

Abstract
Sox9 has gained increasing importance both functionally and as a prognostic factor in cancer. We demonstrate a functional role for Sox9 in inducing a mesenchymal phenotype in lung ADC. We show that Sox9 mRNA and protein are overexpressed in lung ADC, particularly those with KRAS mutations. Sox9 expression correlated with the Notch target gene Hes1, and numerous other Notch pathway components. We observed that Sox9 is a potent inducer of lung cancer cell motility and invasion, and a negative regulator of E-cadherin, a key protein that is lost during epithelial-mesenchymal transition (EMT). Moreover, we show that Notch1 signaling directly regulates Sox9 expression through a SOX9 promoter binding site, independently of the TGF-β pathway, and that Sox9 participates in Notch-1 induced cell motility, cell invasion, and loss of E-cadherin expression. Together, the results identify a new functional role for a Notch1-Sox9 signaling axis in lung ADC that may explain the correlation of Sox9 with tumor progression, higher tumor grade, and poor lung cancer survival. In addition to Notch and TGF-β, Sox9 also acts downstream of NF-κB and Wnt/β-catenin signaling. Thus, Sox9 could potentially act as a hub to mediate cross-talk among key oncogenic pathways in lung ADC. Targeting Sox9 expression or transcriptional activity could potentially reduce resistance to targeted therapy for lung ADC caused by pathway redundancy.
Keywords: Sox9, Notch1, TGF-β, lung cancer, EMT
INTRODUCTION
Sox9 is a member of the high-mobility group-box class DNA-binding protein family of transcription factors and plays a critical role in embryonic development, cell fate determination and lineage commitment. Specifically, Sox9 orchestrates chondrogenesis, bone formation and testis development, among others [1,2]. Germline SOX9 mutations cause campomelic dysplasia, a disorder characterized by numerous skeletal abnormalities and XY sex reversal [3]. Infants with mutant SOX9 usually die due to respiratory distress shortly after birth [4]. This clinical finding likely reflects the role of Sox9 in the developing lung. Sox9 is highly expressed in the developing lung, and is required for branching morphogenesis in the lungs and for controlling alveolar epithelial progenitor cell proliferation [5-6].
Overexpression of Sox9 is found in more than 50% of lung adenocarcinomas (ADCs), the most common histological lung cancer subtype, and is associated with a poor lung ADC survival [8]. Over the past 5 years, it has become increasingly clear that Sox9 plays a major role across numerous human cancers. Sox9 overexpression acts as an oncogene in several cancer types, including breast, colorectal, pancreatic, and prostate cancer and glioma [9-13], although it appears to act as a tumor suppressor in bladder cancer and melanoma [14,15]. In lung cancer, Sox9 overexpression was recently shown to increase cell proliferation and xenograft tumor formation [8,16]. However, the functional role of Sox9 in lung cancer has not been fully elucidated. Furthermore, the upstream oncogenic molecular pathways regulating Sox9 overexpression in lung ADC have not been completely delineated.
The Notch signaling pathway regulates cell fate decisions in nearly every developing tissue and organ, and maintains adult tissue homeostasis. In humans, the Notch receptor is activated after binding to one of 5 Delta or Jagged ligands that are expressed on neighboring cells. Ligand binding initiates a series of cleavage events, and the final cleavage is carried out by the γ-secretase protease complex which frees the intracellular domain portion of Notch (ICD), allowing Notch ICD to translocate to the nucleus. Nuclear Notch ICD complexes with the transcriptional repressor RBP-Jκ, converting it into a transcriptional activator [17]. Notch ICD-induced effects on gene expression are tightly controlled by numerous post-translational modifications and protein co-factors. Several Notch target genes are nearly universally activated across many cell types and developmental processes, such as Hes and Hey, while many other Notch target genes are highly cell-type and context specific [18,19].
Notch1 is a putative oncogene that is aberrantly over-activated in over one-third of lung ADCs through gain-of-function mutations (10%) or altered expression of Notch receptors and/or suppressors (30%) [20]. Notch1 is required for tumor initiation in the KRAS-driven lung ADC mouse model [21]. Moreover, overexpression of constitutively activated Notch1 directly participates in lung carcinogenesis; it induces lung adenomas in mice which progress to ADCs in cooperation with Myc overexpression [22]. Aberrant Notch1 signaling contributes to acquired resistance to EGFR inhibitors, the most widely used targeted therapy for NSCLC [23]. Functionally, Notch increases lung cancer stem cell (CSC) self-renewal and promotes epithelial-mesenchymal transition (EMT) [23,24], reviewed in [25], although the downstream mechanisms mediating these effects have not been fully defined. Few studies have identified specific target genes of oncogenic Notch1 in lung ADC, but those validated thus far include Survivin and IGF1R [26,27]. Because of the exquisite specificity of cell-type and context specific effects of Notch signaling, it is necessary to explore Notch target genes in lung cancer in order to elucidate the genes mediating Notch-induced EMT within this disease.
Sox9 is a well-studied downstream effector of the Notch pathway in murine tissues, especially during development [28-34], through RBP-Jκ binding sites located at positions +40 bp and −325 bp relative to the SOX9 transcriptional start site [35]. However, these binding sites are not conserved in humans. Sox9 expression correlates with Notch signaling in humans [36], but there has been a lack of direct evidence that Notch-induced modulation of Sox9 expression is evolutionarily conserved in humans or that Sox9 is a Notch target in lung cancer. Loss of the TGF-β adaptor β2SP induces TGF-β signaling, resulting in both Notch activation and SOX9 expression in esophageal ADC [37]; although, the specific role of Notch in regulating Sox9 expression was not fully delineated. In this study, we show that the Notch1-Sox9 signaling axis is evolutionarily conserved through a novel RBP-Jκ binding site at -10 bp relative to the SOX9 promoter and that Sox9 expression is regulated by Notch1 in lung ADC independent of TGF-β signaling. Further, we demonstrate that Sox9 mediates Notch1-induced cell motility, invasion, morphological changes, and suppression of E-cadherin, all features of a mesenchymal phenotype.
RESULTS
Sox9 expression is elevated in KRAS-mutant lung ADC
An analysis of publicly available datasets [38-47] on Oncomine (www.oncomine.com) revealed an upregulation of Sox9 mRNA in the majority of lung ADC datasets. Out of 10 available datasets comprising a total of 1,021 samples, 8 datasets with 762 samples showed statistically significant upregulation of Sox9 in lung ADC tumor tissue in comparison to normal adjacent tissue (Fig. 1A). We screened a panel of 14 lung ADC cell lines, and observed detectable levels of Sox9 protein in 10 cell lines. Low Sox9 expression was found in immortalized bronchial epithelial Beas2b cells (Supplementary Fig. S1). We further investigated Sox9 expression at the protein level in 50 human lung ADC surgical samples and paired NATs. Sox9 protein levels were significantly higher in tumors than in NAT (Fig. 1B). Sox9 protein was predominantly nuclear and overexpressed in 70% of human ADCs. In contrast, normal adjacent lung tissue was scored negative, though vascular endothelial cells showed weak positive nuclear and cytoplasmic staining (Fig. 1B), and, when present, bronchial epithelial cells showed moderate cytoplasmic staining with focal nuclear staining.
Figure 1. Sox9 expression in lung adenocarcinoma.
