

Abstract
Estrogen has vascular protective effects in premenopausal women and in women younger than 60 years who are receiving hormone replacement therapy. However, estrogen also increases the risks of breast and uterine cancers and of venous thromboses linked to up-regulation of coagulation factors in the liver. In mouse models, the vasculoprotective effects of estrogen are mediated by the estrogen receptor α (ERα) transcription factor. Here, through next-generation sequencing approaches, we show that almost all of the genes regulated by 17β-estradiol (E2) differ between mouse aorta and mouse liver, ex vivo, and that this difference is associated with a distinct genomewide distribution of ERα on chromatin. Bioinformatic analysis of E2-regulated promoters and ERα binding site sequences identify several transcription factors that may determine the tissue specificity of ERα binding and E2-regulated genes, including the enrichment of NF-κB, AML1, and AP1 sites in the promoters of E2 down-regulated inflammatory genes in aorta but not liver. The possible vascular-specific functions of these factors suggest ways in which the protective effects of estrogen could be promoted in the vasculature without incurring negative effects in other tissues.
Cardiovascular disease (CVD) is the leading cause of death for both men and women in the developed world. However, women of child-bearing age have a much lower incidence of CVD than similarly aged men, and this has been attributed to relatively high circulating levels of the female sex steroid, estrogen (1, 2). Consistent with this observation, a woman's risk of CVD increases markedly after surgical or natural menopause. Treatment with exogenous estrogen reduced the risk of coronary heart disease in women who had undergone ovariectomy and (as part of a combined therapy with progestin) in postmenopausal women younger than age 60 (1, 3–5). Estrogen also decreased the amounts of calcified plaque in the coronary arteries of women aged 50 to 59 (6). The potential cardioprotective effects of estrogen are greatly complicated, however, by the harmful effects it can have in nonvascular tissues, which include increased risk of breast and uterine cancers, especially when administered in the absence of progesterone (3). Estrogen treatment has also been linked to a 2- to 3-fold increased risk of venous thromboembolic events, which probably results from up-regulation of coagulation factors in the liver (7). These and other complications have kept estrogen therapies from being used to prevent CVD (8), despite clear evidence supporting a vascular protective function of circulating estrogen in premenopausal women.
Data from in vitro and animal experimental models indicate a highly protective role for estrogen in the cardiovascular system. Estrogen signaling through estrogen receptor (ER) α attenuates the injury response by promoting vessel reendothelialization, reducing extracellular matrix deposition and inhibiting vascular smooth muscle cell proliferation (9, 10). ERα-mediated estrogen signaling can also reduce serum cholesterol levels and the formation of plaques in a murine atherosclerosis model (11). ERβ is not required for the protective effects of E2 in injury and atherosclerosis; however, ERβ has other vascular effects in mouse models, such as regulation of vascular tone and of ion channel function in vascular smooth muscle cells (12). Human genetic polymorphisms in both ER isoforms have also been linked to CVD (for example, see Refs. 13 and 14 and references therein).
When ERs bind to E2 or other agonists, they move to the nucleus, bind chromatin, and regulate target gene transcription. Accordingly, we expect that the differential physiological responses of tissues to estrogen largely arise from differences in the transcriptional regulatory effects of ERs. ERs can bind directly to estrogen response elements (EREs) or can be recruited to chromatin by binding to other transcription factors (TFs), including SP1, NF-κB, and AP1 (15). Agonist-bound ERs also form signaling complexes on the inner plasma membrane that activate the cellular kinases c-Src, phosphatidylinositol 3-kinase (PI3K), Akt, ERK1/2, and MAPK (16, 17). Because these kinases regulate the activity of many TFs (including c-Myc, c-Fos, NF-κB, and p53), it is expected that part of the transcriptional response to E2 will be driven by downstream effects of this “rapid” or “nongenomic” ER signaling (18), and this hypothesis is supported by recent studies in MCF7 cells (19) and also by our recent studies using a transgenic mouse model that is specifically deficient in rapid signaling (20).
The clinical observations described above, as well as microarray analyses in cell culture and murine models (21–24), suggest that the effect of estrogens on gene regulation can be highly cell type or tissue specific. Here, we examine the mechanisms of tissue-specific estrogen responses in vascular vs other tissues by analyzing the genomewide distributions of ERα binding sites (ERBSs) on chromatin (the ERα cistromes) and the sets of E2-regulated genes in mouse aortas vs mouse livers using next-generation sequencing approaches. We find that the ERα cistromes and the E2-regulated transcriptomes differ greatly between these 2 tissues. Using bioinformatic approaches, we find that the differences in expression and ERα distribution are mirrored by differences in the TF binding sites enriched in the proximal promoter regions of E2-regulated genes and in ERα binding peaks. These results highlight the exceptionally high degree of tissue specificity of transcriptional responses to estrogen and also identify specific TFs that may mediate vascular-specific and hepatic-specific estrogen responses. The observation that E2 may work through distinct mechanisms in different tissues to regulate divergent sets of genes indicates that it may eventually be possible to develop drugs that confer the vasculoprotective effects of estrogen without incurring harmful effects in other tissues.
Materials and Methods
Animals
Wild-type female C57BL/6 mice were ovariectomized at 6 to 7 weeks of age. Ovariectomy reduces circulating E2 to undetectable levels and results in rapid shrinkage of uteri (9). Aortas and livers were harvested from these animals 1 week after surgery and treated with either 10−8 M E2 (the active endogenous form of estrogen) or ethanol vehicle for 45 minutes or 4 hours at 37°C in DMEM (Invitrogen). All animals were handled according to National Institutes of Health standards and protocols approved by the Tufts Medical Center Institutional Animal Care and Use Committee.
RNA sequencing (RNA-Seq)
Whole aortas or liver fragments (the tip of a major lobe, weighing approximately 100 mg) were harvested, placed in DMEM and treated with 100−8 M E2 or ethanol vehicle for 4 hours. Samples were homogenized using a Tissue Tearor model 985370–395 from Biospec Products. Total RNA was extracted from both tissues using the fibrous tissue mini kit (QIAGEN). Four RNA-Seq libraries were prepared from separate biological samples. For 2 of these, RNA was pooled from 4 aortas or 4 liver fragments per sample, and 50 ng of RNA was converted to cDNA using the Ovation RNA-Seq System kit (NuGEN), which uses limited amplification with specialized primers to reduce the rRNA signal. The cDNAs were then prepared essentially as per the Illumina protocol (starting from the end repair step), except that we used the End-It kit (Epicentre) for end repair, Taq polymerase for A addition, Tufts University Core Facility oligos (OLJ131, 5′ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT; and OLJ137, 5′-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG) for adapter ligation, and Easy A thermostable polymerase (Agilent Technologies) with Tufts core oligos (OLJ139, 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA; and OLJ140, 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAAC) for the final limited amplification. Ligated or final amplified products of ∼300-bp size were isolated by an E Gel system or by 1.6% agarose gel electrophoresis followed by a QIAGEN Gel Extraction kit purification. For all other steps, products were purified by addition of 30 μg of glycogen (1286677; Invitrogen) and ethanol precipitation, followed by a 70% ethanol wash. The library was quantified using the Quant-iT dsDNA HS kit (Invitrogen), and approximately 10 ng was submitted for sequencing on an Illumina Genome Analyzer machine (first biological replicate) or an Illumina HiSeq 2000 machine (second replicate). Two additional poly(A+) selected libraries were prepared using 600 ng of RNA extracted from a single aorta or liver fragment. Library preparation was done using the TrueSeq RNA v2 Sample Prep Kit and supplied adapter oligos (RS-122–2001; Illumina Inc) on a Sciclone NGS Workstation (P/N SG3–31020-0300; PerkinElmer) using vendor instrument protocols and 14 cycles of amplification. Libraries were sequenced on an Illumina HiSeq 2000 machine in rapid mode (equal amounts of all 8 libraries, each marked with a distinct adapter tag, were run on a single lane, and demultiplexed reads from 2 lanes were combined). We trimmed the reads to remove the first 8 bases, which tend to be of lower quality, and used Bowtie (25) to map reads to unique locations on the mm9 University of California, Santa Cruz (UCSC) build of the mouse genome (allowing at most 2 mismatches in the first 28 bp of the trimmed read). The numbers of millions of reads mapping to unique locations in the genome, per sample were as follows: aorta (Ao) Vehicle (V): no. 1, 19.6; no. 2, 98.7; no. 3, 35.6; and no. 4, 23.3; AoE2 (E), no. 1, 36.5; no. 2, 83.2; no. 3, 37.4; and no. 4, 37.1; liver (Li) V, no. 1, 36.3; no. 2, 95.7; no. 3, 14.4; and no. 4, 15.9; LiE, no. 1, 36.1; no. 2, 94.4; no. 3, 22.4; and no. 4, 9.8. Read density across mRNA sequences for annotated refseq genes (with the exception of 3′ untranslated region sequences, for which read density has been shown to correlate poorly with mRNA expression level) (26) was determined using HTSeq (27). Significant differential expression was determined using EdgeR (28). To account for potential differences due to library preparation methods, we used EdgeR parameters that allowed for batch effect correction between the first pair (NuGEN) and second pair [poly(A+)] of libraries. Genes with too few counts to allow differential expression analysis in either tissue (fewer than 2 counts in more than half of all samples) were removed from the analysis. Differential gene regulation by E2 between the 2 tissues was assessed using the contrast ([Ao.E2 − Ao.Veh] − [Li.E2 − Li.Veh]) in EdgeR. Genes were called as E2-regulated in aorta or liver or as differentially E2-regulated between tissues, if they had both a P value of <.05 and a fold change of ≥1.5. Using this approach, we found that the data fit well with independent quantitative RT-PCR (qRT-PCR) expression measures for regulated genes and that the combined data from all 4 libraries were better than those for either pair of libraries alone. In particular, the correlation between the log base 2 differential E2 effects between tissues for qRT-PCR compared with the first 2 RNA-Seq samples alone was R2 = 0.80, compared with the second 2 RNA-Seq samples alone was R2 = 0.68 and compared with all 4 RNA-Seq samples was R2 = 0.94 (Supplemental Figure 1A; see also qPCR methods below). A heat map for all genes that were called as P < .05 regulated in aorta or liver, across all 8 libraries is shown in Supplemental Figure 1B. The analysis of associations of E2-regulated genes to biological functions and diseases and their connections in biological networks was performed using Ingenuity Pathway Analysis (IPA) (Ingenuity Systems).
