findings, observing that HT did indeed lead to increased immune infiltrates, even when controlling for changes in immune cell density with prostate involution [49] (figure 2). They found that an increased NK cell infiltrate was associated with a lower risk of progression whereas a high density of macrophages related to an increased risk for recurrence. Taken together these studies support a sort of dual role for androgen-ablation in prostate immunotherapy, first in restoring thymic function and normal T cell production and second, in potentially mitigating the immunological tolerance that develops to prostate cancer.
Figure 2.
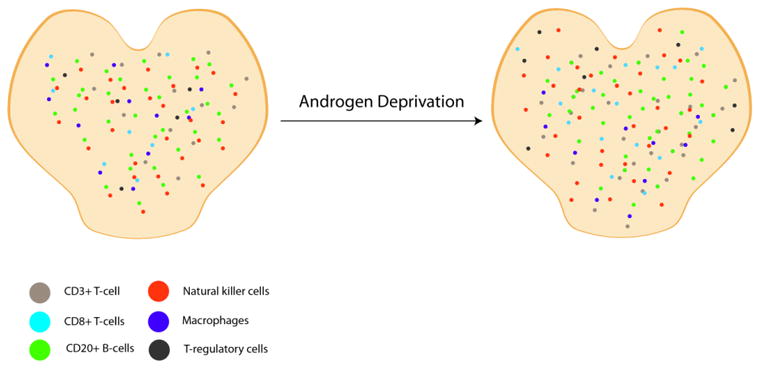
Increase in relative density of immune cells within the prostate following androgen deprivation [49]. After neo-adjuvant androgen deprivation CD3+ T-cells had a 1.87-fold increase in relative density, CD8+ T-cells had approximately 2-fold increase in relative density and macrophages had a 1.78-fold increase in relative density. There was no significant increase in CD20+ B-cells, natural killer cells or T-regulatory cells.
The optimal mode of incorporating HT into immune based treatment strategies has yet to be determined, and to date only a handful of studies have attempted to address this important question. Antonarakis et al, recently reported preliminary results of a randomized trial evaluating sipuleucel-T administered either two weeks before or three weeks after standard HT in men with biochemically recurrent prostate cancer [50]. The primary endpoint of that trial was antigen-specific immune responses to PA2024 (PAP-GM-CSF), the fusion protein used to create sipuleucel-T, as measured through an ELISPOT assay. There were no significant differences in the immune response generated to PA2024 regardless of the sequence of administration; however, there was an augmented cytokine response in the HT followed by sipuleucel-T group. Similarly, that group displayed a higher level of CD8+ T-cell activation compared to the sipuleucel-T first group. These observations led the authors to conclude that HT followed by sipuleucel-T may produce a more robust immunologic response, although long-term data are still pending. Madan et al, reported on a trial evaluating a prostate specific vaccine (vaccinia vector with the PSA gene combined with vaccinia containing the costimulatory molecule B7.1) versus the anti-androgen nilutamide followed by cross over to the other arm at the time of PSA progression. Contradictory to the report by Antonarakis et al, a subgroup analysis of this trial found a non-significant trend toward improved overall survival (OS) in those receiving vaccine alone or vaccine followed by nilutamide as opposed to those receiving nilutamide alone or nilutamide followed by vaccine. At least one set of mouse studies also supports the administration of HT following a cancer vaccine [51]. Suffice it to say, the issue of optimal sequencing of HT with immune based therapies has yet to be resolved. Additional prospective trials, ideally incorporating efficacy outcomes, are needed before any definitive statements regarding how best to incorporate HT with immunotherapies can be made.
Immune checkpoint inhibition
As mentioned above, for individuals who develop clinically apparent tumors, intratumoral T-cell infiltrates can associate with prolonged survival and/or decreased metastatic potential [52]. Unfortunately the control of cancer progression via the immune system is most often transient, largely due to a tumor's ability to produce an immunosuppressive milieu which in turn can lead to a loss of immunologic tumor control. The mechanisms by which this immune escape can occur are varied, and include: 1) upregulation of T-regulatory cells (CD4+CD25+ T-cells) (Tregs) that in turn downregulate effector T-cells and natural killer (NK) cells, 2) induction of myeloid derived suppressor cells (MDSCs) that can suppress T-cell proliferation and induce apoptosis, 3) accumulation of tumor-associated macrophages that can suppress T-cells as well as promote tumor growth and invasion themselves, and 4) enhanced co-inhibitory signaling through the CD28:B7 interactions [18,52,53]. Targeting of this CD28:B7, or immune checkpoint, interaction has garnered great excitement as a means of combating the immunosuppressive effects of tumors. In particular, drugs targeting CTLA-4, PD1 and PD ligand-1 (PD-L1) have experienced a flurry of clinical development over the past several years.
The fully human anti-CTLA-4 monoclonal antibody ipilimumab is the only Food and Drug Administration (FDA) approved immune checkpoint inhibitor. Its approval in advanced, pretreated, melanoma was granted partially on the basis of the landmark trial by Hodi et al which demonstrated a statistically significant increase in median overall survival of approximately 3.5 months compared to those receiving gp100 (a melanoma peptide vaccine) alone [54]. A second phase III trial confirmed a survival benefit in patients with untreated metastatic melanoma given ipilimumab in conjunction with decarbazine versus decarbazine alone [55]. Preclinical data support the use of ipilimumab in prostate cancer, not only as monotherapy, but also as a means to augment the immune response elicited by other therapies (i.e. cancer vaccines, radiation, etc.) [56-58]. To date there are a number of ongoing clinical trials evaluating ipilimumab in men with prostate cancer, however, only a handful have been published. Slovin et al reported the preliminary results of a phase I/II trial evaluating ipilimumab alone or in combination with radiotherapy (given 24-48 hours prior to ipilimumab) in men with metastatic CRPC, either pre- or post-chemotherapy. Ipilimumab was given every 3 weeks at one of 4 dose levels (3, 5 or 10 mg/kg). Thirty-three patients were treated in the dose escalation phase of the trial. Given that no dose limiting toxicities were observed, the 10 mg/kg cohort was expanded to include 50 patients. Overall, the immune-related adverse events (irAEs) were similar to those observed in the melanoma ipilimumab trials, and included: colitis (16%), diarrhea (8%) and hepatitis (10%) – these were generally manageable with immunosuppresion. Eight patients in the 10 mg/kg cohort had a ≥50% decline in PSA, four in the radiation group and four in the monotherapy group. Additionally, in the 28 subjects in the 10 mg/kg cohort with evaluable tumors, one had a complete response and six had stable disease [59]. Based on these results, phase III trials evaluating the combination of ipilimumab with radiation in the post-chemotherapy setting, as well as ipilimumab monotherapy in the pre-chemotherapy setting have completed enrollment. Recently, results of the post-chemotherapy trial were presented [60]. The trial did not reach its primary endpoint of an improved overall survival in the ipilimumab treatment group, but was positive for one of its secondary endpoints, time to progression. A hypothesis-generating, retrospective subgroup analysis suggested that ipilimumab is likely beneficial in mCRPC patients with no visceral metastases and a favorable performance status. Those data likely bode well for the pre-chemotherapy trial, which generally enrolled men with less advanced disease.
