

Abstract
Glucocorticoid receptor-α (GRα) and peroxisome proliferator–activated receptor-γ (PPARγ) are critical regulators of adipogenic responses. We have shown that FK506-binding protein 51 (FKBP51) represses the Akt-p38 kinase pathway to reciprocally inhibit GRα but stimulate PPARγ by targeting serine 112 (PPARγ) and serines 220 and 234 (GRα). Here, this mechanism is shown to be essential for GRα and PPARγ control of cellular adipogenesis. In 3T3-L1 cells, FKBP51 was a prominent marker of the differentiated state and knockdown of FKBP51 showed reduced lipid accumulation and expression of adipogenic genes. Compared with wild-type (WT), FKBP51 knockout (51KO) mouse embryonic fibroblasts (MEFs) showed dramatic resistance to differentiation, with almost no lipid accumulation and greatly reduced adipogenic gene expression. These features were rescued by reexpression of FKBP51 in 51KO cells. 51KO MEFs exhibited reduced fatty acid synthase activity, increased sensitivity to GRα-induced lipolysis, and reduced PPARγ activity at adipogenic genes (adiponectin, CD36, and perilipin) but elevated GRα transrepression at these same genes. A p38 kinase inhibitor increased lipid content in WT cells and also restored lipid levels in 51KO cells, showing that elevated p38 kinase activity is a major contributor to adipogenic resistance in the 51KO cells. In 51KO cells, the S112A mutant of PPARγ and the triple S212A/S220A/S234A mutant of GRα both increased lipid accumulation, identifying these residues as targets of the FKBP51/p38 axis. Our combined investigations have uncovered FKBP51 as a key regulator of adipogenesis via the Akt-p38 pathway and as a potential target in the treatment of obesity and related disorders.
Peroxisome proliferator–activated receptor-γ (PPARγ) is a critical regulator in the differentiation of precursor cells into mature adipocytes and in the proper functioning of adipose tissue as a primary depot for lipid synthesis and storage (1, 2). In vivo, this process is mediated by free fatty acids that bind and activate PPARγ, but it can be greatly augmented both in vivo and in cellular models by use of potent thiazolidinedione agonists, such as rosiglitazone (3). In contrast, adrenal glucocorticoid hormones, such as cortisol and the potent agonist dexamethasone (Dex), serve as antagonists to PPARγ action by activating the antilipogenic and prolipolytic actions of the glucocorticoid receptor (GR) (4–7). Both PPARγ and glucocorticoid receptor-α (GRα) are phosphoproteins. In the case of PPARγ, phosphorylation cascades that regulate the adipogenic actions of the receptor have been identified; these include ERK2 and c-Jun NH2-terminal kinase pathways (8–10), and, more recently, signaling by p38 MAPK (11, 12). The latter mechanism was uncovered by Bost and colleagues (11, 12), who have shown that p38 kinase activity markedly decreases during conversion of preadipocytes to adipocytes and in adipose depots of obese mice. Using p38 inhibitors or knockdown expression, they further showed that p38 served to inhibit PPARγ transcriptional activity. Thus, a key step in maintaining robust PPARγ actions in adipose tissue appears to be attenuation of the inhibitory p38 kinase pathway. In contrast, no reports exist for specific phosphorylation cascades that directly regulate GRα lipolytic actions in adipose tissue.
In our companion article (13), we provided evidence that a key mechanism regulating the antagonistic actions of GRα and PPARγ is control of the Akt-p38 kinase pathway by the molecular cochaperone protein FK506-binding protein (FKBP) 51. Work in the laboratory of Wang and others (14, 15) has shown that FKBP51 serves as a scaffold protein for the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP) that serves to attenuate Akt activation and its downstream consequences. In the companion work, it was found that FKBP51-deficient cells do indeed have elevated Akt activity, leading to increased phosphorylation and activation of p38 kinase, which is known to directly target both GRα and PPARγ (11, 16). In the case of PPARγ, elevated p38 kinase activity in the FKBP51 knockout (51KO) cells caused hyperphosphorylation at serine 112, which is inhibitory to receptor transcriptional activity. In the case of the GR, elevated p38 targeting of serines 220 and 234 was shown to stimulate GR activity.