Chromatin immunoprecipitation sequencing (ChIP-Seq)
Aortas and liver fragments from 5 animals per condition were harvested and treated ex vivo with 10−8 M E2 or ethanol vehicle for 45 minutes in DMEM. Samples were then cross-linked with 1% formaldehyde at 37°C for 15 minutes and quenched with 0.125 M glycine. Tissues were then rinsed with 1× PBS, minced with scissors, and homogenized. Samples were pelleted by brief centrifugation and resuspended in 0.5 mL of lysis buffer (1% SDS, 10 mM EDTA [pH 8.0], 50 mM Tris [pH 8.0], and 1× complete protease inhibitors; Roche 1187358001). DNA was fragmented by 7 rounds of sonication with a Sonifier 250 tip sonicator (Branson Ultrasonics) at output setting 5 for 5 seconds followed by 1 minute on ice. Samples were centrifuged at 13 500 rpm for 15 minutes at 4°C, and the supernatant was collected. Then, 14.5 μL of Protein A Dynabeads and 14.5 μL of Protein G Dynabeads (Invitrogen) were washed by magnetic pull-down 3 times at 4°C with PBS plus 5 mg/mL BSA, and 15 μL of anti-ERα HC-20 antibody (Santa Cruz Biotechnology) and 10 μL of anti-ERα Ab-10 (NeoMarkers) were incubated with the beads for 6 to 8 hours. Beads were washed 2 times with PBS plus BSA to remove unbound antibody and resuspended in 0.5 mL of PBS plus BSA. Then 280 μL of fixed chromatin (in sonication buffer) was mixed with 1.12 mL of dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl [pH 8.1]) and added to the beads, followed by overnight incubation with rocking at 4°C. Beads were collected by magnetic pull-down and washed 6 times with radioimmunoprecipitation assay buffer (50 mM HEPES [pH 7.5], 1 mM EDTA [pH 8.0], 0.7% sodium deoxycholate, 1% NP-40, and 0.5 M LiCl) using 20- and 5-minute alternating wash times and twice with Tris-EDTA buffer (TE) for 5 minutes. DNA was released, and cross-links were reversed by resuspension in elution buffer (100 mM NaHCO3 and 1% SDS) and incubation at 65°C overnight (vortexing every 5 minutes for the first 30 minutes). Samples of input chromatin were also suspended in elution buffer and incubated at 65°C to reverse cross-links. DNA was purified using the QIAquick PCR kit (QIAGEN) and quantified using the Quant-iT dsDNA HS kit (Invitrogen). Then 4 ng of input and ChIP samples from 2 biological replicates were prepared for sequencing essentially as described for RNA-Seq (from the cDNA step onward), except that we used Illumina adapter and amplification oligos. The final library fragment size selected for sequencing was ∼230 bp, consistent with mononucleosome-length input DNA fragments +∼90 bp of linker DNAs. All ChIP-Seq and input chromatin libraries were sequenced on an Illumina HiSeq2000 machine. The first aorta library was also sequenced once on an Illumina Genome Analyzer machine, and these reads were combined with those from the HiSeq run. Reads were trimmed and mapped to the mouse genome as described for RNA-Seq (except that no more than one mismatch was allowed) and deduplicated (keeping only one of any set of reads starting at the exact same base), and an equal number of deduplicated reads were combined (to ensure equal weighting of each replicate). ERα binding peaks that were significantly different from the input control were called using MACs, a Poisson-based algorithm that evaluates the likelihood of ERα binding at a specific site over random chance (29). The final counts of reads that mapped to only one place in the genome, after duplicate removal, were 33.4, 70.1, 44.5, and 73.9 million for the combined aorta ChIP, aorta input, liver ChIP, and liver input samples, respectively.
qPCR
For validation of RNA-Seq experiments, individual aortas and liver fragments from 10 additional ovariectomized female mice were harvested and treated with or without E2 for 4 hours exactly as described for RNA-Seq. RNA was then reverse transcribed using an ImpromII RT kit (Promega). For ChIP-Seq, aortas and liver fragments from 4 independent sets of 3 ovariectomized female mice were treated with or without E2 for 45 minutes, chromatin was prepared, and ChIP against ERα was performed exactly as described for ChIP-Seq. qPCR was performed using SYBR Green Master Mix (QIAGEN), with each PCR performed in triplicate. Gene expression levels were normalized to β2-microglobulin (a well-established normalization control for E2-regulated gene expression in mouse tissues) (30, 31) and ERα binding was normalized to an intergenic region with no detectable ERα binding in mouse tissues (Brown, M., and J. Cook, personal communication). We tested 11 genes that were called by RNA-Seq as at least 1.35-fold differentially regulated by E2 between tissues ([AoE − AoV] − [LiE − LiV]) by qRT-PCR (not all of which also met the P < .05 and fold change >1.5 cutoffs for inclusion in the lists of regulated genes by RNA-Seq). RNA-Seq and qRT-PCR data from all 11 genes were plotted to determine the correlation between these different measures from independent sample sets (Figure 1B). The R2 = 0.94 correlation coefficient from this plot implies that the large majority of genes identified by RNA-Seq are truly regulated by E2. However, no transcriptome-wide analysis is entirely free of false-positive results. Whereas the presence of a small fraction of false-positive results is not expected to alter broad patterns of regulation, gene function, and TF binding site enrichment profiles, we recommend that researchers validate individual gene regulation calls by qRT-PCR before further study. For ERα binding, the ratio of the signal from ChIP vs input DNA is given (normalized to the ChIP/input signal for the nonbound intergenic control region). Primers for qRT-PCR and ChIP qPCR are listed in Supplemental Table 1.
Figure 1.