Another class of immune checkpoint inhibitor that is being evaluated in ongoing clinical trials targets the PD1:PD-L1 interaction. With the exception of cells of the macrophage lineage, PD-L1 is largely unexpressed on normal human tissue [61]. This is in contrast to human carcinomas (i.e. lung, ovary, colon and melanoma) which have been shown to express high levels of PD-L1, most likely pointing to a common mechanism by which tumors induce T-cell apoptosis and evade targeting for immune clearance [61-63]. Indeed, multivariable analysis from melanoma specimens has confirmed that high expression of PD-L1 is associated with poor survival [64]. Prostate cancer, though, expresses low levels of PD-L1 although the T-cells that infiltrate the gland in men with prostate cancer are nearly universally positive for PD-1 [65,66].
Given the strong preclinical rational for blocking the PD-1/PD-L1 interaction, a number of clinical trials evaluating agents designed to disrupt this interaction are underway. Results from two large phase I trials evaluating monoclonal antibodies designed to block this interaction were recently published [67,68]. A Phase Ib trial evaluated a fully human monoclonal anti-PD-1 antibody in patients with advanced melanoma, non-small-cell lung cancer (NSCLC), CRPC, renal-cell carcinoma (RCC) or colorectal cancer [67]. A total of 296 subjects were enrolled in that trial at doses that ranged from 0.1 to 10 mg/kg given IV every 2 weeks. Among the 236 patients who were response evaluable, objective responses (i.e. complete or partial responses) were observed at a cumulative (i.e. at all dose levels) rate of 18% (14/76) in patients with NSCLC, 28% (26/94) in patients with melanoma and 27% (9/33) in patients with RCC. Twenty of 31 (64.5%) were durable, lasting ≥1 year. A planned cohort of 17 CRPC patients was enrolled and treated, but no objective responses were documented in those patients. Interestingly, of the 42 patients who had immunohistochemical analysis performed on their tumors', 9/25 (36%) with PD-L1-positive tumors had objective responses, whereas none of the 17 patients with PD-L1 negative tumors did. Two subjects with CRPC had their tumors stained for PD-L1 expression, both of which were negative.
A second trial targeted the ligand side of the interaction, evaluating an anti-PD-L1 monoclonal antibody at doses ranging from 0.3 to 10 mg/kg given every two weeks [68]. That trial included patients with advanced NSCLC (n=75), melanoma (n=55), colorectal cancer (n=18), RCC (n=17), ovarian cancer (n=17), pancreatic cancer (n=14), gastric cancer (n=7) and breast cancer (n=4). Objective responses were observed at doses ≥1 mg/kg and the cumulative response rates were 17% (9/52) for patients with melanoma, 10% (5/49) for patients with NSCLC, 6% (1/17) for patients with ovarian cancer and 12% (2/17) for patients with RCC. Immunohistochemistry studies were not reported in this trial. Of note, irAE were reported at rates of 39% and 41% in the PD-L1 (MDX-1105) and PD-1(MDX-1106) trials respectively. These included: rash, diarrhea, hypothyroidism, and hepatitis. Additionally, among patients treated with anti-PD-1, 9 (3%) subjects developed an immune mediated pneumonitis, resulting in the death of 3 (1%) patients. Additional, non-tumor specific, early phase trials, as well as a handful of prostate cancer specific trials designed to further evaluate drugs targeting the PD-1/PD-L1 checkpoint are underway.
It is interesting to note that while CTLA-4 and PD-1 blockade should intuitively produce similar clinical responses, this has not been observed. In the previously mentioned ipilimumab trial with or without radiotherapy in patients with mCRPC, the major PSA response (i.e. ≥50% PSA decline) rate among those receiving the maximum dose of ipilimumab (10 mg/kg) was reported at 8/50 (16%) [59]. This is similar to the cumulative objective response rate of 49/203 (24%) for those with NSCLC, melanoma and RCC treated with PD-1 blockade [67]. In contrast, anti-PD-1 treatment produced no objective responses in the 17 patients with CRPC treated on that same trial. While 17 patients is clearly an insufficient number from which to draw definitive conclusions regarding the efficacy of PD-1 blockade in men with CRPC, it does beg the question as to whether CTLA-4 blockade may be the more relevant checkpoint in men with advanced prostate cancer.
Antigen specific immunotherapies
While a number of passive, i.e. antibody based therapies such as rituximab and trastuzumab have led to improved cancer outcomes, treatment with T-cell based immunotherapies (e.g. cancer vaccines and adoptive transfer of ex vivo activated immune cells) have been less successful overall [69,70]. This trend has begun to change in recent years with an improved understanding of how tumors evade T-cell based immune clearance, and the development of more effective immunotherapies [71]. In general, cancer vaccines work through eliciting a tumor specific antigen response, typically leading to activation of effector T-cells and in turn cancer cell lysis [72]. In many ways prostate cancer is an ideal malignancy in which to utilize cancer vaccine strategies. It represents a typical epithelial adenocarcinoma, with insights into immunotherapy gleaned from its study potentially translatable to other malignancies, and, given that both normal and cancerous prostate cells express a handful of unique antigens [i.e. PSA and prostatic acid phosphatase (PAP)], there are several attractive potential targets for said vaccines. Several different vaccination approaches to treating prostate cancer have been evaluated in both the pre-clinical and clinical settings. These include: dendritic cell-based vaccines, DNA and recombinant protein based vaccines, recombinant viral vector vaccines and whole tumor-cell vaccines [13]. Perhaps, the most well-known of these vaccine strategies is sipuleucel-T. Approved in 2010 for the treatment of asymptomatic or mildly symptomatic CRPC, sipuleucel-T represents the only cancer vaccine ever shown to improve overall survival in the phase III setting [7].
Dendritic cell-based vaccines
Dendritic cells are a the most potent type of antigen presenting cell (APC), and are the only ones capable of eliciting a T-cell response among naïve CD4+ and CD8+ T cells [73-75]. Animal models, and in turn early phase clinical trials across several tumor types, have demonstrated the feasibility of this cancer vaccination approach [75]. Sipuleucel-T, an ex vivo autologous immunotherapy product designed to target PAP, provides proof of concept that dendritic cell-based vaccines can lead to improved patient outcomes [7].