To test the relevance of this newly discovered mechanism, we chose cellular adipogenesis models using both 3T3-L1 preadipocytes and wild-type (WT) and 51KO mouse embryonic fibroblasts (MEFs). The adipogenesis model was particularly attractive because FKBP51 is known to be a highly induced protein during adipogenesis, as first shown by McKnight and colleagues (17, 18) and more recently by the laboratory of Piwien-Pilipuk (19). The goal of our study was to evaluate whether FKBP51-mediated Akt-p38 activation would lead to altered adipogenic responses. Because cells deficient in FKBP51 are expected to have elevated GRα-mediated lipolysis activity, with reduced PPARγ-mediated proadipogenic actions, our prediction was that FKBP51-deficient cells would be resistant to adipogenic stimuli, resulting in cells with reduced capacity to accumulate lipid and with a gene profile of decreased lipogenic markers. Our results are highly consistent with this prediction and serve to identify FKBP51 as a potential new drug target in the treatment of obesity and related disorders.
Materials and Methods
Materials
Dex, HEPES, Tris, EDTA, PBS, protease inhibitor cocktail, PD169316, isobutylmethylxanthine (IBMX), Nile Red stain, dithiothreitol, and sodium chloride were all obtained from Sigma-Aldrich. Rosiglitazone was obtained from Alexis Biochemicals. Iron-supplemented bovine calf serum was from HyClone Laboratories Inc. Dialyzed fetal bovine serum (FBS) was from Denville Scientific. The Immobilon-FL polyvinylidene fluoride membrane was obtained from Millipore Corporation. Puromycin and DMEM were from Fisher Scientific. Lipofectamine 2000 transfection reagent and Opti-MEM were obtained from Invitrogen. The S112A mutant of PPARγ was a gift from Dr Mitch Lazar (University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania). The triple serine-to-alanine mutant (3AGR) of GR was a gift from Dr Michael Garabedian (New York University School of Medicine, New York, New York).
Cell lines and adipogenesis
MEFs were isolated from WT and 51KO embryonic day 13.5 embryos, as described previously (20). The knockdown of FKBP51 in 3T3-L1 cells was achieved by viral infection of 3T3-L1 cells with a specific short hairpin RNA (shRNA) for FKBP51 (GGTGAAGATATCACTACGAAGAAAGACAG). A control cell line was made using a scramble shRNA that was not specific to any known RNA sequence. Three hours after infection, cells were washed with fresh medium and allowed to recover for 48 hours, followed by selection with puromycin at 5 ng/μL for 7 days, with medium changes every 24 hours. Confirmation of FKBP51 knockdown was achieved by Western blot analysis. Cells were routinely cultured in DMEM containing 10% bovine calf serum with 1% penicillin-streptomycin. Adipogenic differentiation of 3T3-L1 cells was achieved by treatment with 250 nM Dex, 167 nM insulin, and 100 μM IBMX (DII cocktail) on day 0, 167 nM insulin on day 2, followed by continued incubation in 10% FBS until day 8. MEF differentiation was achieved by treatment with 1 μM Dex, 830 nM insulin, 100 μM IBMX, and 5 μM rosiglitazone (DIIR cocktail) on day 0, 830 nM insulin and 5 μM rosiglitazone on day 2, followed by continued incubation with 5 μM rosiglitazone in 10% FBS until day 8. For p38 MAPK inhibition, 10 μM PD169316 was added to the culture medium, which was renewed every 2 days until the end of the differentiation protocol. Upon differentiation, cells were stained with Nile Red to visualize lipid content. Densitometry was used as a direct measure of lipid content. Total RNA extracted from Nile Red–stained cells was used for real-time PCR analysis (see below).
Transfections and hormone treatments
For PPARγ gene profile studies in undifferentiated cells, WT and 51KO cells were plated on 6-well dishes in DMEM containing 10% bovine calf serum with 1% penicillin-streptomycin. At 85% to 90% confluence, cells were washed with Opti-MEM and transfected with expression plasmids for PPARγ2 and retinoid X receptor using Lipofectamine 2000 according to the manufacturer's protocol. After 6 hours, the medium was replaced with DMEM containing 10% dialyzed FBS with 1% penicillin-streptomycin. Forty eight hours posttransfection, cells were treated with 100 nM rosiglitazone for 2 hours, followed by harvest and total RNA extraction. For GRα gene profile studies, MEFs were untransfected and treated with 100 nM Dex for 2 hours, followed by harvest and total RNA extraction. For the adipogenesis experiments of Figure 7, 51KO MEFs were transfected with the S112A-PPARγ2 or the 3AGR mutants before adipogenic stimulation with DIIR cocktail.
Figure 7.

The S112A-PPARγ2 and 3AGR mutants increase lipid accumulation in 51KO cells. A, 51KO MEFs were transfected with empty vector (Con, Ctrl), WT PPARγ2, or the S112A PPARγ2 mutant, followed by Western blotting for PPARγ protein and adipogenic differentiation and Nile Red staining of lipid content. B, 51KO MEFs were transfected with empty vector, WT GR, or the 3AGR mutant, followed by Western blotting for GRα protein and adipogenic differentiation and Nile Red staining of lipid content. Hsp90 was used as a loading control. Images of stained adipocytes are representative of 3 independent experiments. Quantitation of lipid content by optical density (±SEM; n = 3).