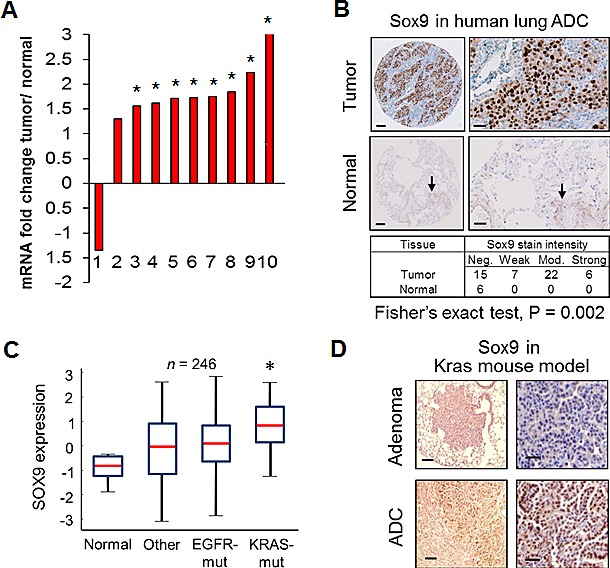
(A) Fold change in log2 median-centered intensity of mRNA expression levels in lung ADC samples, as compared to normal lung tissue samples, in publicly available datasets. Fold change in tumors is relative to 1 for normal tissues; *, P < 0.05. (B) Surgical lung ADC samples were immunohistochemically stained for Sox9. Representative staining is shown in human lung ADC and normal adjacent tissue samples. Quantified scores of staining intensity are shown. Arrows indicate light staining in the vascular endothelium. Scale bar for left panel, 100 μm and for right panel, 25 μm. (C) Normalized SOX9 mRNA expression levels from human lung ADC samples categorized by genetic subgroups were compared to their respective sets of normal adjacent tissues; other, wildtype for both KRAS and EGFR; *, P < 0.05. (D) Representative immunohistochemical staining (N = 4 mice) of lung tissue sections for Sox9 is shown from the LSL-K-rasG12D lung ADC mouse model. Scale bar for left panel, 200 μm and for right panel, 50 μm.
To further investigate Sox9 overexpression in lung ADC, we queried data with annotated mutational information and analyzed Sox9 expression within lung cancer genetic subgroups, including EGFR and KRAS mutations. Sox9 mRNA was significantly higher in lung ADCs with KRAS mutations compared to NAT. EGFR mutant tumors had a trend of higher Sox9 expression than in NAT, but it was not statistically significant (Fig. 1C). These data are consistent with the report that Sox9 overexpression is required for oncogenic Kras-induced pancreatic intraepithelial neoplasia [48]. To investigate if high Sox9 expression is conserved in a GEMM of lung ADC, we stained lung adenoma and ADC sections from the LSL-K-rasG12D lung ADC mouse model for Sox9 and observed Sox9 protein expression to be mostly absent from normal lung and adenoma tissue, but highly expressed in ADC tumors (Fig. 1D). Expression was not uniform in the tumors and was restricted to the more malignant-appearing outgrowths.
Sox9 is a Notch-responsive gene during murine lung carcinogenesis
Notch1 was recently shown to be required for the LSL-K-rasG12D lung ADC GEMM [21]. We analyzed if Sox9 is a downstream effector of Notch1 during lung carcinogenesis. Notch1 overexpression in the mouse lung alveolar epithelium, driven by the Clara cell specific protein (CCSP) promoter, leads to pulmonary adenomas that progress to ADCs when crossed with conditional Myc overexpression [22]. We tested these conditional GEMM of lung ADC to determine if Sox9 expression is an early event downstream of Notch1 signaling (Fig. 2A). As previously reported, within 14 days of doxycycline induced N1ICD expression, hyperplastic regions were clearly discernable in stained lung sections (Fig. 2B) [22]. These hyperplastic regions progressed to adenomas with papillary histology in doxycycline-fed Notch1 mice. Immunohistochemical staining for Notch1 demonstrated high expression levels in the hyperplastic and adenomatous regions. Similarly, Sox9 protein was substantially upregulated in the hyperplastic and adenomatous regions (Fig. 2B). Moreover, multifocal ADCs developed in mice conditionally expressing both N1ICD and Myc driven by the CCSP promoter when induced by doxycycline. Notch protein was highly expressed in this model, and Sox9 displayed a similar pattern (Fig. 2B). To test if enhanced Sox9 transcription was responsible for the increased Sox9 protein levels in these mouse models, we assessed Sox9 mRNA expression and found an 18-fold increase in lung tissue as early as 7 days after Notch1 induction (Fig. 2C). Levels continued to increase over time through to adenoma formation, and also in the ADC samples from the Notch1-Myc mice (Fig. 2C). These data indicate that Sox9 is responsive to Notch signaling as an early event during Notch1-induced lung carcinogenesis in vivo.
Figure 2. Sox9 is downstream of Notch1 during murine carcinogenesis and correlates with the Notch pathway.
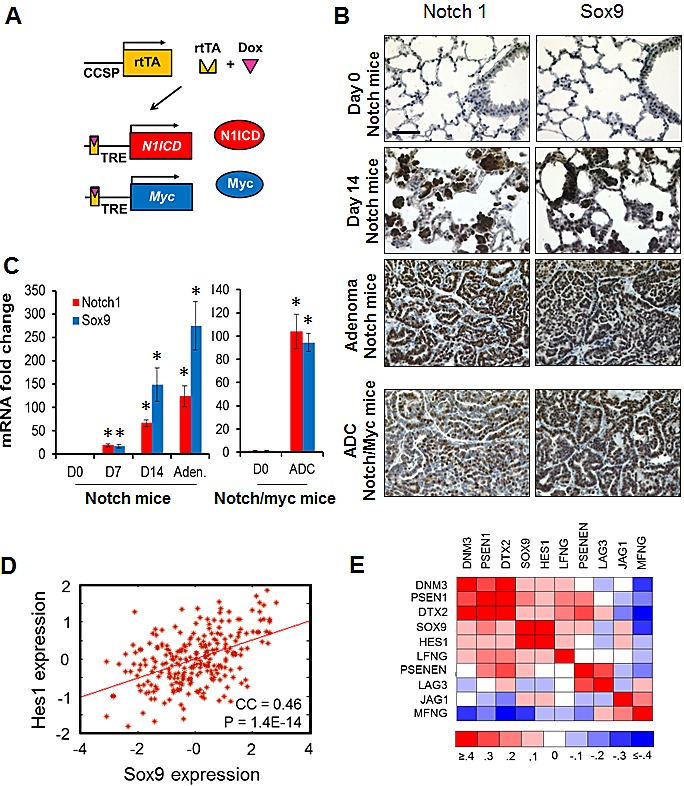
(A) Schematic depicting how Myc and Notch1 transgenes were conditionally expressed in lung epithelial cells with doxycycline (Dox) treatment. (B) Lung tissue sections from the Notch1 (Day 0, Day 14 and adenoma) or Notch1/Myc (Adenocarcinoma) lung cancer mouse models were immunohistochemically stained for Sox9 and Notch1 expression. Scale bar, 50 μm; all pictures are the same magnification. (C) SOX9 mRNA levels were assayed in a set of mouse lung tissues from the indicated conditions (N=3 mice each). Mean fold changes with standard deviations, compared to no doxycycline treatment, are shown; *, P < 0.05. (D) Linear regression of normalized SOX9 and HES1 mRNA levels [41]. (E) Heat map of Pearson correlation coefficients of mRNA gene expression across Sox9 and genes that modulate the Notch pathway. Raw data and P values are detailed in Supplementary Table 1.