Sipuleucel-T is manufactured by first gathering a patient's peripheral blood leukocytes through a leukapheresis procedure. The cells are then shipped to a central processing facility, where they are gradient-density enriched, and then co-cultured along with a fusion protein consisting of PAP fused to granulocyte-macrophage colony-stimulating factor (GM-CSF) [7,75]. After approximately 36 hours of exposure to the PAP/GM-CSF fusion protein, the pheresis product is shipped back for infusion into the patient. The entire procedure (leukapheresis, manufacture, re-infusion) is repeated three times at two week intervals to make up a standard course of Sipuleucel-T therapy [7]. In the pivotal phase III IMPACT trial, it was shown that treatment with sipuleucel-T leads to prolonged median overall survival compared to those receiving a placebo autologous pheresis product (25.8 months versus 21.7 months; Hazard ratio for death, 0.78; P=0.03). Interestingly, there was no observed differences in progression free survival between the groups.
Sipuleucel-T is thought to stimulate dendritic cells which in turn lead to activation of effector T-cells; producing an anti-tumor effect [76]. To ensure the quality of the immunotherapy product, it does undergo CD54+ testing prior to release. This ensures that the product contains ≥40 million CD54+ dendritic cells, the minimum established quality threshold [7]. The pheresis product is a somewhat heterogeneous mixture, however, containing peripheral blood mononuclear cells (PBMCs), APCs and T-cells [77]. It has been shown that individuals receiving products that contain higher CD54+ cells tend to have prolonged overall survival in comparison to those receiving a product with a lower CD54+ cell count; possibly indicating that sipuleucel-T's main antitumor effect is derived from the infused dendritic cells leading to tumor specific effector T-cell activation.
Given the lack of a difference in progression free survival between those receiving sipuleucel-T versus placebo, the results of the IMPACT trial have been called into question [78]. One issue raised was that in the placebo arm, those younger than 65 years of age had considerably prolonged median survival in comparison to those older than 65 (28.2 vs 17.2 months, respectively), a difference not generally observed in other trials. It has also been pointed out that the placebo >65-years-of-age group had shorter survival compared to other studies with similar enrollment criteria. This difference in survival between different age groups and the decreased survival in those over 65 has led some to conclude that the leukapheresis procedure itself may have led to poorer outcomes in the placebo group. Critics have pointed out that the leukapheresis and processing of the cells in the placebo arm results in <12% of the original pheresed cells being re-infused; which, in theory may lead to increased rates of infection or cancer progression and in turn worse outcomes.
The aforementioned issues with the IMPACT trial do raise some interesting concerns – namely the discrepancies in outcomes in the placebo groups with regard to patient age and when compared to historical control. However, there are some issues with the aforementioned arguments. While leukapheresis may remove a large percentage of circulating PBMCs and T-cells, this population is quickly replenished through homeostatic proliferation and migration of peripheral T-cells [79]. The resultant population of immune cells may in fact be different than the original pheresed population; however, it has been shown in both animals and humans that T-cells responding to homeostatic proliferation tend to be more functional than their nonproliferating counterparts [80]. Additionally, others studies have not confirmed that leukapheresis in of itself leads to poor outcomes [81]. Regardless, in the end enthusiasm for sipuleucel-T may not be quite as vociferous as anticipated upon its FDA approval given the lack of objective tumor responses. In addition, since its initial approval, two novel anti-androgen therapies now compete with sipleucel-T in the mCRPC setting [3].
GVAX is a cell-based vaccine which has gone through phase III testing. It consists of two immortalized prostate cancer cells lines, PC-3 and LNCaP, which have been transduced to express GM-CSF [72,82]. Given that many of the antigens on normal prostatic epithelium are also present on prostate cancer cells, immune tolerance likely occurs. By transducing immortalized cells to express GM-CSF, GVAX was developed in an early-treatment model with the intent of breaking this immune tolerance and potentiating an anti-tumor immune response [83]. In spite of early phase trials showing that GVAX had clinical activity, eliciting an immunologic response in patients with advanced prostate cancer, it failed to demonstrate an overall survival advantage in the metastatic CRPC setting [82,84,85]. An explanation for this lack of a survival response may lie in the fact that immunotherapies in general may work best in the minimally metastatic setting. To further explore the notion that GVAX may be more efficacious in those with lower disease burden, a neoadjuvant trial of GVAX pre-prostatectomy has been launched [clinicaltrials.gov, NCT01696877]. That trial randomizes patients between HT alone or low-dose cyclophosphamide, GVAX and HT. Low-dose cyclophosphamide is given the day prior to the GVAX with the intended purpose of potentiating an anti-tumor immune response through the down regulation of Tregs [86].
Virus based vaccines
Viral based cancer vaccines generally involve the insertion of a plasmid encoding tumor proteins into a viral vector, often a poxvirus (e.g.vaccinia, fowlpox) [87]. Following injection, these most likely then go on to infect epithelial cells in the host, and upon lysis, release encoded antigens that are taken up by APC and in turn activate CD4+ and CD8+ T-cells. More sophisticated viral based cancer vaccines not only utilize plasmids encoding tumor associated antigens, but also co-stimulatory molecules [72, 87]. A major drawback of viral based cancer vaccines is that the antibody response to vector antigens is typically more pronounced than the response to the plasmid encoded tumor antigens – effectively neutralizing the viral based vaccine upon multiple administrations [13,88]. In order to overcome this many immunologic treatment strategies involve an immunologic prime followed by a boost. This immunotherapy sequencing is done to enhance the immunologic response. The heterologous vaccine ProstVac-VF for instance utilizes two viral vectors: a vaccinia virus prime followed by a fowlpox virus boost [Madan et al, 2009].
ProstVac-VF is also known as PSA-TRICOM; it consists of two poxvirus vectors (vaccinia and fowlpox) engineered to express PSA as well as the triad of T-cell co-stimulatory molecules (TRICOM): B7.1, ICAM-1 and LFA-3 [89]. The optimal sequencing of these vectors was explored in the phase II setting [90,91]. That trial showed that a single vaccinia PSA-TRICOM (rV-PSA) prime followed by three fowlpox PSA-TRICOM (rF-PSA) boosts led to a trend towards prolonged progression free survival (biochemical and clinical) compared to other dosing strategies. As such, this sequencing was chosen for further testing. In a larger (n = 125) randomized phase II trial evaluating ProstVac-VF, it was found that it led to prolonged overall survival (median overall survival, 25.1 vs 16.6 months, P=0.0061) and, as was the case with sipuleucel-T, no change in progression free survival was observed [7]. At this juncture, ProstVac-VF is undergoing phase III testing in men either asymptomatic or minimally symptomatic metastatic CRPC [clinicaltrials.gov, NCT01322490].