Quantitative real-time PCR analysis
Total RNA was extracted from mouse tissues using a 5-Prime PerfectPure RNA Tissue Kit (Fisher Scientific). Total RNA was read on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and cDNA was synthesized using an iScript cDNA Synthesis kit (Bio-Rad Laboratories). PCR amplification of the cDNA was performed by quantitative real-time PCR using a qPCR core kit for SYBR Green I (Applied Biosystems). The thermocycling protocol consisted of 5 minutes at 95°C, 48 cycles of 15 seconds at 95°C, and 30 seconds at 60°C, finished with a melting curve ranging from 60 to 95°C to allow distinction of specific products. Primers specific to each gene were designed using Primer Express 3.0 software (Applied Biosystems). Normalization was performed in separate reactions with primers to 18S mRNA (TTCGAACGTCTGCCCTATCAA and ATGGTAGGCACGGCGACTA). All primer sequences were uploaded to the primer database at http://www.primerfinder.com/.
Whole-cell extraction
Cells were washed and collected in PBS followed by centrifugation at 1500 × g for 10 minutes. The supernatant was discarded, and the pellet was resuspended in PBS. After a short spin at 20 800 × g for 5 minutes at 4°C, the pellet was rapidly frozen on a dry ice-ethanol mix and stored at −80°C overnight. The frozen pellet was resuspended in 3 volumes of cold whole-cell extract buffer (20 mM HEPES, 25% glycerol, 0.42 M NaCl, and 0.2 mM EDTA, pH 7.4) with protease and phosphatase inhibitors (sodium orthovanadate and sodium fluoride) and incubated on ice for 10 minutes. The samples were centrifuged at 100 000 × g for 5 minutes at 4°C. Protein levels were measured spectrophotometrically by a SpectraMax Plus spectrophotometer (Molecular Devices). The supernatants were used immediately for Western blot analysis.
Gel electrophoresis and Western blotting
Protein samples were resolved by SDS-PAGE and electrophoretically transferred to Immobilon-FL membranes. Membranes were blocked at room temperature for 1 hour in Tris-buffered saline (TBS) (10 mM Tris-HCl [pH 7.4] and 150 mM NaCl) containing 3% BSA plus phosphatase inhibitors. Incubation with the primary antibody was done overnight at 4°C. After 3 washes in TBST (TBS plus 0.1% Tween 20), membranes were incubated with infrared anti-rabbit (IRDye 800, green), anti-mouse (IRDye 680, red), or anti-goat (IRDye 800, green) secondary antibodies (LI-COR Biosciences) at 1:15 000 dilution in TBS for 2 hours at 4°C. Immunoreactivity was visualized and quantified by infrared scanning in the Odyssey system (LI-COR Biosciences). Antibodies against FKBP51 (sc-11518), FKBP52 (sc-1803), GAPDH (sc-32233), and GR (sc-12763) were obtained from Santa Cruz Biotechnologies; protein phosphatase-5 (PP5) was a gift from Michael Chinkers (University of South Alabama College of Medicine, Mobile, Alabama). Antibodies against cyclophilin-40 (Cyp40; PA3-022) were obtained from Thermo Scientific. Anti-FLAG M2 monoclonal IgG (F-3165) was from Sigma-Aldrich.
Fatty acid synthase (FAS) assay
FAS activity was assayed by the incorporation of radiolabeled malonyl-coenzyme A (CoA) into palmitate. In brief, cells were lysed in buffer (20 mm Tris [pH 7.5], 1.0 mm EDTA, and 1.0 mm dithiothreitol) containing phosphatase and protease inhibitors and centrifuged (12 000 × g for 30 minutes at 4°C). Lysates were incubated (20 minutes at 37°C) with 166.6 μM acetyl-CoA, 100 mM potassium phosphate (pH 6.6), 0.1 μCi of [14C]malonyl-CoA, and 25 nM malonyl-CoA in the absence or presence of 500 μM NADPH. The reaction was stopped with a 1:1 chloroform-methanol solution, mixed (30 minutes at 20°C), and centrifuged (12 500 × g for 30 minutes). The supernatant was vacuum dried, and the pellet was resuspended in 200 μL of water-saturated butanol. After addition of 200 μL of double-distilled H2O, vortexing, and spinning (1 minute), the upper layer was removed for reextraction. The butanol layer was dried and counted. Protein was quantified by the Bio-Rad protein assay, and results are expressed as relative counts per minute of 14C incorporated per microgram of cell lysate.