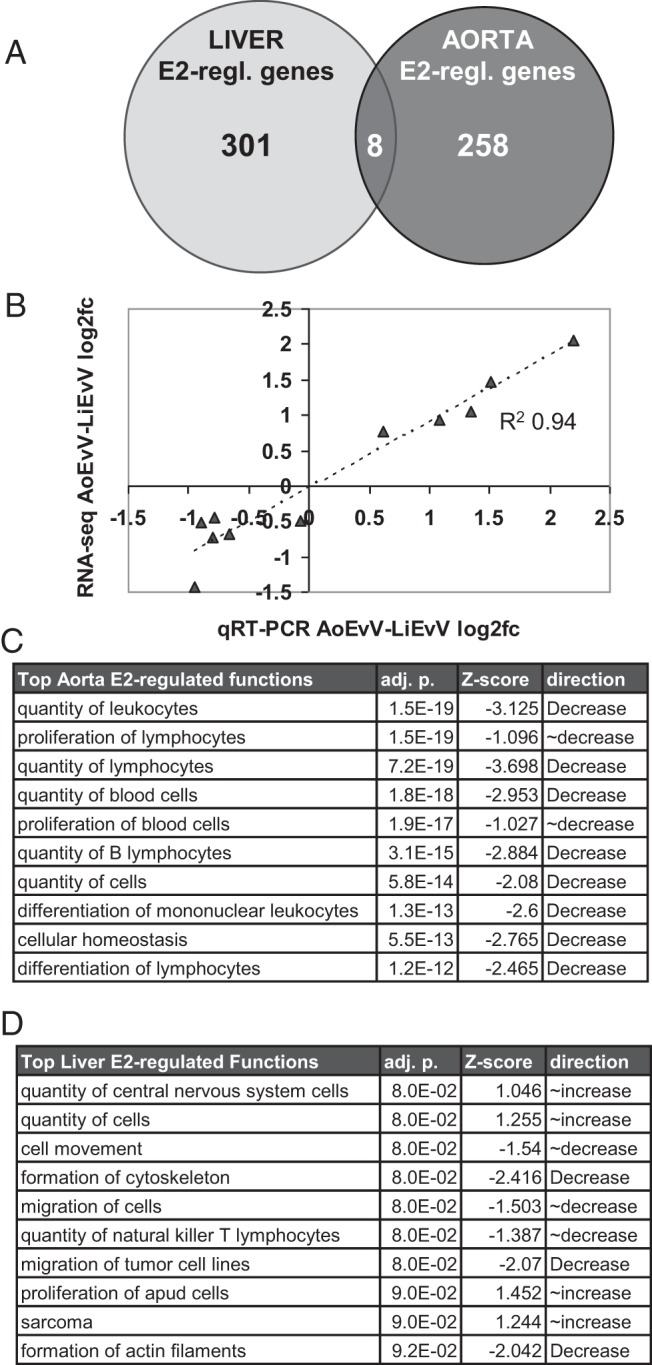
A, Low overlap between genes regulated by 4-hour E2 treatment in mouse liver vs aorta. B, Plot of the qRT-PCR results from separate biological samples vs RNA-Seq results for the differential effect of E2 between aorta and liver [(log2AoE − log2AoV) − (log2LiE − log2LiV)], with the regression line and correlation coefficient as indicated. C, Top 10 IPA function annotations for E2-regulated genes in aorta. D, Top 10 function annotations in liver. To be included on these lists, we required that the association to a function annotation have a multiple testing corrected P ≤ .10 (adj. p.) and also that the direction of regulation of E2 target genes in the tissue be predicted (P < 0.15) to either increase or decrease that function (where Z scores of 1 and 2 correspond to increases with P = .15 and P = .023, respectively, and where negative Z scores correspond to decreases). All of the top 500 function annotations with P ≤ .05 are listed in Supplemental Table 3.
TF Binding Site Analysis
The DNA sequences from sets of E2-regulated promoters and from sets of >1000 expressed but non–E2-regulated control promoters for each tissue (from −1000 to +200 relative to the transcription start site [TSS]) were examined for close matches to TRANSFAC (TRANSFAC Matrix Table 10.3; Biobase GmbH) and JASPAR nonredundant (32) transcription factor binding site consensus sequence (TFBC) matrices with the Search Tool for Occurrences of Regulatory Motifs (STORM) (33), using 2 complementary thresholds, functional depth 0.85, or P = 5e−4. The significance of enrichments relative to control promoters was determined by two-tailed binomial tests adjusted for multiple testing using the Benjamini-Hochberg method (34). The 400-bp sequences surrounding ERBS peak apices or from 2 kb distant flanking sequences were also analyzed by STORM, as for promoters. Significantly enriched matrices were classified into homology groups using similarity, tree-building, and alignment of DNA motifs and profiles (STAMP) (35), and average enrichment and median adjusted enrichment P values were calculated for each group. See Supplemental Methods for details.
Statistical approaches
The statistical approaches used for RNA-Seq and ChIP-Seq are detailed above in the appropriate sections of the Materials and Methods. Otherwise, the significance of differences between continuous variables (eg, ChIP-qPCR values) was determined by two-tailed t tests, and the significance of differences between integral/count variables (eg, gene, TFBC, or ERBS counts) was determined using two-tailed binomial tests. To determine false discovery rates, P values were adjusted for multiple testing using the Benjamini-Hochberg method (34).
Results
Estrogen induces tissue-specific patterns of gene expression
We examined gene regulation by estrogen in mouse aorta and liver using an established ex vivo treatment model (30). Female C57/BL6 mice, aged 6 to 7 weeks, were ovariectomized to remove the principal source of endogenous circulating estrogen (9). Aortas and livers were harvested 1 week later and treated with 10 nM E2 (the active endogenous form of estrogen) or vehicle for 4 hours. This ex vivo approach allows the timing and concentration of E2 to be precisely controlled and eliminates the possibility that the effects of E2 in other parts of the body could indirectly affect gene regulation in aorta or liver. We wished to identify the direct targets of E2 and chose the 4-hour treatment time because, for most genes, this is expected to be too short a time for changes in primary target gene expression (eg, an increase in mRNA resulting in higher levels of a transcriptional regulatory protein) to functionally alter the mRNA levels of indirect downstream genes. However, we cannot entirely rule out this possibility or the possibility that we might not detect those few genes that are either very early direct targets (for which mRNA levels may return to normal by 4 hours) or slow-response direct targets (for which changes in mRNA levels may not be detectable at 4 hours). RNA was isolated from each treatment group, with 4 biological replicates and prepared for next-generation sequencing. Analysis of differences in normalized read counts mapping to exons of known mouse genes identified 266 significantly E2-regulated genes in aorta (64 up and 202 down) and 309 genes in liver (130 up and 179 down, fold change of ≥1.5, P < .05).
We found that there was very little overlap between the E2-regulated genes in aorta and in liver, with only 8 genes regulated by E2 in both tissues (Figure 1A) and with 3 of these being regulated in opposite directions. For each gene that was significantly E2-regulated in either tissue, we looked up how it was regulated in the other tissue (counting 1.5-fold change as regulated, for this contrast, regardless of P value). Using this metric, we found that fewer than one-quarter of genes were regulated in both tissues (22% of genes regulated in aorta were regulated in liver, and 18% of genes regulated in liver were regulated in aorta). Among these, 40 of 63 were regulated in the same direction (P = .04), with 35 of these 40 being down-regulated by E2 in both tissues (P = 1.4e−6). These results indicate that the effects of estrogen on the aorta and liver transcriptomes are extremely divergent and that most E2-regulated genes were affected by estrogen in one tissue and unresponsive in the other. Consistent with these results, we found that 320 genes were significantly differentially regulated by E2 between aorta and liver (135 up-regulated by E2 in aorta relative to liver and 185 down-regulated), more than were regulated by E2 in either tissue alone. Annotated lists of genes that were E2-regulated in each tissue or differentially E2 regulated between tissues are presented in Supplemental Table 2. Raw and processed RNA-Seq data can be found at Gene Expression Omnibus (GEO) accession number GSE57804. We found that the differential E2 effects indicated by RNA-Seq corresponded well with the results of qRT-PCR assays on RNA from independent biological samples: the correlation between differential expression values from qRT-PCR vs RNA-Seq for a set of 11 genes (each called as ≥1.35-fold regulated by RNA-Seq) was R2 = 0.94 (Figure 1B).