DNA Vaccines
Another interesting cancer vaccination strategy employs the use of naked plasmid DNA. Plasmid DNA is typically injected either subcutaneously or intramuscularly. The DNA is then taken up by host cells (e.g. myocytes, keratinocyte, APC, etc) and encoded proteins are subsequently expressed [92]. The relative ease by which these DNA molecules can be constructed makes them an attractive means for engineering a therapeutic vaccine. They are generally thought to be less immunogenic than some of the previously discussed vaccination methods; however, repetitive immunizations may be an effective means by which to circumvent a relatively weak initial immune response [93]. Of the prostate cancer DNA vaccines, the PAP-encoding plasmid (pTVG-HP) has undergone the most clinical development. In the phase I setting it was shown to elicit a PAP-specific T-cell response in men with biochemically recurrent prostate cancer [93,94]. Furthermore, in those individuals with a ≥200% increase in PSA doubling time, 6 of 8 also had durable PAP-specific interferon-γ-secreting T-cell response. In contrast, only 1 of 14 patients lacking a PAP-specific interferon-γ-secreting T-cell response had a ≥200% increase in PSA doubling time (P = 0.001). Based on these early signs of clinical and immunogenic efficacy, several phase II trials have been launched (table 1).
Table 1. Select ongoing immunotherapy trials in prostate cancer patients.
| Ongoing checkpoint blockade trials | |||||
|---|---|---|---|---|---|
| Trial registration number | Estimated enrollment (N) | Phase | Patient population | Intervention | Primary endpoint |
| NCT01530984 | 54 | II | Chemotherapy naïve mCRPC | Ipilimumab vs. Ipilimumab + GM-CSF | Proportion of subjects with >30% PSA decline |
| NCT01688492 | 25 | I/II | Chemotherapy naïve mCRPC | Ipilimumab + abiraterone + prednisone | Safety (phase I) and PFS (phase II) |
| NCT01498978 | 30 | II | mCRPC | Ipilimumab + HT | Fraction achieving an undetectable PSA |
| NCT01377389 | 48 | II | Metastatic castrate-sensitive prostate cancer | Ipilimumab + HT | Proportion achieving a PSA <=0.2 ng/mL at 7 months |
| NCT00323882 | 66 | I/II | Chemotherapy naïve mCRPC | Ipilimumab | Safety |
| NCT01194271 | 20 | Iia | Localized prostate cancer | Neoadjuvant ipilimumab + HT followed by radical prostatectomy | Peripheral blood immune response |
| NCT01057810 | 600 | III | Chemotherapy naïve mCRPC | Ipilimumab vs. Placebo | OS |
| Ongoing therapeutic vaccine trials | |||||
| NCT01696877 | 32 | I/II | High-risk men pre-prostatectomy | Neoadjuvant GVAX + cyclophophamide (200 mg/mˆ2) + HT vs. HT | Safety, prostatic CD8+ T-cell infiltrate quantification |
| NCT01322490 | 1200 | III | Chemotherapy naïve mCRPC | ProstVac-VF TRICOM + GM-CSF vs. ProstVac-VF TRICOM + placebo vs placebo + placebo | OS |
| NCT00450463 | 65 | II | non-metastatic CRPC | ProstVac-VF TRICOM + GM-CSF + flutamide vs. flutamide | Time to treatment failure |
| NCT01867333 | 76 | II | mCRPC | ProstVac-VF TRICOM + enzalutamide vs. enzalutamide | Time to progression |
| NCT01145508 | 144 | II | Chemotherapy naïve mCRPC | ProstVac-VF TRICOM + docetaxel + prednisone vs. docetaxel + prednisone | OS |
| NCT01875250 | 38 | II | Non-metastatic castration sensitive prostate cacner | ProstVac-VF TRICOM + enzalutamide vs. enzalutamide | Decrease in tumor re-growth rate |
| NCT00849S121 | 34 | II | Non-metastatic CRPC | pTVG-HP + GM-CSF, multiple schedules being tested | Safety, durable immune response |
| NCT01341652 | 56 | II | Biochemically recurrent without metastasis | PTVG-HP + GM-CSF vs. GM-CSF | MFS |
| Combination therapeutic vaccine/checkpoint blockade trials | |||||
| NCT01420965 | 57 | II | Chemotherapy naïve mCRPC | Sipuleucel-T vs. Sipuleucel-T + CT-011 (anti-PD1 antibody) vs. Sipuleucel-T + CT-011 + cyclophosphamide (125 or 250 mg/mˆ2) | Feasibility and changes in immune response |
| NCT01832870 | 9 | I | Advanced prostate cancer otherwise eligible to receive sipuleucel-T | Sipuleucel-T followed by ipilimumab | Ag specific memory T-cell response, T-cell proliferation to PA2024, PAP and PHA and Ab responses to PA2024 and PAP |
| NCT01804465 | 66 | II | Chemotherapy naïve mCRPC | Sipuleucel-T + immediate vs. delayed ipilimumab (3 weeks post-sipuleucel-T) | Safety, impact of timing on PAP/PA2024 specific immune responses |
| NCT01706458 | 30 | II | CRPC | Sipuleucel-T vs. sipuleucel-T + pTVG-HP | Immune response |
Passive Immunotherapy
Perhaps the greatest advances in the area of immune based cancer therapeutics have been in the development of passive immunotherapies (i.e. monoclonal antibodies). Rituximab, a chimeric monoclonal antibody directed at CD20+, was the first antibody approved for the treatment of any malignancy. Since it approval for relapsed or refractory low grade or follicular non-Hodgkin lymphomas in 1997 it has been followed by several additional antibody therapies, including: Trastuzumab, bevacizumab and cetuximab [69]. This class of drug has varied mechanisms of action [69]. For instance, cetuximab works by inhibiting cellular growth signals (i.e. epidermal growth factor receptor) while bevacizumab inhibits pro-angiogeneic signalling (i.e. vascular endotherlial growth factor) [3,95]. Antibodies can also exert a direct cytotoxic effect. Both rituximab and trastuzumab are thought to primarily exert their antitumor effect through antibody-dependent cell-mediated cytotoxicity (ADCC) in which the antibody's Fc portion interacts with the Fc receptor present on natural killer (NK) cells, macrophages and neutrophils, ultimately leading to the antibody coated cell's death through a number of different mechanisms [96]. Some antibodies that elicit an ADCC effect also activate the complement cascade [69]. Still other antibodies are conjugated to toxins, such as trastuzumab emtansine, acting more as a means to traffic a cytotoxic agent to cancer deposits [97].