Statistical analysis
Data were analyzed with Prism 5 (GraphPad Software) using ANOVA combined with the Tukey posttest to compare pairs of group means or unpaired t tests. Values of P ≤ .05 were considered statistically significant.
Results
FKBP51 is essential to cellular adipogenesis
FKBP51 has been identified as a highly induced protein during the proliferative phase of 3T3-L1 cell differentiation into adipocytes (17). However, the functional role of FKBP51 in adipocyte generation has yet to be defined. To establish the relevance of FKBP51 in our system, Western blot and real-time PCR analyses of several steroid receptor–associated cochaperones were performed in 3T3-L1 cells before and after terminal differentiation with the standard Dex, insulin, and IBMX (DII) cocktail. Figure 1 shows highly elevated expression of FKBP51 protein and moderate expression of protein phosphatase-5 (PP5) upon differentiation but dramatically reduced FKBP52 and Cyp40. Like FKBP51 (see the companion article [13]), PP5 has also been shown to negatively regulate GRα, while stimulating PPARγ. Thus, 2 cochaperones with similar functionality on GRα and PPARγ receptors appear to be necessary components of mature adipocytes. In WT MEFs, a large increase in FKBP51 mRNA expression was also seen upon differentiation (data not shown).
Figure 1.
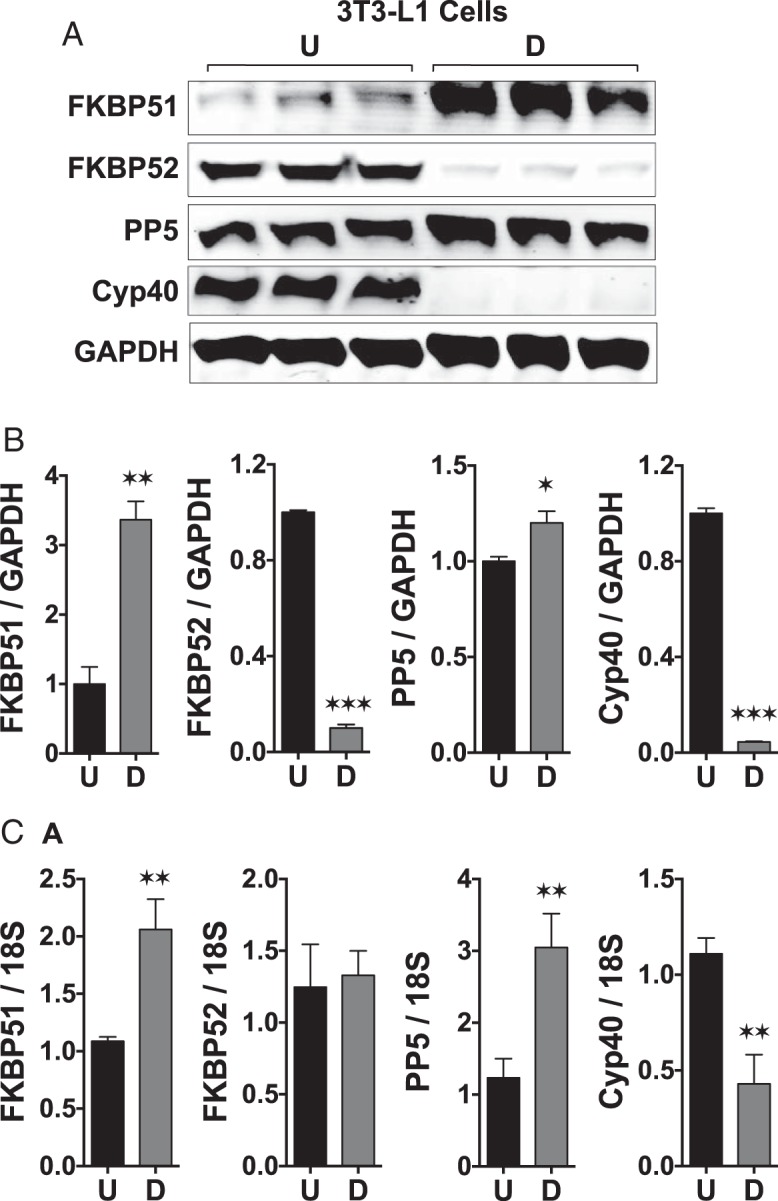
FKBP51 is a marker of adipocyte differentiation. A, Western-blot analysis of FKBP51, FKBP52, PP5, Cyp40, and GAPDH in undifferentiated (U) and differentiated (D) 3T3–L1 cells. B, Densitometry analysis of panel A. C, Real-time PCR analysis of the indicated genes. Transcript expression was normalized to 18S mRNA. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001. All data represent ±SEM (n = 3).