Estrogen regulates distinct biological functions in aorta vs liver
We used IPA to investigate potential functional relationships between the E2-regulated genes identified by RNA-Seq in each tissue. The 10 most significant enrichments of aorta or liver E2-regulated genes with groups of genes associated with specific biological functions or diseases (IPA function annotations) are shown in Figure 1, C and D. In liver, the top 10 function annotations associated with gene regulation by E2 were connected to increased cellular proliferation (increased proliferation, quantity of cells, or cancer) and also connected to decreased cell movement/migration. In contrast, almost all of the functions associated with E2-dependent gene regulation in aorta were related to decreased proliferation or differentiation of immune cells. Only 1 of the top 10 and only 54 of the top 500 function annotations were shared between aorta and liver (Figure 1, C and D, and Supplemental Table 3), indicating that the functions and processes controlled by estrogen are likely to differ greatly between these 2 tissues.
The correlation between gene regulation by E2 in aorta and immune cell function is particularly strong, with more than two thirds of the 500 top significantly associated function annotations being related to quantity, proliferation, migration, adhesion, viability, and differentiation of leukocytes (generally decreased) or with the overall immune response (generally decreased) or infection (generally increased). Consistent with this observation, several genes associated with increased vascular inflammation are >2-fold down-regulated by E2 in aorta, including AGER, the advanced glycation end-product receptor, which induces inflammation in vascular cells and is strongly associated with atherosclerosis (36), CRP, C-reactive protein, a well-established serum marker for atherosclerotic CVD risk (37), SELPLG, the selectin P ligand expressed on vascular endothelial cells and critical for capture of leukocytes in early inflammatory reactions (38), STAT4, a TF controlling inflammatory gene expression in response to several inflammatory signals, which is expressed at high levels in atherosclerosis-prone aortas and promotes smooth muscle cell proliferation in response to IL-12 (39), and IL1RL1/ST2L, interleukin 1 receptor-like 1, an interleukin receptor that mediates the inflammatory response of endothelial cells to IL-33 (40), as well as 6 other chemokine and interleukin receptors (Cxcr4, Ccr7, Il1r2, IL2rb, IL2rg, and Il27ra) and the chemokine Xcl1.
Patterns of ERα binding to chromatin are unique to each tissue
One potential mechanism for estrogen to regulate different genes in aorta vs liver is recruitment of ERα to different DNA binding sites in each tissue. To test whether this was the case, we performed ChIP-Seq to map the cistrome of ERα in aorta and liver in the same ex vivo model as was used for RNA-Seq. ERα has been shown to be recruited to its sites on chromatin within 45 minutes of estrogen treatment (41). Hence, to identify the immediate binding sites of ERα, which are likely to control the regulation of first-wave target genes, we performed ChIP-Seq on chromatin from aortas and livers treated with E2 for 45 minutes. Equal numbers of reads from 2 biological replicate samples were combined, and ERBSs were distinguished using MACS to identify regions of significant enrichment relative to input control libraries (29). We found 1754 high-quality ERBSs in aorta, and 33 273 in liver (all with ≥3-fold enrichment over background, P ≤ 10−5, and an empirical false discovery rate of ≤10%). BED formatted peak coordinates can be found in Supplemental Tables 4A and 4B. Raw and processed data are available at GEO accession number GSE52351. Peaks from aorta and liver had very similar characteristics, including similar median fold enrichment values (6.3- vs 8.8-fold) and nearly equal evolutionary conservation scores at peak centers (0.065 over background for each, Supplemental Figure 2). This finding indicates that the larger number of significant peaks in the liver is unlikely to arise, for instance, from a lower background signal in the liver, allowing the detection of weaker, less well-conserved ERBSs (because this would have resulted in a lower conservation score for liver peaks). We found that 700 of 1754 (39.9%) of the ERBSs in aorta overlapped those in liver (Figure 2A). Although this number was statistically significant (P < 5e−12), the fact that >60% of aortic ERBSs differ from liver ERBSs indicates that ERα cistromes differ considerably between these 2 tissues. A representative region showing differential ERα binding (2 liver-specific peaks and 1 aorta-specific peak) is shown in Figure 2B. We performed ChIP-qPCR, using independent biological samples, to validate the differences in ERBSs identified by ChIP-Seq between liver and aorta (Figure 2C). In every tested case we saw ERα binding enrichments, relative to a background nonbound intergenic location, that were consistent with the ChIP-Seq results, with greater than 3-fold enrichment by qPCR only in the tissue or tissues where a greater than 3-fold enriched peak was also called by ChIP-Seq.
Figure 2.

A, Venn diagram showing the low overlap of ERBSs in mouse aorta vs liver. B, Plot of normalized read count density (reads per million reads) in a 20-kb region displaying both aorta- and liver-specific ERBSs. C, ERα ChIP on new samples followed by qRT-PCR with primers to ChIP-Seq peaks confirm the tissue specificity of ERα binding. Dotted line represents 3-fold enrichment, which was the threshold used to call a ChIP-Seq peak. Error bars show SEM. *, Significantly higher than the control region, P = .05; ∧, significantly different between aorta and liver, P = .05. The enrichment in liver at AGT and TFGB2 vs control approached significance at P = .058 and P = .073. D, Extreme differences between aorta and liver are seen for ERBS distributions relative to those for transcribed regions. Chance is the percentage of peaks that would overlap each set of regions if they were randomly distributed. TSS→3kb, regions range from the transcription start site to 3 kb upstream; TTS→3kb, regions range from the transcription termination site to 3 kb downstream. All aorta and liver distributions were significantly different from chance (P < .05), with the exception of aorta mRNA, TTS→3kb, and intergenic peaks. E, background-normalized fraction of E2-regulated genes in each indicated set that have an ERBS within the indicated ranges relative to their start sites (percentage of genes with ERBS in that range − percentage expected for a random distribution of ERBSs). F, Average read density for aorta or liver ERα ChIP-Seq relative to the promoter sequences of the indicated gene sets (Ao, aorta; Li, liver; U, E2–up-regulated; D, E2–down-regulated) was normalized to reads per million, and adjusted for background by subtracting reads per million values for input chromatin from each tissue.
Using CEAS (cis-regulatory element annotation system) (42, 43), we found that ERBSs in aorta were enriched within 3 kb upstream of annotated TSSs (2.3-fold over chance, P = 2e−16) and that ERBSs in liver were even more highly enriched (6.2-fold, P = 1e−322) (Figure 2D). This enrichment was even more striking within 1 kb upstream of TSSs (4.2-fold in aorta and 12.2-fold in liver). Conversely, liver ERBSs were antienriched in the distal intergenic regions (>3 kb from the transcribed regions), whereas aorta ERBSs were not significantly enriched or antienriched in the distal intergenic regions. Compared with our E2-regulated gene sets, we found that a large fraction of liver E2–down-regulated genes had an ERBS within 1 kb upstream of their start sites (27.4%, P < 2.2e−16) and that this fraction was even larger for liver E2–up-regulated promoters (47.7%, P < 2.2e−16). In addition to being much greater than expected by chance, this value is considerably greater than the 13.4% enrichment of liver ERBSs within 1 kb of all annotated TSSs, indicating a specific enrichment of ERBSs near E2-regulated promoters in liver. In contrast, a smaller fraction of aorta E2–up-regulated genes had an ERBS within 1 kb upstream of their TSSs (6.3%, P = 2e−6), and this value was even less for E2–down-regulated promoters (1%, P = .03). The background-adjusted percentage of aorta or liver ERBSs at different ranges (upstream and downstream) for the TSSs of each indicated subset of E2-regulated genes is plotted in Figure 2E. For liver up- and down-regulated genes, the high enrichment within 1 kb of the promoter drops off between 1 and 2 kb and is associated with antienrichment at greater distances. In contrast, the moderate enrichment of ERBSs near aorta E2–up-regulated genes increases at greater distances, whereas there was no significant enrichment of ERBSs within any distance further than 1 kb away from aorta E2–down-regulated promoters. The enrichment of ERα ChIP-Seq read density relative to the TSSs of each class of E2-regulated promoter is graphed in Figure 2F.