In regard to passive immunotherapies for prostate cancer, there have been no major successes thus far. The humanized monoclonal antibody, J591, has gone through perhaps the most clinical testing [98]. The initial pilot study of J591 was geared at determining its utility as an unlabeled monoclonal antibody [99]. While it was found to elicit a dose dependent ADCC effect, it did not produce a robust anti-tumor effect in patients. Its current development has therefore focused more on developing J591 radioimmunotherpay conjugates as well as combining it with other agents with the intent of enhancing its activity. For instance, a phase I study evaluating 177lutetium-labeled J591 (177Lu-J591) has demonstrated enough biologic activity that a number of additional phase I/II studies have been launched [100]. 177Lu-J591 is currently being tried as monotherapy [clinicaltrials.gov, NCT00195039], in combination with docetaxel [clinicaltrials.gov, NCT00916123] and in combination with ketoconazole [clinicaltrials.gov, NCT00859781]. Unconjugated J591 is currently being tested in combination with low-dose interleukin-2 (IL-2) [clinicaltrials.gov, NCT00040586].
Combination approaches
Multiple immunotherapies
Given the varied mechanisms of action of the aforementioned immunotherapies, there is a strong rational for combining different approaches with the goal of augmenting an anti-tumor immune response. Several murine models have demonstrated that combination therapy can lead to an enhanced immunologic anti-tumor response [23,57,101-103]. To date, the primary focus of combinatorial clinical trials has been on evaluating how best to pair checkpoint inhibition, therapeutic vaccines and cytokines with one another. Only a few combination immunotherapy trials in prostate cancer trials have been reported to date. For the most part these have all focused on incorporating ipilimumab alongside with other immuntherapies, such as GM-CSF or GVAX [104-107]. Combining ipilimumab with GM-CSF or GVAX was generally well tolerated, with an acceptable side effect profile. A few patients were reported to have had clinical responses (typically PSA declines), all of whom received at least 3 mg/kg of ipilimumab. None of the aforementioned trials incorporated single agent control arms, so conclusions regarding the additive effect of combining therapies cannot be made. A number of other combinatorial trials are in process, these include Prostvac-VF plus ipilimumab, interleukin 21 (IL-21) in combination with an anti-PD1 antibody, sipuleucel-t in combination with an anti-PD1 antibody plus cyclophosphamide and, as previously mentioned, J591 in combination with IL-2 [clinicaltrials.gov: NCT00113984, NCT01629758, NCT01420965, NCT00040586].
Chemotherapy and immunotherapy
Chemotherapeutics have historically been viewed as immunosuppressive since they can inhibit or kill effector cells, or lead to anergy. However, more recent data show that the effects of chemotherapy on the immune system are more complicated and heterogeneous. Indeed, certain chemotherapy agents may in fact stimulate the immune system through various mechanisms, including: inhibition of cells mediating immune tolerance (e.g. Tregs and myeloid derived suppressor cells), activation of immune effector cells (e.g. NK cells, cytotoxic T-cells, B-cells), and cytotoxicity resulting in the uptake and cross presentation of tumor antigens by APC [108,109]. Depending on the dose and mechanism of the chemotherapeutic in question, the immune response can be quite different, with certain agents (e.g. microtubule inhibitors and topoisomerase inhibitors) leading to more robust APC maturation [110]. This further dissection of the effect of chemotherapy on the immune system has led to the realization that all cancer cell death is not equal, with some modes of cytotoxicity leading to immune cell activation, termed an “immunogenic cell death”, while others allow for immune tolerance to develop [111]. Preclinical work supporting the aforementioned immunologic effects of chemotherapy exists, however, data in humans are still lacking. A number of ongoing clinical trials are currently trying to determine the optimal way to meld chemotherapeutic and immunotherapeutic strategies. Several dedicated reviews on this topic provide a strong conceptual framework for these often overlooked immunologic anticancer mechanisms [108,109].
Given that persistent levels of tumor antigens can lead to T-cell tolerance, it has been postulated that patients with lower tumor burdens would be less tolerized to said tumor antigens and theoretically more susceptible to immune based therapies [112]. Chemotherapy may also therefore serve as a neoadjuvant means of debulking tumors prior to subsequent immunotherapies [113]. Some data supporting the notion that a low tumor burden leads to better immunologic responses comes from a retrospective analysis of a phase II ProstVac VF trial [114]. In that report, patients with longer predicted survival at the time of enrollment (i.e. ≥18 months), and presumably less disease burden, appeared to benefit from the ProstVac VF. Those with shorter predicted survival on the other hand did not demonstrate a clear benefit from this therapeutic vaccine.
Some of the more interesting work being done in combining chemotherapy along with immunotherapy seeks to exploit the immunomodulatory effect produced by certain chemotherapeutics. For instance, a recently opened neoadjuvant trial in men with localized prostate cancer is evaluating the combination of low-dose cyclophosphamide followed by GVAX vaccination then androgen deprivation therapy prior to prostatectomy [clinicaltrials.gov, NCT01696877]. This trial is based on preclinical work showing that low-dose cyclophosphamide can abrogate immune tolerance through enhancement of CD8+ T-cell infiltration into the prostate, depleting Tregs and increasing the expression of dendritic cell maturation markers [109,115]. The primary objective of this trial is to quantify the CD8+ T-cell infiltrate in those receiving GVAX and low-dose cyclophosphamide compared to those only receiving hormonal therapy. Additional human studies are needed to fully understand the implication of incorporating chemotherapy into an immune based treatment strategy.
Radiation and immunotherapy
Somewhat analogous to the immunologic effects of chemotherapy, radiation not only exerts a local cytotoxic effect, but also has the ability to augment a host's anti-cancer immune response. Like chemotherapy, radiation-induced cytotoxicity can lead to an “immunologic cell death” and in turn an immune mediated antitumor effect as well [111]. The most well described immunologic effect of radiation is the abscopal effect in which local radiation can lead to regression of metastatic tumors outside of the radiation field [15]. The abscopal effect was first described in the 1950s [116], and while it was long postulated to be an immune mediated phenomenon, the first direct evidence to implicate the immune system was not reported until 2004 [117]. Demaria and colleagues showed that an abscopal effect could be elicited in mice harboring two 67NR breast cancer cell line tumors when radiation was applied to one of the tumors in conjunction with FLT-3 ligand, a dendritic cell growth factor [117]. Further evidence for an immunologic mediated mechanism lies in the fact that: 1) no tumor regression was observed when FLT-3 ligand was given alone, 2) the effect was tumor specific (i.e. growth of an A20 lymphoma cell line was not affected by irradiating a 67NR tumor in the same animal) and 3) T-cell deficient mice (i.e. nude mice) did not demonstrate an abscopal effect. Generally speaking though, the abscopal effect has been relatively uncommon; likely because of the challenges involved in overcoming tolerance to tumor antigens in cancer patients [111]. Recent trials either completed or underway, are seeking to combine radiation with immune based therapeutics in an attempt to tip the balance towards more robust anti-tumor immune responses [59,118-120]. As previously mentioned one combination regimen that has been explored in the phase I/II setting, and more recently in an ongoing phase III trial, is ipilimumab plus radiation therapy in patients with metastatic CRPC [59,60].