As a first step of FKBP51 functionality, stable knockdown of FKBP51 expression (51KD) in 3T3-L1 cells was achieved via lentiviral delivery of shRNA. An approximately 40% reduction in FKBP51 protein was observed (see Figure 3A in the companion article [13]). In response to adipogenic stimuli, an approximately 40% reduction in lipid accumulation was also seen in 51KD cells compared with that in WT controls (Figure 2A). Real-time PCR analysis (Figure 2B) revealed that most adipogenic markers were also reduced (adiponectin, adipocyte P2 lipid-binding protein, CD36, phosphoenolpyruvate carboxykinase, perilipin, and lipoprotein lipase), whereas one (FAS) was not significantly reduced. Interestingly, uncoupling protein (UCP) 1 mRNA was significantly increased in the stimulated 51KD cells, suggesting a state of elevated energy expenditure.
Figure 3.

51KO cells are resistant to lipid accumulation and have elevated Dex-induced lipolysis. A, Western blot analysis of WT and 51KO MEFs demonstrating a complete lack of FKBP51 in the 51KO cells and restoration of FKBP51 expression after rescue expression of FLAG-FKBP51 (KO-R). Hsp90 was used as loading control. B, Detection of lipid accumulation in undifferentiated (U) and differentiated (D) WT, 51KO, and KO-R cells by Nile Red staining. Phase-contrast images are also shown. C, Quantitation of intracellular lipid in cells treated in panel B by optical density (±SEM; n = 3–6). D, FAS enzymatic activity in differentiated WT and 51KO cells (±SEM; n = 6). E, Nile Red staining of lipid content in WT and 51KO cells subjected to Dex treatment every medium change during differentiation (Dex ET). F, Quantitation of intracellular lipid in cells treated in panel E by optical density (±SEM; n = 6). Note the reduced y-axis range compared with that in panel C. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO.
Figure 2.

Stable knockdown of FKBP51 decreases 3T3–L1 differentiation and adipogenic marker expression. A, 3T3–L1 preadipocytes were infected with lentiviral constructs for scrambled (Scr) or FKBP51 shRNA to achieve stable knockdown of FKBP51 protein expression (51KD). Infected cells were induced to differentiate with DII cocktail, followed by Nile Red staining for lipid content at day 8 postinduction. Lipid content was quantified by optical density. Phase-contrast images are also shown. B, Real-time PCR analysis of the indicated adipogenic markers of Scr and 51KD 3T3–L1 cells after differentiation. Transcript levels were normalized to 18S mRNA. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001. All data represent ±SEM (n = 3). aP2, adipocyte P2 lipid-binding protein; LPL, lipoprotein lipase; PEPCK, phosphoenolpyruvate carboxykinase.
As a next step, WT and 51KO MEFs were stimulated to differentiate using the DII cocktail plus rosiglitazone (DIIR). 51KO cells were even more resistant to stimuli-induced lipid accumulation than 51KD 3T3-L1 cells, and this resistance was completely reversed by rescue expression of FKBP51 (Figure 3, A–C). Interestingly, 51KO cells had reduced FAS enzymatic activity (Figure 3D). Gene profiling in these cells revealed greatly reduced expression of adiponectin, CD36, and perilipin in the stimulated 51KO compared with WT cells (Figure 4), as well as moderately reduced lipoprotein lipase and FAS. Interestingly, PPARγ levels were higher in the treated KO cells, suggesting a possible compensatory mechanism for reduced PPARγ activity.
Figure 4.