To determine if Sox9 expression correlates with Notch signaling in human lung ADC, we performed gene co-expression analyses across several datasets. There was a signficant correlation between Sox9 and Hes1 expression, a common Notch-target gene, in 3 of 3 examined studies (Fig. 2D and Supplementary Fig. S2). Both Hes1 and Sox9 positively correlated with the catalytic enzyme that cleaves Notch PS
EN1, the Notch ligand JAG1, and positive modulators of Notch signaling, DTX2 and LFNG. Hes1 and Sox9 both negatively correlated with MFNG, which inhibits ligand-dependent Notch activation (Fig. 2E and Supplementary Table S1).
Sox9 is responsive to Notch1 signaling in human lung adenocarcinoma
Because Sox9 was a reported target gene of the Notch pathway during murine development and we found a significant correlation between the Notch pathway and Sox9 expression at the mRNA level across microarray datasets of lung ADC samples, we set out to determine if Sox9 is a key gene downstream of Notch in lung ADC. We treated lung ADC cells with EDTA, known to induce Notch activation by chelating calcium [49]. EDTA-induced Notch activation consistently upregulated expression of Hes1 at least 5-fold at both the RNA and protein levels (Fig. 3A and Supplementary Fig. S3). We observed dose-dependent inhibition of Notch by the gamma-secretase inhibitor (GSI) RO4929097 (Supplementary Fig. S4). The GSI substantially blocked EDTA-induced Hes1 expression (Fig. 3A, left panel). Similarly, Sox9 expression was significantly increased after EDTA-induced Notch activation in 3 of 4 lung ADC cell lines examined, and Sox9 upregulation was significantly eliminated in the presence of the GSI (Fig. 3A, right panel), suggesting that Sox9 is highly responsive to Notch signaling in lung ADC. The intensities of upregulated Sox9 and Hes1 expression induced by Notch signaling were different, as is typical of Notch-target genes [28,50].
Figure 3. Sox9 is downstream of Notch signaling in human lung cancer.
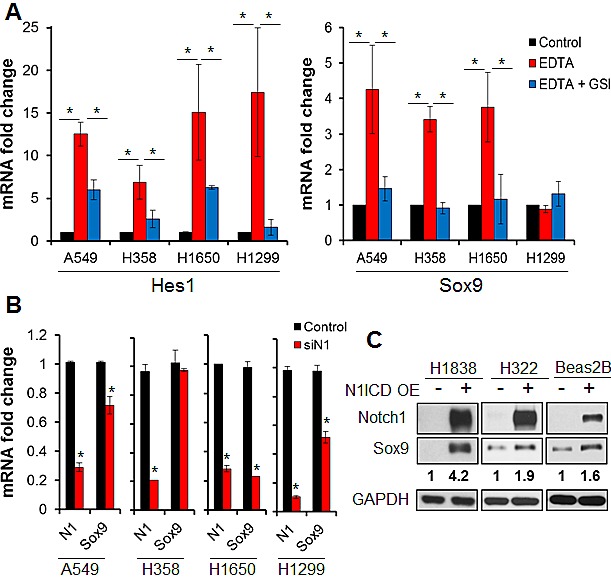
(A) Mean mRNA levels and standard deviations of HES1 and SOX9 assessed by quantitative RT-PCR are shown in untreated lung ADC cells (Control), after EDTA-induced Notch activation (EDTA), and after Notch activation was blocked by a gamma secretase inhibitor (EDTA + GSI); *, P < 0.05. (B) mean mRNA fold change and standard deviations of Notch1 and Sox9 are shown in lung adenocarcinoma cells 72 hours after transient transfection with siRNA against Notch1 as compared to transfection with a scrambled siRNA (Control); *, P < 0.05. (C) Expression of Notch1 and Sox9 were analyzed by western blot in lung ADC and immortalized bronchial epithelial cells 48 hours after transfection with vector or Notch1 intracellular domain. Quantification of Sox9 by densitometry, normalized to GAPDH protein levels, is shown.
We next set out to determine if Sox9 is downstream of Notch1 or Notch3, the two Notch receptors implicated in lung ADC [20,24,26]. We knocked down Notch1 expression in H1299 cells, which had high basal Sox9 protein levels among the screened cell lines, and Sox9 mRNA and protein expression were repressed (Fig. 3B, Supplementary Fig. S5A). Similar results were observed in A549 and H1650 cells, but not in H358 cells (Fig. 3B), suggesting that other upstream pathways might also drive Sox9 expression in lung cancer. Conversely, knockdown of Notch3 failed to decrease Sox9 expression in any of the 4 cell lines tested. However, in two cell lines, Notch3 silencing increased Sox9 expression, concomitant with an increase in Notch1 mRNA caused by Notch3 knockdown (Supplementary Fig. S5B and C). This was likely due to a compensatory increase in Notch1 secondary to decreased Notch3 signaling, a known Notch1 antagonist [51]. Overexpression of the active form of Notch1 (N1ICD) induced Sox9 expression in lung ADC cell lines and immortalized bronchial epithelial cells that had low basal Sox9 levels (Fig. 3C). These data confirmed that Notch1 signaling influences Sox9 expression in human lung ADC.
Notch1 directly regulates Sox9 gene expression
Recently, two RBP-Jκ binding sites in the murine SOX9 promoter were identified at approximately −325 bp (S2) and +40 bp (S5) relative to the transcriptional start site (Fig. 4A) [35,52]. Alignment of the mouse and human Sox9 promoters revealed that these mouse RBP-Jκ binding sites are not conserved in humans (Fig. 4A). We queried −950 to +50 bp relative to the human transcriptional start site using the LASAGNA algorithm [53] and identified two novel RBP-Jκ binding motifs with potential high affinity located at positions −81 bp (S3) and −10 bp (S4) (Fig. 4A). To test whether direct binding of N1ICD to these RBP-Jκ binding sites in the human Sox9 promoter is necessary for induction of Sox9 expression in human NSCLC, we used a luciferase reporter under the control of a 1 kb-fragment of the Sox9 promoter, transiently expressed in the Sox9-low H1838 cells. Activity of the wild-type reporter was induced 4.5-fold, when co-transfected with N1ICD (Fig. 4C). We mutated the four putative RBP-Jκ binding sites, as well as one additional site (Site 1) proposed by Song et al. [37] to cooperate with the Smad3 SBE (Smad binding element) upstream of Sox9 in esophageal adenocarcinoma (Fig. 4B). Only mutation of Site 4, that we identified by LASAGNA, abolished activation of the luciferase reporter activity by N1ICD, showing that this −10 bp site is, indeed, a functional site responsible for the induction of Sox9 by Notch1 in human lung ADC (Fig. 4C). Consistent with these data, we performed ChIP-QPCR assays on H1838 cells transiently transfected with N1ICD, using primers designed to amplify Site 4, and found that N1ICD physically associated with the RBP-Jκ binding site in the human Sox9 promoter at or near the −10 bp site (Fig. 4D and E). Taken together, these results establish Sox9 as a direct transcriptional Notch1 target in human lung ADC.
Figure 4. Sox9 is a Notch1 target gene in lung adenocarcinoma.