Conclusions
Until recently, immunotherapies had largely not lived up to their promise of providing a robust, clinically relevant, antitumor effect. With the approval of sipuleucel-t, however, this trend has begun to reverse. Since then, a number of additional agents, specifically immune checkpoint inhibitors and newer therapeutic vaccines, have shown early signs of clinical activity. While monotherapy trials continue to be conducted, it seems like the most effective means of inducing a robust antitumor response will be through the combination of immunotherapies with one another, or with other immune augmenting treatments (e.g. HT, chemotherapy or radiation therapy). Indeed, it was recently reported that the combination of PD-1 and CTLA-4 blockade led to a profound anti-melanoma effect with 53% of those enrolled at the maximum tolerated dose having an objective response, all of whom had tumor reductions of ≥80% [121]. Given the marked anti-tumor effect observed on this combination immunotherapy trial, one has to question whether or not the medical community now has a mandate to focus more seriously on combination trials. Furthermore, the low rate of objective responses reported in older immunotherapy trials may no longer be sufficient to drive ongoing clinical development. While it may be the case that less active immunotherapies lead to an initial increase in tumor burden, possibly as a result of a lag between the start of therapy and the development of an adequate immune response versus the development of a tumoral immune-cell infiltrate, as demonstrated with some of the newer highly effective therapies (e.g. checkpoint inhibitors) a rapid observable response is attainable [121,123]. With a higher bar, perhaps the era of relying on so called immune-related response criteria as a clinical trial endpoint may also come to an end [122]. Much work is still needed, but advances over the past few years have given reason for optimism. Lingering issues in need of resolution include: how best to combine treatments, in what disease state (i.e. advanced, adjuvant, neoadjuvant) to test them, how to incorporate said treatments into our current treatment standards (e.g. determining the optimal sequence of immunotherapies with HT) and which therapy is most appropriate for which disease (e.g. apparent efficacy of anti-CTLA-4 therapy in prostate cancer in contrast to the lack thereof with PD-1 blockade). These issues are not insignificant, but with continued work, our current trajectory of progress will surely continue. This renaissance in immune therapy development is reason for excitement - the realization of fully harnessing the immune system to combat cancer is within sight.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;168(1):9–12. doi: 10.1016/s0022-5347(05)64820-3. [DOI] [PubMed] [Google Scholar]
- 3.Schweizer MT, Antonarakis ES. Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: a new era of hormonal therapies is born. Ther Adv Urol. 2012;4(4):167–178. doi: 10.1177/1756287212452196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–1159. doi: 10.1200/jco.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 6.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 7.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 8.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. doi: 10.1016/s0140-6736(10)61389-x. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 12.Parker C, Nilsson S, Heinrich D, Helle SI, O'Sullivan JM, Fossa SD, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213–223. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 13.Aragon-Ching JB, Williams KM, Gulley JL. Impact of androgen-deprivation therapy on the immune system: implications for combination therapy of prostate cancer. Front Biosci. 2007;12:4957–4971. doi: 10.2741/2441. [DOI] [PubMed] [Google Scholar]
- 14.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 15.Drake CG. Radiation-Induced Immune Modulation. In: DeWeese TLL, Laiho M, editors. Molecular Determinants of Radiation Response. Springer; New York: 2011. pp. 251–263. [Google Scholar]
- 16.Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med. 2000;343(1):37–49. doi: 10.1056/nejm200007063430107. [DOI] [PubMed] [Google Scholar]
- 17.Delves PJ, Roitt IM. The immune system. Second of two parts. N Engl J Med. 2000;343(2):108–117. doi: 10.1056/nejm200007133430207. [DOI] [PubMed] [Google Scholar]
- 18.Simeone E, Ascierto PA. Immunomodulating antibodies in the treatment of metastatic melanoma: the experience with anti-CTLA-4, anti-CD137, and anti-PD1. J Immunotoxicol. 2012;9(3):241–247. doi: 10.3109/1547691x.2012.678021. [DOI] [PubMed] [Google Scholar]
- 19.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/s0065-2776(06)90008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwek SS, Dao V, Roy R, Hou Y, Alajajian D, Simko JP, et al. Diversity of antigen-specific responses induced in vivo with CTLA-4 blockade in prostate cancer patients. J Immunol. 2012;189(7):3759–3766. doi: 10.4049/jimmunol.1201529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwek SS, Cha E, Fong L. Unmasking the immune recognition of prostate cancer with CTLA4 blockade. Nat Rev Cancer. 2012;12(4):289–297. doi: 10.1038/nrc3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 23.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116(7):1935–1945. doi: 10.1172/jci27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol Res. 2013;1(1):32–42. doi: 10.1158/2326-6066/CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 25.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol. 2006;6(10):728–740. doi: 10.1038/nri1939. [DOI] [PubMed] [Google Scholar]
- 27.Sheikh NA, Petrylak D, Kantoff PW, Dela Rosa C, Stewart FP, Kuan LY, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother. 2013;62(1):137–147. doi: 10.1007/s00262-012-1317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 29.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Yu J, Sreekumar A, Varambally S, Shen R, Giacherio D, et al. Autoantibody signatures in prostate cancer. N Engl J Med. 2005;353(12):1224–1235. doi: 10.1056/NEJMoa051931. [DOI] [PubMed] [Google Scholar]
- 31.Drake CG, Doody AD, Mihalyo MA, Huang CT, Kelleher E, Ravi S, et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell. 2005;7(3):239–249. doi: 10.1016/j.ccr.2005.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mercader M, Bodner BK, Moser MT, Kwon PS, Park ES, Manecke RG, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci U S A. 2001;98(25):14565–14570. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs WJ, Olsen NJ. Androgen receptors in human thymocytes. J Immunol. 1987;139(2):490–493. [PubMed] [Google Scholar]
- 34.Viselli SM, Olsen NJ, Shults K, Steizer G, Kovacs WJ. Immunochemical and flow cytometric analysis of androgen receptor expression in thymocytes. Mol Cell Endocrinol. 1995;109(1):19–26. doi: 10.1016/0303-7207(95)03479-q. [DOI] [PubMed] [Google Scholar]
- 35.Cohen JH, Danel L, Cordier G, Saez S, Revillard JP. Sex steroid receptors in peripheral T cells: absence of androgen receptors and restriction of estrogen receptors to OKT8-positive cells. J Immunol. 1983;131(6):2767–2771. [PubMed] [Google Scholar]