Differences in TF consensus sequences between up- and down-regulated genes in aorta and liver
We used the STORM binding site analysis program (33) together with custom downstream analyses to identify enriched TFBCs in the promoters of the 4 sets of E2-regulated genes: liver up-regulated, liver down-regulated, aorta up-regulated and aorta down-regulated. We found that each of these 4 sets of promoters had different sets of enriched TFBCs, as summarized for “homology groups” of TFBCs for TFs with highly similar binding sites (Figure 3A; complete data for all enriched matrices can be found in Supplemental Table 5). Interestingly, almost all of these TFBCs were significantly enriched primarily or exclusively in aorta down-regulated promoters. Moreover, TFBCs with enrichment in several promoter sets were generally most strongly enriched in aorta down-regulated promoters (such as SMAD and CP2/RREB). Only the PBX homology group, which was enriched in liver down-regulated promoters, showed no enrichment in aorta E2–down-regulated promoters. Interestingly, the ER group composed of EREs and closely homologous matrices was not significantly enriched in most classes of promoters, and its enrichment was only 1.16-fold in aorta down-regulated promoters. We compared these results with those from IPA upstream regulator analysis, which identifies likely associations with upstream TFs based on how the known targets of these TFs are regulated by E2 and assigns a Z score for strength and direction of these effects. Interestingly, this IPA indicates that transcriptional regulation by E2 in aorta is associated with decreased activities of several of the factors whose sites are enriched in aorta down-regulated promoters, including SPI1 (Z score = −2.4), HoxA7 (Z score = − 0.6), TCF3 (Z score = −1.2), the RELA NF-κB component (Z score = −1.7), and the NF-κB complex (Z score = −2.9). These results suggest that E2 and ER may down-regulate genes in the aorta by decreasing the activities of TFs whose sites are enriched in down-regulated promoters.
Figure 3.

A, Homology groups of TFs whose TFBCs are enriched by ≥1.1-fold, with median Benjamini-Hochberg adjusted P ≤ .05 for enrichment for one or more classes of E2-regulated promoters (Ao, aorta; Li, liver; Up, E2–up-regulated; Down, E2–down-regulated). *,c Adjusted P < .05; **, adjusted P < 1e−4. # mats is the number of separate TFBC matrices in that homology group, and Pattern provides a summary of the set or sets of regulated promoters that, individually, show significant enrichment for that homology group. B and C, Average site frequencies per kilobase for representative matrices from the NF-κB (B) and AML1 (C) groups (TRANSFAC matrices V$NFKAPPAB_Q6 and V$AML1_Q6) were plotted relative to the TSSs of the indicated promoter sets. Dotted lines, background frequencies for expressed but not E2-regulated control genes in liver and aorta. Insets, WebLogo diagrams (44) of these position frequency matrices (with base height related to the information content associated with enrichment of that base).
The observed TFBC enrichments have the potential to be relevant to the regulation of a large fraction of promoters in each set. For instance, there are 73 more NF-κB sites (on average for all matrices in the NF-κB group) in aorta E2–down-regulated promoters relative to unregulated control promoters, indicating that NF-κB site enrichment could potentially be involved in the regulation of more than one third of the 202 aorta E2–down-regulated promoters. The frequencies for matches to a representative NF-κB matrix (enriched only in aorta down-regulated promoters) and the acute myeloid leukemia 1 (AML1) matrix (enriched in aorta up- and down-regulated promoters), relative to the TSSs of each E2-regulated gene set and each nonregulated control gene set, are shown in Figure 3, B and C. Note that the fold enrichment values listed in Figure 3A are for the whole 1.2-kb promoter region and thus may underestimate the functional enrichment of TFBCs at regions of the greatest regulatory importance. For instance, NF-κB group TFBCs (enriched on aorta down-regulated promoters by 1.16-fold overall) are 1.32-fold enriched over the proximal promoter region (−250 to +0) and only 1.06-fold enriched further upstream (−1000 to −250). In other cases, enrichment is more broadly localized or favors upstream over immediate promoter sequences, as for AML-1 group TFBCs on aorta up-regulated promoters (fold enrichments of 1.08 overall, 1.11 upstream, and 0.98 over the proximal promoter). Note also that the degree of enrichment depends on how frequently matches to a TFBC occur by chance. In addition, within each group of up- or down-regulated promoters, only a fraction may be regulated by any given TF, and the degree of enrichment of TFBCs for that TF in the whole group would be correspondingly smaller than the enrichment at this subset of promoters.
Distinct sets of TF consensus elements are associated with aorta vs liver ERBSs
To determine what might underlie the differences in ERBS localization between aorta and liver, we performed a second TFBC analysis, applying the STORM approach we used on promoters to the 400-bp sequences surrounding the apices of ERBS peaks. TFBC homology groups with significant enrichment in aorta ERBSs, liver ERBSs, or both are summarized in Figure 4A (with all data for individual enriched matrices presented in Supplemental Table 6). Some of these groups of related matrices, such as AP2, CREB, and NF1 show roughly equal enrichment in liver and aorta ERBSs. Others, showed markedly higher enrichment in liver (including the CDP/CLOX, CEBP/HLF, and HNF1/LHX3/POU3 groups) or in aorta (including the AP1 and AR groups). TFBC enrichment relative to peak centers for the shared CREB group and liver-specific CDP/CLOX/HNF6 group are shown in Figure 4, B and C. Interestingly, we found that the matrices with homology to EREs fell into 2 closely related but separable groups, the ER group (containing TFBCs for ER, T3R, and Pax2 and some TFBCs for PPARG), and the HNF4/MYB/RORA/DR/COUP group (containing TFBCs for HNF4, MYB, RORA, DR, COUP, Esrrb, HIC1, NR2F1, and VDR and most of the TFBCs for PPARs). A homology tree showing the separation of ERE-related matrices into subgroups is shown in Supplemental Figure 3. Interestingly, the HNF4 group was more highly enriched in liver and the ER group was much more highly enriched in aorta (Figure 4, D and E). Use of STAMP (35) to derive a familial binding profile (eg, consensus matrix) from all matrices in the ER and HNF4 subgroups revealed that both are composed of 2 close matches to ERE half sites (AGGTCA). However, in the ER group these half sites are arranged in head-to-head orientation with 3 bp separation, whereas the in the HNF4 group these half sites are arranged in a head-to-tail orientation with 1 bp separation (WebLogo [44] matrix representations on the bottom of Figures 4, D and E). Taken together, these results indicate that ERα may be recruited to chromatin by a considerably different set of factors in liver vs aorta.
Figure 4.

A, Enrichment of the indicated homology groups of TFBCs in aorta or liver ERBS peaks, as per Figure 3A. **, P < 1e−10. Pattern indicates the relative enrichment between aorta and liver ([Ao_enrichment_ratio − 1]/[Li_enrichment_ratio − 1]), where ≫ indicates relative enrichment of >1.9-fold, > indicates relative enrichment of >1.3 fold, and ∼= indicates relative enrichment of <1.3 fold. * and **, adjusted P values for the difference in enrichment between aorta and liver. B–E, Average match frequencies per kilobase for all matrices for the indicated homology groups were plotted vs peak apexes. Dashed lines, input chromatin background (BKG) frequencies for the same regions in aorta or liver. D and E, bottom: WebLogo representations of consensus matrices derived from all matrices for the ER and HNF4 homology groups.