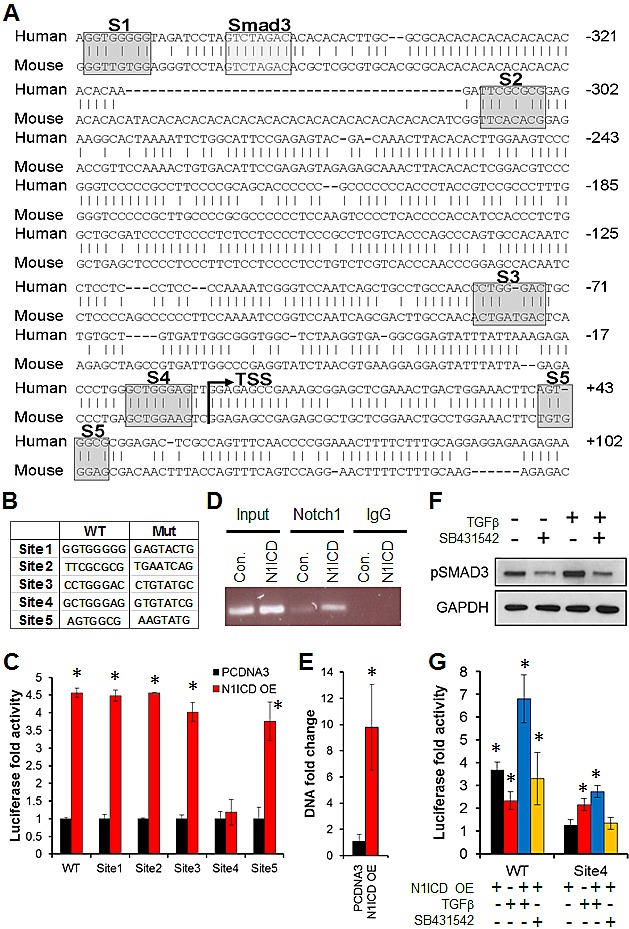
(A) Alignment of the human and mouse SOX9 promoter sequences is shown, with indicated putative RBP-Jκ binding Sites 1 - 5 and Smad3 binding element, relative to the SOX9 transcriptional start site (TSS). Sites 1, 2, and 5 were previously reported (See text), and we identified Sites 3 and 4 through LASAGNA, (B) Wild-type and mutated primer sequences used for site-directed mutagenesis of Sites 1 - 5 are shown. (C) Normalized luciferase fold activity (to Renilla luciferase) and standard deviations in cells co-transfected with Notch1 intracellular domain (N1ICD OE) and a luciferase reporter driven by a 1 kb fragment of the wild-type or mutated SOX9 promoter in sites 1 – 5, compared to the vector control (PCDNA3); *, P < 0.05. (D) ChIP was performed using either a Notch1 or IgG control antibody on cells transfected with N1ICD or a control vector (PCDNA3), and the SOX9 promoter was amplified by PCR, using primers that were designed to amplify Site 4. (E) Quantitative PCR was performed after ChIP; *, P < 0.05. (F) pSmad3 was analyzed by western blot after treatment with vehicle controls, recombinant human TGF-β or the TGF-β inhibitor, SB431542. (G) Normalized luciferase fold activity (to Renilla luciferase) is shown in cells transfected with N1ICD and a luciferase reporter driven by a 1 kb fragment of the wild-type or mutated SOX9 promoter at Site 4, and treatment with TGF-β or SB431542, each compared to its empty vector control, which was set to 1 (Not shown); *, P < 0.05.
Because Smad3 was recently shown to interact with Notch1 in esophageal adenocarcinoma cells after treatment with TGF-β, we tested if N1ICD binding to the Sox9 promoter RBP-Jκ site was dependent on TGF-β signaling in lung ADC. TGF-β increased Smad3 phosphorylation (Fig. 4F) and significantly induced Sox9 reporter luciferase activity in both the wild-type and mutated Site 4 promoters by 2-fold (Fig. 4G). Combined TGF-β treatment with transient overexpression of N1ICD resulted in an additive increase in luciferase activity, compared to either one individually, indicating that Sox9-promoter driven gene expression does not require cooperativity of N1ICD and TGF-β in lung ADC. Further, TGF-β treatment induced an additive increase in the basal luciferase activity even with a mutation at Site 4Inhibition of TGF-β signaling by SB431542 abolished TGF-β-induced Smad3 phosphorylation (Fig. 4F) but had no effect on N1ICD-induced luciferase activity (Fig. 4G). These data confirm that Notch1 and TGF-β induce Sox9 expression independently in lung ADC.
Sox9 mediates Notch1-induced cell motility and suppression of epithelial-like cellular morphology
To determine if Sox9 affects cell motility, we carried out in vitro wound-healing assays with H1299 and A549 cells. Knockdown of either Notch1 or Sox9 significantly reduced migration, as compared to cells transfected with a scrambled siRNA sequence. Overexpression of Sox9 rescued the reduced cell motility caused by loss of Notch1 signaling, in both H1299 (Fig. 5A and B) and A549 cells (Supplementary Fig. S6A). We then tested if silencing Sox9 could block Notch-enhanced migration. Transient overexpression of activated Notch1 increased A549 cell motility, though not significantly. However, silencing of Sox9 expression reversed the increased cell motility caused by activated Notch1, down to similar levels seen with silenced Notch1 (Supplementary Fig. S6B). These data demonstrate that Sox9 at least partly mediates Notch1-induced cell migration.
Figure 5: Sox9 mediates Notch1-induced cell migration and morphological changes.
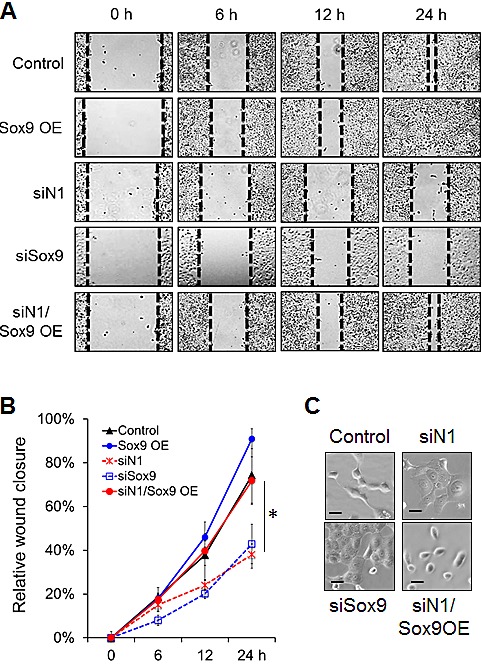
(A) Migration ability of lung adenocarcinoma cells was examined with a wound healing assay starting at 48 hours after transient knockdown of Notch1 or Sox9, or overexpression (OE) of Sox9. The control sample was transfected with both a scrambled siRNA sequence and empty vector. Representative images are shown at 0, 6, 12 and 24 hours. Black dotted lines indicate the wound edge closure of monolayer cells. (B) Mean migration distance of the wound edge, and standard error of the mean, is shown in all treatment compared to cells transfected with both a scrambled siRNA sequence and empty vector (Control); *, P < 0.05. (C) Representative brightfield images of lung ADC cells 72 hours after knockdown of Notch1 or Sox9, or Sox9 overexpression, compared to cells transfected with both a scrambled siRNA sequence and empty vector (Control). Scale bar, 10μm.
H1299 cells are characterized by a mesenchymal gene expression signature and spindle-shaped morphology with few cell-cell contacts. Compared with control cells, H1299 cells with knocked down Notch1 or Sox9 expression underwent profound morphologic changes, which included a larger, flattened phenotype and tighter, more numerous cell-cell contacts. When Sox9 was overexpressed in cells with knocked down Notch1, the mesenchymal appearance of the cells was rescued, with characteristic spindle-like shape and loss of tight cell-cell contacts (Fig. 5C).