- 36.Benten WP, Lieberherr M, Giese G, Wrehlke C, Stamm O, Sekeris CE, et al. Functional testosterone receptors in plasma membranes of T cells. FASEB J. 1999;13(1):123–133. doi: 10.1096/fasebj.13.1.123. [DOI] [PubMed] [Google Scholar]
- 37.Pearce P, Khalid BA, Funder JW. Androgens and the thymus. Endocrinology. 1981;109(4):1073–1077. doi: 10.1210/endo-109-4-1073. [DOI] [PubMed] [Google Scholar]
- 38.Kumar N, Shan LX, Hardy MP, Bardin CW, Sundaram K. Mechanism of androgen-induced thymolysis in rats. Endocrinology. 1995;136(11):4887–4893. doi: 10.1210/endo.136.11.7588221. [DOI] [PubMed] [Google Scholar]
- 39.Brelinska R. Thymic epithelial cells in age-dependent involution. Microsc Res Tech. 2003;62(6):488–500. doi: 10.1002/jemt.10410. [DOI] [PubMed] [Google Scholar]
- 40.Sutherland JS, Goldberg GL, Hammett MV, Uldrich AP, Berzins SP, Heng TS, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J Immunol. 2005;175(4):2741–2753. doi: 10.4049/jimmunol.175.4.2741. [DOI] [PubMed] [Google Scholar]
- 41.Olsen NJ, Olson G, Viselli SM, Gu X, Kovacs WJ. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology. 2001;142(3):1278–1283. doi: 10.1210/endo.142.3.8032. [DOI] [PubMed] [Google Scholar]
- 42.Dulos GJ, Bagchus WM. Androgens indirectly accelerate thymocyte apoptosis. Int Immunopharmacol. 2001;1(2):321–328. doi: 10.1016/s1567-5769(00)00029-1. [DOI] [PubMed] [Google Scholar]
- 43.Wilson CA, Mrose SA, Thomas DW. Enhanced production of B lymphocytes after castration. Blood. 1995;85(6):1535–1539. [PubMed] [Google Scholar]
- 44.Viselli SM, Reese KR, Fan J, Kovacs WJ, Olsen NJ. Androgens alter B cell development in normal male mice. Cell Immunol. 1997;182(2):99–104. doi: 10.1006/cimm.1997.1227. [DOI] [PubMed] [Google Scholar]
- 45.Roden AC, Moser MT, Tri SD, Mercader M, Kuntz SM, Dong H, et al. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol. 2004;173(10):6098–6108. doi: 10.4049/jimmunol.173.10.6098. [DOI] [PubMed] [Google Scholar]
- 46.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, et al. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol. 2000;165(11):6047–6055. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 47.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188(12):2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol. 2013;3:63. doi: 10.3389/fonc.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gannon PO, Poisson AO, Delvoye N, Lapointe R, Mes-Masson AM, Saad F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J Immunol Methods. 2009;348(1-2):9–17. doi: 10.1016/j.jim.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Antonarakis ES, Kibel A, Tyler RC, McCoy C, Wang Y, Sheikh NA, et al. Randomized phase II trial evaluating the optimal sequencing of sipuleucel-T and androgen-deprivation therapy (ADT) in patients (pts) with biochemically recurrent prostate cancer (BRPC) [abstract] J Clin Oncol. 2013;31(suppl 6) abstr 34. [Google Scholar]
- 51.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/s0065-2776(06)90008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/s0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 54.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robert C, Thomas L, Bondarenko I, O'Day S, M DJ, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 56.Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94(15):8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hurwitz AA, Foster BA, Kwon ED, Truong T, Choi EM, Greenberg NM, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60(9):2444–2448. [PubMed] [Google Scholar]
- 58.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2 Pt 1):728–734. [PubMed] [Google Scholar]
- 59.Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013 doi: 10.1093/annonc/mdt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerritsen WR, Kwon ED, Fizazi K, Bossi A, Van den Eertwegh A, Logothetis C, et al. CA184-043: A randomized, multicenter, double-blind phase 3 trial comparing overall survival (OS) in patients (pts) with post-docetaxel castration-resistant prostate cancer (CRPC) and bone metastases treated with ipilimumab (ipi) vs placebo (pbo), each following single-dose radiotherapy (RT) [abstract] European Cancer Congress. 2013 abstr 2850. [Google Scholar]
- 61.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 62.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra137. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010;116(7):1757–1766. doi: 10.1002/cncr.24899. [DOI] [PubMed] [Google Scholar]
- 65.Barach YS, Lee JS, Zang X. T cell coinhibition in prostate cancer: new immune evasion pathways and emerging therapeutics. Trends Mol Med. 2010 doi: 10.1016/j.molmed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, Drake CG. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+ Prostate. 2009;69(15):1694–1703. doi: 10.1002/pros.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29(36):4828–4836. doi: 10.1200/jco.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10(8):580–593. doi: 10.1038/nri2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 74.Shurin MR. Dendritic cells presenting tumor antigen. Cancer Immunol Immunother. 1996;43(3):158–164. doi: 10.1007/s002620050317. [DOI] [PubMed] [Google Scholar]
- 75.Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18(23):3894–3903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 76.Madan RA, Gulley JL, Kantoff PW. Demystifying immunotherapy in prostate cancer: understanding current and future treatment strategies. Cancer J. 2013;19(1):50–58. doi: 10.1097/PPO.0b013e31828160a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sheikh NA, Petrylak D, Kantoff PW, Dela Rosa C, Stewart FP, Kuan LY, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother. 2013;62(1):137–147. doi: 10.1007/s00262-012-1317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huber ML, Haynes L, Parker C, Iversen P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104(4):273–279. doi: 10.1093/jnci/djr514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mackall CL, Hakim FT, Gress RE. Restoration of T-cell homeostasis after T-cell depletion. Semin Immunol. 1997;9(6):339–346. doi: 10.1006/smim.1997.0091. [DOI] [PubMed] [Google Scholar]
- 80.Drake CG. Re: interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104(18):1422. doi: 10.1093/jnci/djs340. author reply 1422-1423. [DOI] [PubMed] [Google Scholar]
- 81.Gulley JL, Leitman SF, Dahut W, Schlom J. Re: interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104(14):1106. doi: 10.1093/jnci/djs280. author reply 1109-1112. [DOI] [PubMed] [Google Scholar]
- 82.Simons JW, Carducci MA, Mikhak B, Lim M, Biedrzycki B, Borellini F, et al. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naive prostate cancer. Clin Cancer Res. 2006;12(11 Pt 1):3394–3401. doi: 10.1158/1078-0432.ccr-06-0145. [DOI] [PubMed] [Google Scholar]
- 83.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13(6):1810–1815. doi: 10.1158/1078-0432.ccr-06-2318. [DOI] [PubMed] [Google Scholar]