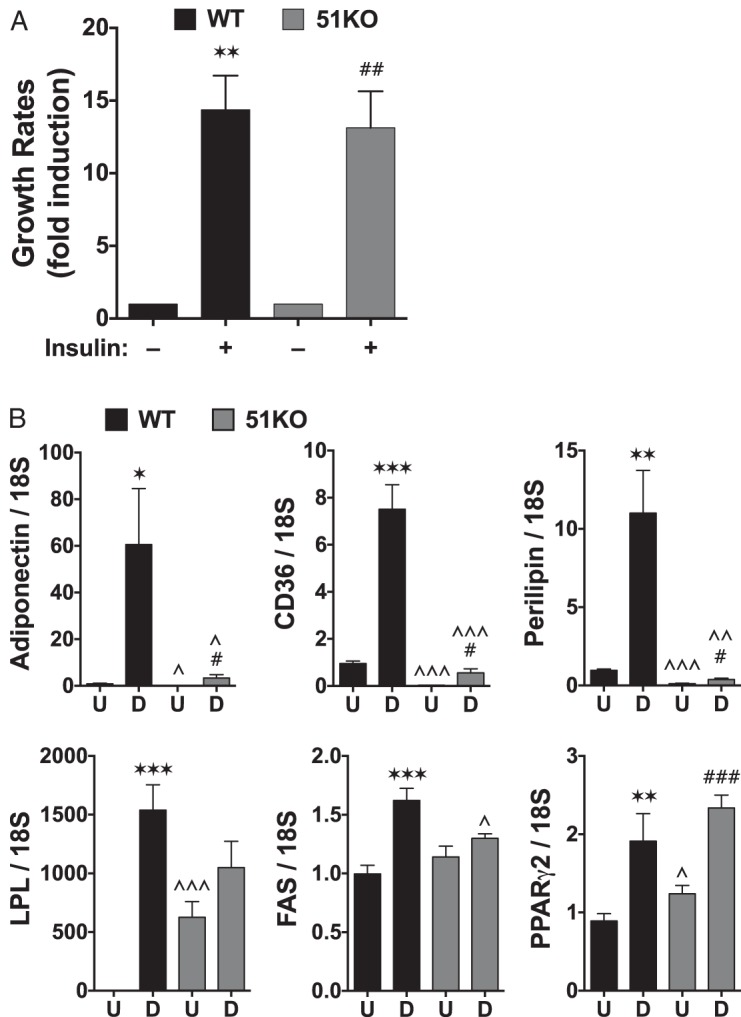
FKBP51 does not affect insulin-induced proliferation but is required for expression of adipocytic genes. A, WT and 51KO MEFs were equally plated and grown in dialyzed FBS for 24 hours before treatment with 100 nM insulin. Growth was measured 24 hours later by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoline bromide] assay, as described previously (32) (±SEM; n = 3). B, Real-time PCR analysis of undifferentiated (U) and differentiated (D) WT and 51KO cells at the indicated genes (±SEM; n = 6). Transcript expression was normalized to 18S mRNA. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO. LPL, lipoprotein lipase.
51KO cells exhibit elevated GR but reduced PPARγ activities at metabolic genes
Although Dex is thought to stimulate the early steps of adipogenic differentiation (21), in MEFs, we have shown that lipid accumulation is unaffected if Dex is left out of stimulatory cocktails that contain rosiglitazone (7). Indeed, in mature adipose tissue and cells, Dex (glucocorticoid) exposure causes lipolysis as the primary response (4–7). This was tested in our system by exposing differentiating WT and 51KO MEFs to continual Dex exposure (Figure 3, E and F). The results show greatly reduced lipid levels in WT cells exposed to chronic Dex compared with those in WT cells with the single acute Dex treatment at day 0 (0.4-fold vs 10-fold lipid, respectively). In 51KO cells, chronic Dex reduced lipid content even further than in control 51KO cells (0.05-fold vs 0.1-fold lipid, respectively). These results suggest elevated GRα lipolytic activity in the 51KO cells.
To accurately assess the contribution of FKBP51 to both GRα and PPARγ activities at metabolic genes, we used WT and 51KO MEFs grown in charcoal-stripped and dialyzed serum. In this way, the confounding effects of serum steroids and growth factors present in the DII cocktail media can be avoided. In response to Dex, GR control of progluconeogenic genes (pyruvate dehydrogenase kinase-4, phosphoenolpyruvate carboxykinase, glucose-6-phosphatase) occurs by the transactivation mechanism and elevated transactivation of these genes is seen in the 51KO cells (Figure 5A). In WT cells, GR genomic control of lipolysis appears to occur principally by transrepression of proadipogenic genes (adiponectin, CD36, and perilipin). In KO cells, increased transrepression activity by GR was observed for 2 of these genes (CD36 and adiponectin), and a reduction in basal expression was also found for these genes. In response to rosiglitazone, WT cells showed increased expression of adiponectin, CD36, and perilipin, but this activity was almost completely lost in the 51KO cells (Figure 5B). A similar pattern of reduced expression in 51KO cells was seen for rosiglitazone induction of uncoupling proteins (UCP1, UCP2, and UCP3).
Figure 5.