Sox9 mediates Notch1-induced cell invasion and loss of epithelial marker expression
Because our data suggested that Sox9 mediates the adoption of a mesenchymal phenotype induced by Notch1, we analyzed cell invasion, a hallmark of mesenchymal cells. Transient Notch1 or Sox9 knockdown significantly reduced invasion of H1299 cells through Matrigel-coated Transwell inserts, and transient overexpression of Sox9 rescued the cells' invasive ability (Fig. 6A and B).
Figure 6. Sox9 mediates Notch1-induced cell invasion and expression of E-cadherin.
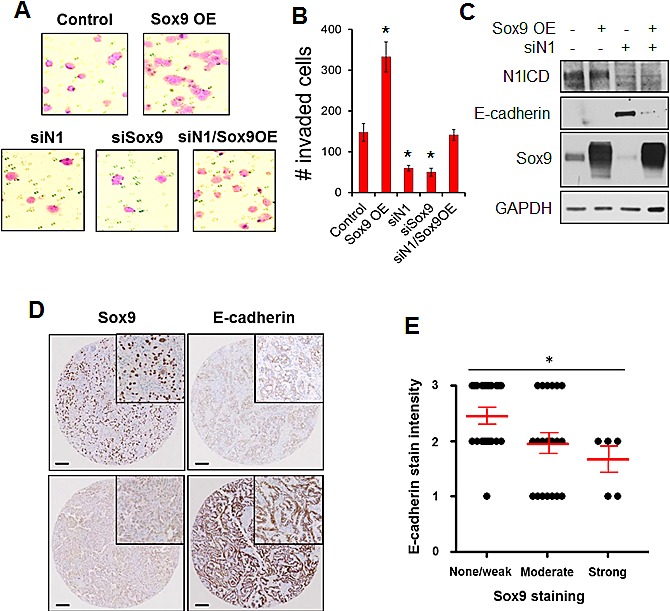
(A) invasion assays were performed on H1299 lung ADC cells 48 hours after transient knockdown of Notch1 or Sox9, or overexpression (OE) of Sox9 compared to cells transfected with both a scrambled siRNA sequence and empty vector (Control). Representative images show crystal violet-stained invaded cells. (B) Total number of invaded cells from experiments in A were quantified and compared to the control samples; *, P < 0.05. (C) Intracellular Notch1 (N1ICD), E-cadherin and Sox9 were analyzed by western blot 72 hours after transient knockdown of Notch1 or overexpression of Sox9, or transfection with a scrambled siRNA sequence and empty vector, in H1299 cells. (D) Lung ADC tumors were immunohistochemically stained for Sox9 and E-cadherin. Representative staining is shown in human lung adenocarcinoma samples with high (upper panel) or low (lower panel) Sox9 staining. Scale bar, 100 μm. (E) Quantified Sox9 and E-cadherin immunohistochemical stain intensity scores were graphed; *, P < 0.05.
The cell-cell adhesion molecule E-cadherin is a major determinant of epithelial features. In line with the morphological changes, E-cadherin expression was substantially increased after knockdown of Notch1 expression in H1299 cells. Overexpression of Sox9 substantially, though not completely, blocked the upregulation of E-cadherin expression caused by knock-down of Notch1 (Fig. 6C and Supplementary Fig. S6C). Similarly, in A549 cells, repression of Sox9 upregulated E-cadherin protein expression, even in the presence of activated Notch1 ICD overexpression (Supplementary Fig. S6D). Knockdown of Notch1 significantly reduced expression of MMP9, Slug and Snail, however, overexpression of Sox9 did not rescue expression of these genes after Notch1 knockdown. Furthermore, modulation of Sox9 expression did not affect mRNA levels of Vimentin, CEACAM1, Twist, Zeb1 or Zeb2 (Data not shown). To establish the correlation between Sox9 and E-cadherin expression in vivo, we stained the human lung ADC tumors for E-cadherin. We found a significant inverse correlation between E-cadherin and Sox9 staining intensities (Fig. 6D and E). Together, these findings suggest that Sox9 may be a gatekeeper partly mediating Notch's role in EMT by inducing loss of epithelial features, blocking E-cadherin expression, and increasing cell motility and invasion.
DISCUSSION
Sox9 is a developmental gene required for lineage commitment in numerous tissues and organs, including the lung. It is well-accepted that genes regulating development also play critical roles in cancer development and progression, and thus it is not surprising that Sox9 expression is dysregulated in cancer. Sox9 is overexpressed in several tumor types and associated with poor survival [16,54,55]. Upregulation of Sox9 expression was reported to increase cell proliferation [9,11,16], and induce invasion in pancreatic, prostate, and urothelial cancer [12,56,57]. Sox9 cooperates with Slug to induce EMT in mammary stem cells, and tumor progression in breast cancer [10]. These reports in total indicate the importance of examining the pathways inducing Sox9 dysregulation and to fully delineate the mechanisms through which Sox9 participates in lung carcinogenesis and tumor progression.
We demonstrate that Sox9 is overexpressed at the mRNA level across the majority of publicly available datasets and at the protein level in 50 primary lung ADC tumors, consistent with previous reports [16,55]. We show that Sox9 is overexpressed within the lung ADC subgroup that harbors KRAS mutations, and confirm that Sox9 is overexpressed within the more malignant portions of tumor from the LSL-K-rasG12D lung ADC mouse model [9]. The Notch pathway cooperates with Kras in development of murine pancreatic intraepithelial neoplasia (PanIN) [58] and lung ADC [21]. Furthermore, Sox9 overexpression induces acinar-to-ductal reprogramming and cooperates with Kras during PanIN formation [48].
In KRAS-mutant lung ADCs, Sox9 overexpression may be secondary event associated with tumor progression [9], rather than directly downstream of the Kras pathway. Conversely, in the murine model of Notch1-induced lung cancer, Sox9 is overexpressed as early as 7 days after induction of Notch1 overexpression in the alveolar epithelium, specifically confined to the alveolar hyperplastic regions, and thus is likely to be directly downstream of Notch. We propose that Sox9 participates as a primary downstream Notch effector early in Notch1-induced carcinogenesis, but as a secondary event during KRAS-mutant lung carcinogenesis, and both Kras and Notch contribute significantly to lung tumor formation. Further studies are needed to determine if, 1) Sox9 overexpression is required for tumor formation in the Kras and Notch1 lung cancer mouse models, 2) if Sox9 acts downstream of Kras or Notch in the Kras lung cancer mouse model, and 3) if Sox9 alone can drive the development of premalignant or malignant lesions.
Aberrantly activated Notch signaling is associated with poor survival, CSCs, EMT, and chemoresistance in lung cancer [23,24,59,60]; however, very little is known about the downstream genes mediating the effects of Notch in lung ADC [26,27]. We show that Sox9 is not only a direct target of Notch1 in lung ADC but also functionally mediates Notch1-induced cell motility, invasion, and mesenchymal-like characteristics. While we observed that Sox9 overexpression decreased E-cadherin, the mode by which this happens is unclear. A search of 5 kb upstream of the E-cadherin transcription start site revealed no Sox9 binding sites. Future studies are needed to investigate the mechanisms by which Sox9 regulates E-cadherin expression. Work is also needed to examine if Sox9 mediates Notch-induced chemo- and radio-resistance, as well as CSC self-renewal.