Networks of E2-regulated genes show links to enriched TFBCs and nongenomic signaling
We explored networks of E2-regulated genes (connected through protein-protein or regulatory interactions) using IPA. Nodes in these networks with outward pointing arrows (indicating regulatory direction) to E2-regulated genes suggest factors that may work together with ERα to contribute to the regulatory pattern observed, whereas nodes with inward pointing arrows from E2-regulated genes suggest ways in which E2-mediated changes in gene expression could have downstream effects. For example, NF-κB is a central node in the top network in aorta (Figure 5A), and arrows pointing from the NF-κB node to E2-regulated genes indicate target genes (generally down-regulated), whereas arrows pointing from E2-regulated genes to NF-κB suggest ways in which initial E2-responsive genes may alter NF-κB activity. In the top liver network, ER is a node, as are the GATA2 and STAT5 TFs and the Akt kinase (a target of rapid signaling through ERα) (Figure 5B). The other top 5 aorta and liver networks are shown in Supplemental Figures 4 to 7. Notably, many of the major nodes in these networks, in both aorta and liver, are kinases that are activated by rapid/nongenomic signaling by E2-bound ERα, including Akt, PI3K, ERK, p38 MAPK, and the upstream MAPK-activating kinases MAP2K and MAP3K. This observation suggests that rapid, nongenomic signaling mediated by ERα could be important for the gene regulatory effects of E2 in both tissues.
Figure 5.

IPA networks showing protein-protein or regulatory interactions between E2-regulated genes in aorta (A) or liver (B). Shown are the top networks by number of incorporated E2-regulated genes and P value. P values indicate the probability that a network with the same number and character of interaction lines would occur by chance. Numbers give the log2 fold change values for each significantly E2-regulated gene. Dotted boxes highlight E2 down-regulation (negative log2 values).
Discussion
Here we have compared the genes regulated by E2 and the chromatin binding sites for ERα in mouse liver vs mouse aorta. To our knowledge, this is the first study to directly compare E2-regulated genes and ERBSs, genomewide, in different whole tissues. We find that both the E2-regulated transcriptomes and the ERα cistromes differ greatly between liver and aorta. Furthermore, different TF consensus elements are enriched in E2-regulated promoters and ERBSs in each tissue, providing clues for possible mechanisms underlying the tissue-specific gene regulatory effects of E2. We also find that the E2-regulated gene sets in liver and aorta are predicted to have highly divergent biological functions. The most striking connection was between aorta E2–down-regulated genes and the reduction in functions related to inflammation. Whereas some of the function annotations in liver also suggest an anti-inflammatory effect of E2 (including several functions shared with aorta), the Z scores generally indicate a much weaker association with decreased inflammatory function and sometimes indicate that an inflammatory function that is decreased in aorta is increased in liver (Supplemental Table 3). Thus, these data suggest that, although E2 may have anti-inflammatory effects in both tissues, the strength of these affects appears to be much greater in aorta. Interestingly, we found that, of the 13 genes that were strongly regulated by E2 in liver and aorta (regulated at least 1.9-fold in both, and P < .05 in at least one), 11 were down-regulated in both tissues (more than expected by chance, P = 1.1e−5). Most of these genes were involved in inflammation and immune cell function, including Ccr7, Cd79b, Dok3, Dtx1, Il2rb, Ncf4, Tlr1, and Traf1, suggesting that E2 may have a tissue-nonspecific strong down-regulatory effect on a core set of immune modulator genes.
We find that >60% of ERBSs differ between aorta and liver. This difference is similar in magnitude to the greatest documented difference between ERα cistromes: the ∼84% difference observed between U2OS osteosarcoma and MCF7 breast cancer cell lines (21). To test whether the low overlap between ERα cistromes in aorta and liver might be peculiar to these 2 tissues or potentially indicative of a more general phenomenon, we compared our results with those of a recent ERα ChIP-Seq study in mouse uterus (45). We found that the large majority of uterus ERα binding peaks differed from both our aorta and liver peak sets. Of the 16 939 uterus peaks, 33 273 liver peaks, and 1754 aorta peaks, only 474 were found in all 3 tissues (27% of aorta, 2.8% of uterus, and 1.4% of liver peaks), 2830 were in liver and uterus (17% of uterus peaks), 327 were in aorta and uterus (19% of aorta peaks) and 225 were in aorta and liver (13% of aorta peaks) (Supplemental Figure 8). To get a sense of the portion of this difference that might arise from differences between model systems (eg, the uterus ChIP-Seq study used in vivo E2 administration rather than ex vivo treatment), we compared our liver ChIP-Seq results with those from a recent study that used a ChIP microarray (ChIP2) to measure ERα binding in mouse liver after in vivo E2 administration (46). Despite the differences in models, we found that 5017 of 5439 peaks from that study (92%) overlapped our 33 723 top liver peaks. Accordingly, differences in model systems are unlikely to explain the small overlap observed between the uterus ERα ChIP-Seq dataset and our liver and aorta datasets. This finding implies that ERα cistromes vary greatly among different tissues and that this is not just an effect that is peculiar to liver or aorta.
We found a strong enrichment of ERBSs near E2–up- or down-regulated TSSs in liver, a weaker but still highly significant enrichment near E2–up-regulated TSSs in aorta, but only a barely significant enrichment near E2–down-regulated TSSs in aorta. Analysis of the DNA sequences in ERα binding peaks identified several TF homology groups that were either exclusively enriched or much more highly enriched in liver ERBSs relative to aorta ERBSs, as well as some that were most highly enriched in aorta ERBSs (Figure 4A). We noted that whereas the liver- or aorta-specific TFBC groups often showed sharp peaks at the center of ERBS sequences, the TFBC groups that were at least 1.4-fold enriched in both aorta and liver ERBSs (including AP2, CREB, E2F, and SP1) often displayed a relatively broad peak of enrichment stretching for >400 bp to either side of the peak apex (as shown for E2F and SP1 in Supplemental Figure 9). Given their broad enrichment profile, these factors may not be directly involved in recruiting or stabilizing discrete ERα binding events but may rather be characteristic of larger regulatory regions that are also enriched in ERα binding sites, such as promoters (which tend to be enriched in E2F and SP1 sites and which also have enrichment of nearby ERBSs). Consistent with this finding, E2F and SP1 sites are more highly enriched near liver ERBSs than near aorta ERBSs, similar to the relative enrichment of liver and aorta ERBSs near annotated TSSs. Note that, because the aorta is primarily composed of smooth muscle cells, ERBSs and E2-regulated genes in aorta are likely to reflect binding events and gene regulation in smooth muscle cells. It is possible, however, that some weak ERα binding sites or weakly E2–up-regulated promoters may instead reflect strong binding/up-regulatory events occurring in the minority aortic endothelial cell population, adventitial fibroblasts, or even infiltrating immune cells (although, given the lack of blood circulation in this ex vivo model system, any immune cells would have to have been present in the healthy aorta at the time of harvest).
Interestingly, we find that the standard ERE (2 head-to-head ERE half sites with 3 bp spacing) was highly enriched in aorta ERBSs, but that a variant element similar to HNF4 sites (2 head-to-tail ERE half sites with 1 bp spacing) was enriched in liver ERBSs. This could indicate a novel function of HNF4 group factors in recruiting or stabilizing ERα on chromatin. Alternatively, ERα has recently been shown to bind to and transactivate through pairs of half sites in a variety of orientations and spacings (47), suggesting that liver- and aorta-specific complements of cofactors or posttranslational modifications may alter ERα binding site preferences, such that what constitutes a functional ERE might differ between these 2 tissues.
Many of the TFs whose sites were enriched in liver and/or aorta ERBSs have been shown in other systems to be associated with ERα, either by coimmunoprecipitation or ChIP studies or both, including AP1/jun/fos, AP2, AR, C/EBP, E2F, ETS/ELK, HNF1, MYC/USF, and SP1 (18, 41, 45, 46, 48–51) (although none of these have previously been associated with ERα function in vascular cells or tissues). Other TFBC enrichments, including CDP/CLOX/HNF6 and NF1/XVENT, suggest potentially novel ERα interacting or recruiting factors. In human MCF7 breast cancer cells, FoxA1 is a major ERα-cooperating factor that is present at about half of all ERα-binding sites (41, 52). We found only a weak association of FoxA1 sites with liver ERBSs (1.07-fold enriched, well below our 1.4-fold initial cutoff) probably because FoxA1 expression is ∼20- or ∼1000-fold higher in MCF7 cells than in mouse liver or mouse aorta (average reads per million reads from 2 previous RNA-Seq studies in MCF7 cells, each with 2 replicates (53, 54), was 105, compared with our measurements of 5 for liver and 0.1 for aorta). Furthermore, TFBCs for most other factors in the HNF3/FOX/FREAC family were either nonenriched or antienriched in both liver and aorta ERBSs, suggesting that this particular mechanism of establishing functional ERBSs is not present or is much reduced in mouse aorta and mouse liver. On the other hand, 4 members of the large HNF3/FOX/FREAC homology group (including both matrices for FoxF2: Jaspar$FOXF2 and V$FREAC2) were enriched an average of 1.51-fold in liver, raising the possibility that HNF3/FOX/FREAC factors may be important for directing ERα distribution in liver but that the specific factors involved are different from those in MCF7 cells.