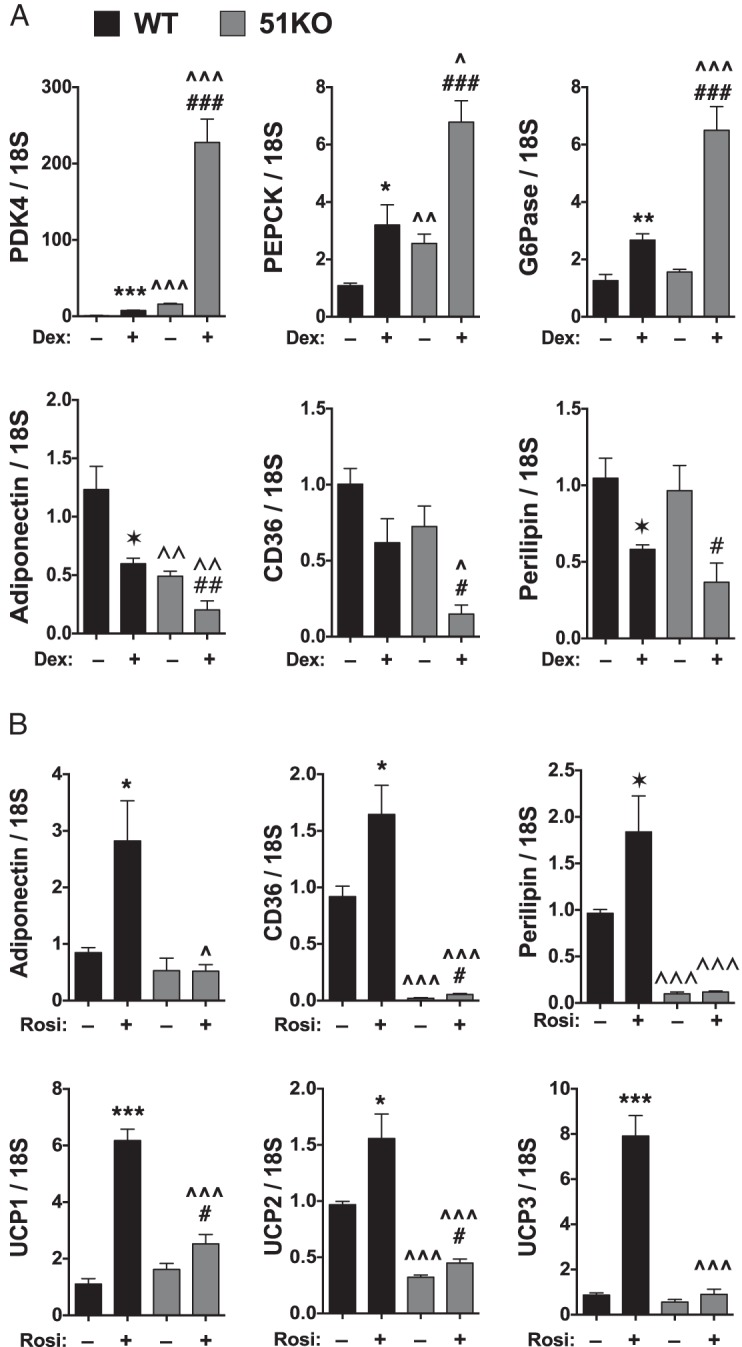
FKBP51 is a reciprocal regulator of GRα and PPARγ at metabolic genes. A, WT and 51KO MEFs were treated with or without 100 nM Dex for 2 hours followed by real-time PCR analysis of the indicated genes (±SEM; n = 6). B, WT and 51KO MEFs transfected with PPARγ2 were treated with or without rosiglitazone (Rosi) for 2 hours followed by real-time PCR of the indicated genes (±SEM; n = 6). All transcript expression was normalized to 18S mRNA. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO. PDK4, pyruvate dehydrogenase kinase; PEPCK, phosphoenolpyruvate carboxykinase; G6Pase, glucose-6- phosphatase.
Adipogenic resistance in 51KO cells is mediated by p38 kinase targeting serine 112 of PPARγ and serines 220 and 234 of GRα
In our companion article (13), we demonstrated that loss of FKBP51 caused activation of Akt and its downstream target p38 kinase, leading to elevated phosphorylation of GRα at serines 220 and 234 and PPARγ at serine 112 to promote increased GRα but decreased PPARγ activities, respectively. To determine the role of this mechanism in adipogenic resistance in 51KO cells, we first asked whether elevated p38 kinase activity was a contributing factor by treating WT and 51KO cells with the p38 kinase inhibitor PD169316 (Figure 6). The results show elevated lipid accumulation in WT cells treated with PD169316, confirming that basally active p38 kinase activity is an attenuating signal in adipogenesis and lipid accumulation (11, 12). More importantly, PD169316 treatment of 51KO cells resulted in a complete rescue of lipid accumulation to the levels seen in untreated WT cells, showing that elevated p38 kinase activity in the KO cells is a major contributor to the resistant phenotype.
Figure 6.