This study has important clinical implications since Notch signaling is aberrantly over-activated not only in 40% of lung ADCs but across numerous tumor types [17]. Gamma secretase inhibitors that inhibit Notch activation, and monoclonal antibodies are in clinical trials, but they are largely ineffective due to dose-limiting toxicities or development of resistance [61-63]. Clearly, novel methods to target the tumorigenic effects of Notch while minimizing toxicities are necessary. The results of this study could be an impetus for investigations for inhibiting Sox9 transcriptional activity or expression.
MATERIALS AND METHODS
Cell culture and drug treatments
Human bronchial epithelial Beas2b cells were a kind gift of Dr. John Langenfeld, A549, H1299, H358, H1650, H322, H441, H1975, H838, H1838, H23, and H1755 cell lines were obtained from the American Type Culture Collection (ATCC), and Sk-Lu-1, H2030, and HCC-44 cell lines were a gift from Bristol Myers Squibb (Princeton, NJ). Cells were cultured in media as recommended by ATCC and maintained at 37° with 5% CO2 and ambient O2. For EDTA-induced Notch activation, cells were exposed to 1 mM EDTA or culture media for 30 min, then placed back in culture media for 1 hour. For inhibition of Notch activation, cells were treated with 1 μM RO4929097 or DMSO for 48 hours. Human recombinant TGF-β (PeproTech) was used at a concentration of 10 ng/ml for 48 hours. TGF-β inhibitor S431542 (Sigma) was used at a concentration of 10 μM for 48 hours.
Human patient samples
Formalin-fixed paraffin embedded tissue specimens were obtained through the Tissue Retrieval Service, Rutgers Cancer Institute of New Jersey, from patients who underwent surgery at Robert Wood Johnson University Hospital. This study was approved by the Institutional Review Board of Rutgers Robert Wood Johnson Medical School.
Genetically engineered mouse models (GEMM)
Animal experimentation was conducted with the approval of the Institutional Animal Care and Use Committee of the University of California, San Francisco, CA. Mice expressing the doxycycline-responsive reverse tetracycline transactivator (rtTA) driven by the rat Clara cell secretory protein (CCSP) promoter, mice with a tetracycline-responsive promoter element controlling expression of human MYC, and mice with a tetracycline-responsive promoter element controlling expression of human N1ICD, have been previously described [22]. Transgenic mice were fed with a supplemental diet containing doxycycline (200 mg/kg). Tissue sections were obtained and RNA samples were extracted as previously reported [22].
Immunohistochemical analysis and scoring
Surgical specimens were collected by the Tissue Retrieval Services, and a tissue array comprising 50 lung ADC and 6 normal adjacent tissue (NAT) samples were prepared as 1 mm diameter cores in duplicate on two slides, totaling 4 cores per tissue, by the Histopathology and Imaging Shared Resource, at Rutgers Cancer Institute of New Jersey. Tumor/tissue quality was monitored under the guidance of a clinical pathologist. Immunohistochemical staining was done by permeabilizing with 0.1% Triton X-100 in PBS for 10 minutes, quenching endogenous peroxides with 3% hydrogen peroxide for 10 minutes, followed by blocking, as well as primary and secondary antibody incubation. Immunoreactivity was visualized with 3,3'-diaminobenzidine (DAB, Sigma). The primary antibodies used were: Sox9 (1:50, HPA001758, Sigma), E-cadherin (pre-diluted, 760-4440, Ventana), Notch1 (1:100, EP1238Y, Epitomics).
Samples for which at least 25% of the tumor cells stained positive were considered positive. Staining intensity was scored as 0, negative; 1, weak; 2, moderate; and 3, strong. Scoring was performed in a blinded manner by two clinical pathologists and discordant results were discussed until a consensus was reached.
Western blot analyses
Monolayer cells were washed and lysed with freshly prepared lysis buffer. Total cell lysates (20 μg) were assayed by western blot using standard procedures. Cleaved Notch1 and E-cadherin were visualized using high-intensity ECL. All others were visualized with standard ECL. Protein levels in all experiments were normalized to either GAPDH or α-tubulin. The following antibodies were used: Sox9 (AB5535, Millipore), cleaved Notch1 (Val1744, 4147S, Cell Signaling), full-length Notch1 (C20, 6014, Santa Cruz), Hes1 (25392, Santa Cruz), E-cadherin (21791, Santa Cruz), GAPDH (MAB374, Millipore) and α-tubulin (T9026, Sigma).
Real-time quantitative RT-PCR (Q-PCR)
Total RNA was isolated using TRIzol (Invitrogen) and further purified using Qiashredder columns then by the RNeasy Mini kit (both from Qiagen Ltd., Germany) following standard protocols. RNA quantity and quality was assessed using a Nanodrop spectrophotometer. Reverse transcription was carried out using the Qiagen QuantiTect Reverse Transcription Kit in accordance with the manufacturer's instructions, followed by Q-PCR on a Stratagene Mx3005P machine using SYBR Green according to standard procedures. Primers were synthesized by IDT and results were analyzed using Microsoft Excel. Each assay was done at least in biological and PCR triplicate. Specificity of PCR amplification was assessed regularly using a melting-curve analysis. β-actin was used as the internal standard for normalization.
siRNA and plasmid transfections
For siRNA transfection, a mixture of 20 pM siRNA (3 different siRNA sequences pooled) and 1 μL RNAiMax was diluted in Optimem. Experiments were performed 48- or 72-hours post-transfection. siRNA oligos were obtained from Santa Cruz (siNotch1- sc-36095; siNotch2- sc-40137; siNotch3- sc-37135; siNotch4- sc-40135; siSox9- sc-36533; scr- sc-37007; FITC-A- sc-36869). Plasmid transfections were performed using Fugene transfection reagent. N1ICD cloned into pcDNA3.0 (Invitrogen) was a kind gift of Dr. Lucio Miele. The SOX9-pCMV-Tag2V expression vector was a kind gift of Dr. I-Shou Chang. In all experiments, scrambled siRNA sequence, or vector-only plasmids, or both, were used as controls.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) assay was done according to the manufacturer's protocol (Upstate Biotechnology) in triplicate and on 2 separate occasions. In short, chromatin was cross-linked to DNA by adding formaldehyde to tissue culture dishes to a final concentration of 0.75%. Formaldehyde was neutralized and cells were scraped, washed, and resuspended in lysis buffer. Samples were sonicated to 200-500 bp fragments, diluted in ChIP dilution buffer, pre-cleared with salmon sperm DNA–protein A agarose slurry, centrifuged, and the supernatant was collected. Samples were incubated with immunoprecipitating Notch1 or control IgG antibody overnight at 4°C with rotation. Immune complexes were collected with salmon sperm DNA–protein A agarose slurry, then washed with low-salt immune complex buffer, high-salt immune complex buffer, LiCl immune complex buffer, and twice with TE buffer. Input DNA was removed for later normalization, then remaining samples were eluted, cross-links were reversed by incubation at 65°C overnight, and DNA was purified using the Qiagen Gel Extraction kit. Quantitative PCR was performed as outlined above, using the following primer to amplify the Sox9 Site 4 (Fig. 4A) promoter region: forward primer: 5'- GGACTGCTGTGCTG-TGATTG-3', reverse primer: 5'- AGTTTCGAGCTCCGCTTTCG -3'. Samples were subsequently run on a 1.5% gel.