Interestingly, the strength of associations of enriched TFBC groups with each class of E2-regulated promoter appears to be the inverse of what is seen for the association of ERα-binding sites with that promoter class. For example, liver up-regulated promoters were strongly associated with nearby ERBSs and weakly associated with promoter-enriched TFBCs, whereas aortic E2–down-regulated promoters showed strong TFBC enrichment and poor correlation with nearby ERBSs. Taken together with the observation that there are fewer ERBSs in mouse aorta vs liver, this suggests that, for each class of E2-regulated genes, regulation can occur either by nearby ERα binding (in which case the number and locations of ERBSs are essential, but the promoter sequence is relatively unimportant, eg, liver up-regulated genes) or by specific promoter-bound TFs (in which case the number and locations of ERBSs are unimportant, eg, aorta down-regulated genes) or by a combination of both (in which case weaker associations to both ER binding and promoter-enriched TFBCs are seen, eg, up-regulated genes in aorta and down-regulated genes in liver).
In liver, where ERBSs are highly enriched near regulated TSSs, some of the promoter-enriched TFBCs could correspond to TFs that recruit ERα to promoters. Alternatively, promoter-enriched TFBCs could correspond to promoter-bound TFs that cooperate with ERα bound at nearby or distal enhancer elements. The very low levels or lack of ERBS enrichment at all ranges up to 50 kb from E2–down-regulated promoters in aorta, however, indicates that the regulation of these genes is generally not directed by ERα bound to chromatin. Instead, our data suggest that these genes may be regulated by the effects of rapid/nongenomic signaling through membrane-bound ERα on the TFs whose sites are enriched in their promoters. This could also be the case for members of the other E2-regulated gene sets whose TSSs are >50 kb away from the nearest ERBSs (16, 32, and 42% of liver up-regulated, liver down-regulated, and aorta up-regulated genes). Interestingly, even though aorta up-regulated promoters show enrichment of nearby ERBSs, the pattern of this enrichment (moderate promoter enrichment, but also significant enrichment at distances up to 50 kb away from the TSS) is quite different from that seen in liver E2-regulated promoters (very strong promoter enrichment dropping to antienrichment at distances of >2 kb). This finding suggests that up-regulation by E2 in aorta may be generally mediated by ERα binding to distal enhancers, whereas up- and down-regulation in liver may be mediated by ERα binding in or near immediate promoter regions.
The importance of transcriptional regulation by rapid/nongenomic signaling is supported by our recent observations that expression of a peptide that blocks the interaction between ERα and striatin, which is necessary for rapid signaling, greatly alters gene regulation by E2 in mouse aorta. The peptide also removes the protective effect of E2 in vascular injury, similar to a whole-body knockout of ERα expression (20). In addition, approximately 25% of E2-regulated genes in MCF7 breast cancer cells were found to be regulated by a compound that only activates rapid signaling (19). We find that network IPA indicates possible regulatory functions for rapid/nongenomic signaling kinases in the E2-dependent regulation of genes in aorta and in liver (including MAPKs, Akt, Erk, Jnk, p38, and PI3K). Furthermore, several of the TFs identified as specifically enriched in aorta E2–down-regulated promoters are known targets for rapid-signaling kinases, including AP1 (regulated by MAPKs and ERK) and NF-κB (regulated by PI3K and Akt) (18, 55). Differences in rapid signaling could also contribute to tissue-specific distributions of ERα binding to chromatin, because many of the TFs whose sites are enriched in aorta or liver ERBSs (including AP1, C/EBP, CREB, and ERα itself) are also targets of rapid signaling. Note that it is possible that some of the E2-regulated promoters that are >50 kb away from the nearest ERα binding site could be controlled by chromatin-bound ERβ, which might be consistent with the moderate enrichment of EREs in aorta down-regulated promoters.
Using normalized RNA-Seq count data to estimate relative mRNA levels, we examined whether the differential enrichment of TFBCs in aorta vs liver E2-regulated promoters or ERBSs could be explained by differences in expression of the corresponding TFs (Supplemental Table 7). In several cases, enrichment did correlate with tissue expression levels. For instance, AML1 group TFBCs show specific enrichment in aorta E2-down-regulated promoters, consistent with an 8.3- and 3.7-fold higher mRNA level for the Runx1 and Cbfb AML1 subunits in aorta relative to liver. Similarly, higher expression of AP4, HEB and KAISO, NF-κB, and OLF1 proteins in aorta and of HNF4 in liver may help to explain the tissue specific enrichments of TFBCs for these factors in E2-regulated promoters or ERBSs. In most other cases, though, expression differences do not appear to directly relate to TFBC enrichment, and in a few cases TF expression anticorrelates with TFBC enrichment (such as the promoter enrichment of PPARα sites in aorta despite the higher expression of PPARα in liver). Effects such as this could potentially be mediated by tissue-specific differences in rapid signaling pathways downstream of membrane bound ERα. Indeed, we find generally higher levels of striatin, Akt, ERK, jnk, and most PI3K mRNAs in aorta, consistent with the hypothesis that rapid signaling may play a greater role in gene regulation in aorta. On the other hand, specialized rapid signaling pathways may be stronger in liver, as suggested by the much greater expression of mapk15 and pik3c2g mRNAs in liver.
The strongest gene regulatory effect we observed was the down-regulation of inflammatory genes by E2 in aorta, with weaker or mixed effects in liver. Because inflammation plays a central role in promoting CVD, particularly atherosclerosis, this aorta-specific effect could be a major part of the mechanism by which estrogen protects against CVD. We found enrichment of TFBCs for 3 functionally interacting inflammatory TFs in aorta down-regulated genes, NF-κB, AP1 (a mediator of inflammation that can work in concert with NFκB) (56) and AML1 (which cooperates with NF-κB in myeloid tumors) (57). These observations imply that E2 mediates aorta-specific repression of the AP1/NF-κB/AML1-mediated induction of inflammatory genes. Because there was low or no enrichment of ERBSs near aorta E2–down-regulated promoters, this effect probably does not involve ERα recruitment to NF-κB sites but, rather, may be mediated by rapid signaling to PI3K, Akt, and MAPK pathways, which have been shown to mediate further activation of NF-κB after its release from IκBs and which also regulate AP1 activity (18, 55, 58–60).
In summary, we find that the genes that respond to estrogen in mouse aorta differ almost completely (in identity and likely function) from those that respond to estrogen in mouse liver. These distinct E2-regulated transcriptomes are likely to be mediated, in part, by differences in the number and distribution of in ERα binding sites between these 2 tissues. This difference in distribution is likely to be mediated by differences in factors that recruit ERα to its binding sites, because distinct sets of TFBCs are found to be associated with liver and aorta ERBSs. E2-regulated genes in aorta are generally far away from ERBSs but show high enrichment for the binding sites of several factors that are regulated by kinases activated via rapid signaling through membrane-bound ER. This and other observations suggest that many of the differences between ERα cistromes and E2-regulated transcriptomes in aorta vs liver may arise from differential effects of rapid signaling on the activities of ER-recruiting factors, and on promoter-bound TFs that may regulate transcription independent of ERα binding to chromatin. Importantly, by showing that the transcriptional response to estrogen is likely to be mediated by different mechanisms in different tissues, this study gives support to the idea that the beneficial effects of estrogen in the vasculature can be uncoupled from its harmful effects in other tissues. This study also identifies TFs that may mediate the aorta- and liver-specific responses to estrogen, which may represent targets for pharmacological therapies designed to confer the positive effects of estrogen in preventing CVD without also increasing the risk of thrombosis and cancer.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr Jennifer Cook (Dana Farber, Boston, Massachusetts) for helpful suggestions for the study and articles. We also thank Mark Aronovitz, Heather Nickerson, and Tanya Kershaw at the Mouse Physiology Core (Tufts Medical Center, Boston, Massachusetts) for their expert assistance with mouse surgeries.