- 85.Small EJ, Demkow T, Gerritson WR, Rolland F, Hoskin P, Smith DC, et al. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel vs. docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC) [Abstract] American Society of Clinical Oncology Genitourinary Cancers Symposium. 2009 Abstr 9. [Google Scholar]
- 86.Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen HR, Goldberg MV, et al. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res. 2009;69(10):4309–4318. doi: 10.1158/0008-5472.can-08-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arlen PM, Gulley JL, Madan RA, Hodge JW, Schlom J. Preclinical and clinical studies of recombinant poxvirus vaccines for carcinoma therapy. Crit Rev Immunol. 2007;27(5):451–462. doi: 10.1615/critrevimmunol.v27.i5.40. [DOI] [PubMed] [Google Scholar]
- 88.Harrington LE, Most Rv R, Whitton JL, Ahmed R. Recombinant vaccinia virus-induced T-cell immunity: quantitation of the response to the virus vector and the foreign epitope. J Virol. 2002;76(7):3329–3337. doi: 10.1128/JVI.76.7.3329-3337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madan RA, Arlen PM, Mohebtash M, Hodge JW, Gulley JL. Prostvac-VF: a vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs. 2009;18(7):1001–1011. doi: 10.1517/13543780902997928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaufman HL, Wang W, Manola J, DiPaola RS, Ko YJ, Sweeney C, et al. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2004;22(11):2122–2132. doi: 10.1200/jco.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 91.Kaufman HL, Wang W, Manola J, DiPaola RS, Ko YJ, Sweeney CJ, et al. Phase II prime/boost vaccination using poxviruses expressing PSA in hormone-dependent prostate cancer: follow-up clinical results from ECOG 7897 [abstract] J Clin Oncol 2005. 2005;23(16S):4501. [Google Scholar]
- 92.Alam S, McNeel DG. DNA vaccines for the treatment of prostate cancer. Expert Rev Vaccines. 2010;9(7):731–745. doi: 10.1586/erv.10.64. [DOI] [PubMed] [Google Scholar]
- 93.Becker JT, Olson BM, Johnson LE, Davies JG, Dunphy EJ, McNeel DG. DNA vaccine encoding prostatic acid phosphatase (PAP) elicits long-term T-cell responses in patients with recurrent prostate cancer. J Immunother. 2010;33(6):639–647. doi: 10.1097/CJI.0b013e3181dda23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McNeel DG, Dunphy EJ, Davies JG, Frye TP, Johnson LE, Staab MJ, et al. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27(25):4047–4054. doi: 10.1200/jco.2008.19.9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 96.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 97.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Akhtar NH, Pail O, Saran A, Tyrell L, Tagawa ST. Prostate-specific membrane antigen-based therapeutics. Adv Urol. 2012;2012:973820. doi: 10.1155/2012/973820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morris MJ, Divgi CR, Pandit-Taskar N, Batraki M, Warren N, Nacca A, et al. Pilot trial of unlabeled and indium-111-labeled anti-prostate-specific membrane antigen antibody J591 for castrate metastatic prostate cancer. Clin Cancer Res. 2005;11(20):7454–7461. doi: 10.1158/1078-0432.ccr-05-0826. [DOI] [PubMed] [Google Scholar]
- 100.Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Oncol. 2005;23(21):4591–4601. doi: 10.1200/jco.2005.05.160. [DOI] [PubMed] [Google Scholar]
- 101.Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95(17):10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi: 10.1158/0008-5472.can-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fong L, Kwek SS, O'Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69(2):609–615. doi: 10.1158/0008-5472.can-08-3529. [DOI] [PubMed] [Google Scholar]
- 105.van den Eertwegh AJ, Versluis J, van den Berg HP, Santegoets SJ, van Moorselaar RJ, van der Sluis TM, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):509–517. doi: 10.1016/s1470-2045(12)70007-4. [DOI] [PubMed] [Google Scholar]
- 106.Harzstark AL, Fong L, Weinberg VK, Ryan CJ, Lin AM, Sun J, et al. Final results of a phase I study of CTLA-4 blockade in combination with GM-CSF for metastatic castration resistant prostate cancer (mCRPC) J Clin Oncol. 2010;28:15s. Abstract. 2010 (suppl; abstr 4689) [Google Scholar]
- 107.Gerritsen WR, van den Eertwegh AJ, de Gruijl TD, Giaccone G, Scheper RJ, Sacks N, et al. Biochemical and immunologic correlates of clinical response in a combination trial of the GM-CSF-gene transduced allogeneic prostate cancer immunotherapy and ipilimumab in patients with metastatic hormone-refractory prostate cancer (mHRPC) [Abstract] J Clin Oncol. 2007;25:18s. suppl; abstr 5120. [Google Scholar]
- 108.Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118(6):1991–2001. doi: 10.1172/jci35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22(3):113–124. doi: 10.1016/j.smim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 110.Tanaka H, Matsushima H, Mizumoto N, Takashima A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009;69(17):6978–6986. doi: 10.1158/0008-5472.can-09-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265. doi: 10.1093/jnci/djs629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.den Boer AT, van Mierlo GJ, Fransen MF, Melief CJ, Offringa R, Toes RE. The tumoricidal activity of memory CD8+ T cells is hampered by persistent systemic antigen, but full functional capacity is regained in an antigen-free environment. J Immunol. 2004;172(10):6074–6079. doi: 10.4049/jimmunol.172.10.6074. [DOI] [PubMed] [Google Scholar]
- 113.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3(8):630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 114.Gulley JL, Arlen PM, Madan RA, Tsang KY, Pazdur MP, Skarupa L, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59(5):663–674. doi: 10.1007/s00262-009-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Motoyoshi Y, Kaminoda K, Saitoh O, Hamasaki K, Nakao K, Ishii N, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep. 2006;16(1):141–146. [PubMed] [Google Scholar]
- 116.Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26(305):234–241. doi: 10.1259/0007-1285-26-305-234. [DOI] [PubMed] [Google Scholar]
- 117.Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–870. doi: 10.1016/j.ijrobp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 118.Chi KH, Liu SJ, Li CP, Kuo HP, Wang YS, Chao Y, et al. Combination of conformal radiotherapy and intratumoral injection of adoptive dendritic cell immunotherapy in refractory hepatoma. J Immunother. 2005;28(2):129–135. doi: 10.1097/01.cji.0000154248.74383.5e. [DOI] [PubMed] [Google Scholar]
- 119.Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys. 2012;82(2):924–932. doi: 10.1016/j.ijrobp.2010.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28(28):4324–4332. doi: 10.1200/jco.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N Engl J Med. 2013 doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. doi: 10.1158/1078-0432.ccr-09-1624. [DOI] [PubMed] [Google Scholar]
- 123.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma. N Engl J Med. 2013 doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]