Site directed mutagenesis and luciferase assay
The pGL2 firefly luciferase reporter plasmid under the control of the Sox9 promoter was a kind gift of Dr. William L. Farrar. Mutations were induced using site-directed-mutagenesis primers in conjunction with the Agilent QuikChange II Site Directed Mutagenesis kit, according to the manufacturer's protocol. Cells were co-transfected with one of these plasmids and either the N1ICD or empty vector in triplicate. After 48 hrs, cells were harvested for determination of luciferase activity by a luminometer. A Renilla luciferase plasmid was used for normalization. Mutagenesis primers are listed in Supplementary Methods.
Wound healing motility assay
Cells were plated such that they would be 95-100% confluent at the start of the assay. Cells were transfected 48 hours prior to creating a single scratch wound in triplicate wells. The wound closure experiment was performed in 2% fetal bovine serum to reduce potential differences in cell proliferation. Images were obtained at various time-points after wounding.
Matrigel invasion assay
H1299 cells were transfected 48 hours prior to trypsinization and plated onto Matrigel invasion chambers (BD Biosciences) without serum. The chemoattractant in the lower chamber was 10% fetal bovine serum. The invasion assay was conducted for a minimum of 24 hours to reduce confounding due to potential differences in cell proliferation. Uninvaded cells were removed, and invaded cells were fixed with 4% PFA for 10 minutes, and stained with 0.05% crystal violet. Invaded cells were manually scored.
Statistical analysis
All reported values represent the mean and standard deviation (SD) of at least three independent experiments, unless otherwise noted. All experiments were performed in biological triplicate and repeated at least once. Student's t-tests were used for statistical comparisons. Correlation between Sox9 and E-cadherin staining was done by a Kruskal-Wallis test. Correlation coefficients between mRNA levels in microarray datasets were obtained by Pearson correlation. A P value ≤ .05 was considered statistically significant. All tests were done using Microsoft Excel or Stata version 12.
SUPPLEMENTARY FIGURES AND METHODS
Acknowledgments
We thank Drs. Lucio Miele, I-Shou Chang, and William L. Farrar for their kind gifts of plasmids used in this study. We thank Dr. Karin Cohen-Solal for her advice on the TGF-β experiments and for the SB431542 TGF-β inhibitor. We are thankful to Sohrab Amiri for technical assistance. We thank Julie Friedman from the Tissue Retrieval Shared Resource, and David Foran from the Histopathology and Imaging Shared Resource, Rutgers Cancer Institute of New Jersey. We are thankful to Marina Chekmareva for her oversight during TMA building and IHC optimization. We thank D. John Langenfeld for the Beas2b cell line. We are thankful to Dr. Shridar Ganesan for advice on the ChIP assays. We are appreciative to Dr. Hatem Sabaawy for continued advice and for critical review of this manuscript.
Grant Support
This work was supported by a career development grant from the NCI, National Institutes of Health K22CA140719 to S.R.P.
REFERENCES
- 1.Akiyama H, Lefebvre V. Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner Metab. 2011;294:390–395. doi: 10.1007/s00774-011-0273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee YH, Saint-Jeannet JP. Sox9 function in craniofacial development and disease. Genesis. 2011;494:200–208. doi: 10.1002/dvg.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schafer AJ, Foster JW, Kwok C, Weller PA, Guioli S, Goodfellow PN. Campomelic dysplasia with XY sex reversal: diverse phenotypes resulting from mutations in a single gene. Ann N Y Acad Sci. 1996;785:137–149. doi: 10.1111/j.1749-6632.1996.tb56252.x. [DOI] [PubMed] [Google Scholar]
- 4.Schafer AJ, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovic M, Weissenbach J, Mansour S, Young ID, Goodfellow PN. The role of SOX9 in autosomal sex reversal and campomelic dysplasia. Philos Trans R Soc Lond B Biol Sci. 1995;3501333:271–277. doi: 10.1098/rstb.1995.0161. [DOI] [PubMed] [Google Scholar]
- 5.Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MA, Kopp JL, Sander M, Wellik DM, Spence JR. Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc Natl Acad Sci U S A. 2013;11047:E4456–E4464. doi: 10.1073/pnas.1311847110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turcatel G, Rubin N, Menke DB, Martin G, Shi W, Warburton D. Lung mesenchymal expression of Sox9 plays a critical role in tracheal development. BMC Biol. 2013;11:117. doi: 10.1186/1741-7007-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, Okubo T, Rawlins E, Hogan BL. Epithelial progenitor cells of the embryonic lung and the role of microRNAs in their proliferation. Proc Am Thorac Soc. 2008;53:300–304. doi: 10.1513/pats.200710-162DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou CH, Ye LP, Ye SX, Li Y, Zhang XY, Xu XY, Gong LY. Clinical significance of SOX9 in human non-small cell lung cancer progression and overall patient survival. J Exp Clin Cancer Res. 2012;31:18. doi: 10.1186/1756-9966-31-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, Lopez de MA, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012;725:1301–1315. doi: 10.1158/0008-5472.CAN-11-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, Tam WL, Mani SA, van OA, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;1485:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, He S, Yuan J, Mao X, Cao Y, Zong J, Tu Y, Zhang Y. Oncogenic role of SOX9 expression in human malignant glioma. Med Oncol. 2012;295:3484–3490. doi: 10.1007/s12032-012-0267-z. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, Mathews LA, Cabarcas SM, Zhang X, Yang A, Zhang Y, Young MR, Klarmann KD, Keller JR, Farrar WL. Epigenetic Regulation of SOX9 by the NF-kappaB Signaling Pathway in Pancreatic Cancer Stem Cells. Stem Cells. 2013;318:1454–1466. doi: 10.1002/stem.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang G, Lunardi A, Zhang J, Chen Z, Ala U, Webster KA, Tay Y, Gonzalez-Billalabeitia E, Egia A, Shaffer DR, Carver B, Liu XS, Taulli R, et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat Genet. 2013;457:739–746. doi: 10.1038/ng.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passeron T, Valencia JC, Namiki T, Vieira WD, Passeron H, Miyamura Y, Hearing VJ. Upregulation of SOX9 inhibits the growth of human and mouse melanomas and restores their sensitivity to retinoic acid. J Clin Invest. 2009;1194:954–963. doi: 10.1172/JCI34015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aleman A, Adrien L, Lopez-Serra L, Cordon-Cardo C, Esteller M, Belbin TJ, Sanchez-Carbayo M. Identification of DNA hypermethylation of SOX9 in association with bladder cancer progression using CpG microarrays. Br J Cancer. 2008;982:466–473. doi: 10.1038/sj.bjc.6604143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang SS, Fang WT, Hou YH, Huang SF, Yen BL, Chang JL, Li SM, Liu HP, Liu YL, Huang CT, Li YW, Jang TH, Chan SH, et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin Cancer Res. 2010;1617:4363–4373. doi: 10.1158/1078-0432.CCR-10-0138. [DOI] [PubMed] [Google Scholar]
- 17.Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;347:1420–1430. doi: 10.1093/carcin/bgt127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakano D, Kato A, Parikh N, McKnight K, Terry D, Stefanovic B, Kato Y. BCL6 canalizes Notch-dependent transcription, excluding Mastermind-like1 from selected target genes during left-right patterning. Dev Cell. 2010;183:450–462. doi: 10.1016/j.devcel.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;79:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 20.Westhoff B, Colaluca IN, D'Ario G, Donzelli M, Tosoni D, Volorio S, Pelosi G, Spaggiari L, Mazzarol G, Viale G, Pece S, Di Fiore PP. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A. 2009;10652:22293–22298. doi: 10.1073/pnas.0907781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S, Keith B, Simon MC, Pure E, Radtke F, Capobianco AJ, Kissil JL. Notch1 Is Required for Kras-Induced Lung Adenocarcinoma and Controls Tumor Cell Survival via p53. Cancer Res. 2013. [DOI] [PMC free article] [PubMed]