This work was supported by the National Institutes of Health (Grant HL107964 to G.R.S. and T32 postdoctoral fellowship support to F.K.G. and C.V. [Grant HL69770, Richard H. Karas, Principal Investigator]).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ao
- aorta
- E
- E2
- E2
- 17β-estradiol
- ChIP-Seq
- chromatin immunoprecipitation sequencing
- ER
- estrogen receptor
- ERBS
- ERα binding site
- ERE
- estrogen response element
- IPA
- Ingenuity Pathway Analysis
- Li
- liver
- PI3K
- phosphatidylinositol 3-kinase
- qRT-PCR
- quantitative RT-PCR
- RNA-Seq
- RNA sequencing
- STAMP
- similarity, tree-building, and alignment of DNA motifs and profiles
- STORM
- Search Tool for Occurrences of Regulatory Motifs
- TF
- transcription factor
- TFBC
- transcription factor binding site consensus sequences
- TSS
- transcription start site
- V
- vehicle.
References
- 1. Mendelsohn ME, Karas RH. HRT and the young at heart. N Engl J Med. 2007;356:2639–2641. [DOI] [PubMed] [Google Scholar]
- 2. Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. [DOI] [PubMed] [Google Scholar]
- 3. Shifren JL, Schiff I. Role of hormone therapy in the management of menopause. Obstet Gynecol. 2010;115:839–855. [DOI] [PubMed] [Google Scholar]
- 4. Parker WH, Broder MS, Chang E, et al. Ovarian conservation at the time of hysterectomy and long-term health outcomes in the nurses' health study. Obstet Gynecol. 2009;113:1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivera CM, Grossardt BR, Rhodes DJ, et al. Increased cardiovascular mortality after early bilateral oophorectomy. Menopause. 2009;16:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591–2602. [DOI] [PubMed] [Google Scholar]
- 7. Studd J. Ten reasons to be happy about hormone replacement therapy: a guide for patients. Menopause Int. 2010;16:44–46. [DOI] [PubMed] [Google Scholar]
- 8. North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause 2010;17:242–255. [DOI] [PubMed] [Google Scholar]
- 9. Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. Estrogen receptor-α mediates the protective effects of estrogen against vascular injury. Circ Res. 2002;90:1087–1092. [DOI] [PubMed] [Google Scholar]
- 10. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-α but not estrogen receptor-β. Circulation. 2001;103:423–428. [DOI] [PubMed] [Google Scholar]
- 11. Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor α is a major mediator of 17β-estradiol's atheroprotective effects on lesion size in Apoe−/− mice. J Clin Invest. 2001;107:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Y, Bian Z, Lu P, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science. 2002;295:505–508. [DOI] [PubMed] [Google Scholar]
- 13. Demissie S, Cupples LA, Shearman AM, et al. Estrogen receptor-α variants are associated with lipoprotein size distribution and particle levels in women: the Framingham Heart Study. Atherosclerosis. 2006;185:210–218. [DOI] [PubMed] [Google Scholar]
- 14. Peter I, Kelley-Hedgepeth A, Huggins GS, et al. Association between arterial stiffness and variations in oestrogen-related genes. J Hum Hypertens. 2009;23:636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer MR, Haas E, Prossnitz ER, Barton M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol Cell Endocrinol. 2009;308:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–14743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. [DOI] [PubMed] [Google Scholar]
- 19. Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernelot Moens SJ, Schnitzler GR, Nickerson M, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. 2012;126:1993–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M. Unique ERα cistromes control cell type-specific gene regulation. Mol Endocrinol. 2008;22:2393–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe H, Suzuki A, Goto M, Lubahn DB, Handa H, Iguchi T. Tissue-specific estrogenic and non-estrogenic effects of a xenoestrogen, nonylphenol. J Mol Endocrinol. 2004;33:243–252. [DOI] [PubMed] [Google Scholar]
- 23. Ahlbory-Dieker DL, Stride BD, Leder G, et al. DNA binding by estrogen receptor-α is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. 2009;23:1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boverhof DR, Fertuck KC, Burgoon LD, Eckel JE, Gennings C, Zacharewski TR. Temporal- and dose-dependent hepatic gene expression changes in immature ovariectomized mice following exposure to ethynyl estradiol. Carcinogenesis. 2004;25:1277–1291. [DOI] [PubMed] [Google Scholar]
- 25. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramskold D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 2009;5:e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schnoes KK, Jaffe IZ, Iyer L, et al. Rapid recruitment of temporally distinct vascular gene sets by estrogen. Mol Endocrinol. 2008;22:2544–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Lone R, Knorr K, Jaffe IZ, et al. Estrogen receptors α and β mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol. 2007;21:1281–1296. [DOI] [PubMed] [Google Scholar]
- 32. Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schones DE, Smith AD, Zhang MQ. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 35. Mahony S, Auron PE, Benos PV. DNA familial binding profiles made easy: comparison of various motif alignment and clustering strategies. PLoS Comput Biol. 2007;3:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci (Lond). 2011;121:43–55. [DOI] [PubMed] [Google Scholar]
- 37. Karakas M, Koenig W. CRP in cardiovascular disease. Herz. 2009;34:607–613. [DOI] [PubMed] [Google Scholar]
- 38. da Costa Martins P, García-Vallejo JJ, van Thienen JV, et al. P-selectin glycoprotein ligand-1 is expressed on endothelial cells and mediates monocyte adhesion to activated endothelium. Arterioscler Thromb Vasc Biol. 2007;27:1023–1029. [DOI] [PubMed] [Google Scholar]
- 39. Guo F, Zarella C, Wagner WD. STAT4 and the proliferation of artery smooth muscle cells in atherosclerosis. Exp Mol Pathol. 2006;81:15–22. [DOI] [PubMed] [Google Scholar]
- 40. Aoki S, Hayakawa M, Ozaki H, et al. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol Cell Biochem. 2010;335:75–81. [DOI] [PubMed] [Google Scholar]
- 41. Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. [DOI] [PubMed] [Google Scholar]
- 42. Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25:2605–2606. [DOI] [PubMed] [Google Scholar]
- 43. Ji X, Li W, Song J, Wei L, Liu XS. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 2006;34:W551–W554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hewitt SC, Li L, Grimm SA, et al. Research resource: Whole-genome estrogen receptor α binding in mouse uterine tissue revealed by ChIP-seq. Mol Endocrinol. 2012;26:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gao H, Fält S, Sandelin A, Gustafsson JA, Dahlman-Wright K. Genome-wide identification of estrogen receptor α-binding sites in mouse liver. Mol Endocrinol. 2008;22:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Joshi SR, Ghattamaneni RB, Scovell WM. Expanding the paradigm for estrogen receptor binding and transcriptional activation. Mol Endocrinol. 2011;25:980–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grober OM, Mutarelli M, Giurato G, et al. A global analysis of estrogen receptor β binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor α for target gene regulation. BMC Genomics. 2011;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin CY, Vega VB, Thomsen JS, et al. Whole-genome cartography of estrogen receptor α binding sites. PLoS Genet. 2007;3:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stender JD, Kim K, Charn TH, et al. Genome-wide analysis of estrogen receptor α DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Welboren WJ, van Driel MA, Janssen-Megens EM, et al. ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28:1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. [DOI] [PubMed] [Google Scholar]
- 53. Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cunningham M, Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. 2011;40:66–73. [DOI] [PubMed] [Google Scholar]
- 56. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4:a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nakagawa M, Shimabe M, Watanabe-Okochi N, et al. AML1/RUNX1 functions as a cytoplasmic attenuator of NF-kappaB signaling in the repression of myeloid tumors. Blood. 2011;118:6626–6637. [DOI] [PubMed] [Google Scholar]
- 58. Müller M, Morotti A, Ponzetto C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol Cell Biol. 2002;22:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vanden Berghe W, Plaisance S, Boone E, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. [DOI] [PubMed] [Google Scholar]
- 60. Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.