- 22.Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 2011;7118:6010–6018. doi: 10.1158/0008-5472.CAN-11-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie M, He CS, Wei SH, Zhang L. Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo. Eur J Cancer. 2013. [DOI] [PubMed]
- 24.Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013;198:1972–1980. doi: 10.1158/1078-0432.CCR-12-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;3411:41–45. doi: 10.1016/j.canlet.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 26.Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, Miele L, Bocchetta M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene. 2010;2917:2488–2498. doi: 10.1038/onc.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Li D, Liu H, Xu H, Zheng H, Qian F, Li W, Zhao C, Wang Z, Wang X. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol Ther. 2011;111:14–21. doi: 10.4161/cbt.11.1.13730. [DOI] [PubMed] [Google Scholar]
- 28.Shih HP, Kopp JL, Sandhu M, Dubois CL, Seymour PA, Grapin-Botton A, Sander M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;13914:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakanishi K, Chan YS, Ito K. Notch signaling is required for the chondrogenic specification of mouse mesencephalic neural crest cells. Mech Dev. 2007;1243:190–203. doi: 10.1016/j.mod.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Martini S, Bernoth K, Main H, Ortega GD, Lendahl U, Just U, Schwanbeck R. A critical role for Sox9 in notch-induced astrogliogenesis and stem cell maintenance. Stem Cells. 2013;314:741–751. doi: 10.1002/stem.1320. [DOI] [PubMed] [Google Scholar]
- 31.Muto A, Iida A, Satoh S, Watanabe S. The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Muller glial cell development in mouse retina. Exp Eye Res. 2009;894:549–558. doi: 10.1016/j.exer.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Taylor MK, Yeager K, Morrison SJ. Physiological Notch signaling promotes gliogenesis in the developing peripheral and central nervous systems. Development. 2007;13413:2435–2447. doi: 10.1242/dev.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujimaki R, Toyama Y, Hozumi N, Tezuka K. Involvement of Notch signaling in initiation of prechondrogenic condensation and nodule formation in limb bud micromass cultures. J Bone Miner Metab. 2006;243:191–198. doi: 10.1007/s00774-005-0671-y. [DOI] [PubMed] [Google Scholar]
- 34.Mead TJ, Yutzey KE. Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc Natl Acad Sci U S A. 2009;10634:14420–14425. doi: 10.1073/pnas.0902306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meier-Stiegen F, Schwanbeck R, Bernoth K, Martini S, Hieronymus T, Ruau D, Zenke M, Just U. Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS One. 2010;57:e11481. doi: 10.1371/journal.pone.0011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Candy PA, Phillips MR, Redfern AD, Colley SM, Davidson JA, Stuart LM, Wood BA, Zeps N, Leedman PJ. Notch-induced transcription factors are predictive of survival and 5-fluorouracil response in colorectal cancer patients. Br J Cancer. 2013;1094:1023–1030. doi: 10.1038/bjc.2013.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song S, Maru DM, Ajani JA, Chan CH, Honjo S, Lin HK, Correa A, Hofstetter WL, Davila M, Stroehlein J, Mishra L. Loss of TGF-beta adaptor beta2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res. 2013;737:2159–2169. doi: 10.1158/0008-5472.CAN-12-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA, Jr, Johnson GL, Hirsch FR, Merrick DT, Franklin WA, Baron AE, Keith RL, Nemenoff RA, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005;1676:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;9824:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei TY, Juan CC, Hisa JY, Su LJ, Lee YC, Chou HY, Chen JM, Wu YC, Chiu SC, Hsu CP, Liu KL, Yu CT. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012;1039:1640–1650. doi: 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;721:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 42.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;88:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 43.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;9824:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, Huang CY. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landi MT, Consonni D, Rotunno M, Bergen AW, Goldstein AM, Lubin JH, Goldin L, Alavanja M, Morgan G, Subar AF, Linnoila I, Previdi F, Corno M, et al. Environment And Genetics in Lung cancer Etiology (EAGLE) study: an integrative population-based case-control study of lung cancer. BMC Public Health. 2008;8:203. doi: 10.1186/1471-2458-8-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, Laird-Offringa IA. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;227:1197–1211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou J, Aerts J, den HB, van IW, den BM, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;54:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopp JL, von FG, Mayes E, Liu FF, Dubois CL, Morris JP, Pan FC, Akiyama H, Wright CV, Jensen K, Hebrok M, Sander M. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;226:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;205:1825–1835. doi: 10.1128/mcb.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van dW I, De SG, De SM, Vandekerckhove B, Leclercq G, Plum J, Taghon T. An early decrease in Notch activation is required for human TCR-alphabeta lineage differentiation at the expense of TCR-gammadelta T cells. Blood. 2009;11313:2988–2998. doi: 10.1182/blood-2008-06-164871. [DOI] [PubMed] [Google Scholar]
- 51.Beatus P, Lundkvist J, Oberg C, Lendahl U. The notch 3 intracellular domain represses notch 1-mediated activation through Hairy/Enhancer of split (HES) promoters. Development. 1999;12617:3925–3935. doi: 10.1242/dev.126.17.3925. [DOI] [PubMed] [Google Scholar]
- 52.Haller R, Schwanbeck R, Martini S, Bernoth K, Kramer J, Just U, Rohwedel J. Notch1 signaling regulates chondrogenic lineage determination through Sox9 activation. Cell Death Differ. 2012;193:461–469. doi: 10.1038/cdd.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee C, Huang CH. LASAGNA-Search 2. 0: integrated transcription factor binding site search and visualization in a browser. Bioinformatics. 2014. [DOI] [PubMed]
- 54.Wang H, He L, Ma F, Regan MM, Balk SP, Richardson AL, Yuan X. SOX9 regulates low density lipoprotein receptor-related protein 6 (LRP6) and T-cell factor 4 (TCF4) expression and Wnt/beta-catenin activation in breast cancer. J Biol Chem. 2013;2889:6478–6487. doi: 10.1074/jbc.M112.419184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou CH, Ye LP, Ye SX, Li Y, Zhang XY, Xu XY, Gong LY. Clinical significance of SOX9 in human non-small cell lung cancer progression and overall patient survival. J Exp Clin Cancer Res. 2012;31:18. doi: 10.1186/1756-9966-31-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai C, Wang H, He HH, Chen S, He L, Ma F, Mucci L, Wang Q, Fiore C, Sowalsky AG, Loda M, Liu XS, Brown M, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest. 2013;1233:1109–1122. doi: 10.1172/JCI66666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling S, Chang X, Schultz L, Lee TK, Chaux A, Marchionni L, Netto GJ, Sidransky D, Berman DM. An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res. 2011;7111:3812–3821. doi: 10.1158/0008-5472.CAN-10-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;10548:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, Lai TC, Lee CH, Hsiao YW, Lu J, Shen CN, Lu PJ, Hsiao M. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;731:406–416. doi: 10.1158/0008-5472.CAN-12-1733. [DOI] [PubMed] [Google Scholar]
- 60.Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, Honorio S, Xie Y, Scaglioni PP, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010;7023:9937–9948. doi: 10.1158/0008-5472.CAN-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, Zhang L, Higgins GA, Parker EM. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;27913:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 62.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;4357044:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 63.O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;2048:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.