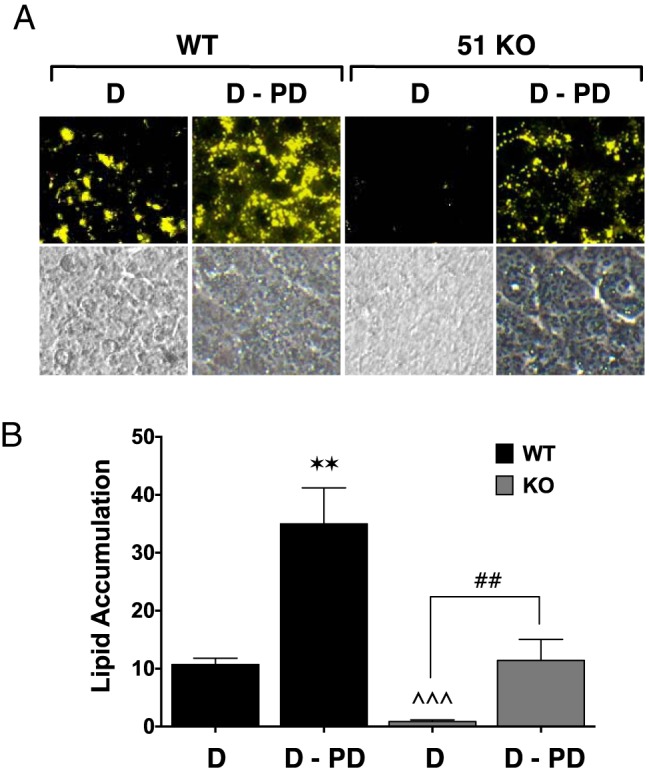
Rescue of lipid accumulation in 51KO cells by the p38 kinase inhibitor PD169316. A, WT and 51KO MEFs were differentiated by treatment with the DIIR mixture in the absence (D) or presence of PD169316 inhibitor (D-PD) specific to p38 MAPK, followed by staining for lipid content with Nile Red. Phase-contrast images are also shown. Images of stained adipocytes are representative of 3 independent experiments B, Optical density quantitation of intracellular lipid in cells treated in panel A (±SEM; n = 3). Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO.
To determine the roles of the phosphorylated residues targeted by p38, serine-to-alanine mutants of each receptor were used. In Figure 7A, 51KO cells were transfected with WT PPARγ or the S112A mutant known to have elevated basal and rosiglitazone-induced adipogenic activity (22, 23). After transfection, the 51KO cells were subjected to adipogenic stimulation with DIIR cocktail. It can be seen that transfection of WT PPARγ did not increase lipid accumulation in the 51KO cells, demonstrating that even overexpression levels of WT PPARγ are inactive in these cells. In contrast, the S112A mutant caused a large increase in the lipid content in the 51KO cells, showing that phosphorylation at serine 112 is the basis for PPARγ inactivity in the absence of FKBP51. Similar experiments were performed with the human GR mutant (3AGR) in which 3 serine phosphorylation sites, 203, 211, and 226 (equivalent to mouse serines 212, 220, and 234), were mutated to alanine (Figure 7B). The GR3A mutant is known to have reduced hormone-induced transcriptional activity (24, 25). Like S112A PPARγ, expression of 3AGR in the 51KO cells also caused lipid accumulation to increase.
Discussion
In this work, we show that FKBP51 is a important regulator of cellular adipogenesis that is necessary for the complete transformation of preadipocytes into mature, lipid-bearing cells. Key data supporting this conclusion include the inability of 2 cell lines (3T3-L1 and MEF) to differentiate into lipid-laden cells in the absence of FKBP51 that correlated with reduced enzymatic activity by FAS and a corresponding decrease in the expression of lipogenic genes. Of particular interest is the reduced expression observed for CD36, an essential importer of fatty acids in mature adipocytes. After adipogenic stimulation, expression of CD36 was greatly reduced in FKBP51-deficient 3T3-L1 cells and MEFs. In this work and in our companion study (13), we also show that FKBP51 exerts this control over adipogenesis by serving as a positive regulator of PPARγ, the dominant proadipogenic factor controlling this process, and as a negative regulator of GR, whose activities are known to antagonize PPARγ actions by promoting lipid mobilization and secretion from adipocytes. Observations supporting this conclusion include increased susceptibility of 51KO cells to the lipolytic actions of glucocorticoids and the reciprocal effect that FKBP51 loss had on Dex-induced GR and rosiglitazone-induced PPARγ activities at 3 important lipometabolic genes, adiponectin, perilipin, and CD36. In 51KO cells, PPARγ induction of the 3 genes was greatly reduced, whereas transrepression by GR was greatly increased. It is not surprising, therefore, that 51KO MEFs are almost completely incapable of importing and/or storing lipid due to the 2-fold effect that FKBP51 exerts over GR and PPARγ. This study also suggests that the dominant mechanism by which glucocorticoids promote adipose lipolysis is through the transrepressive actions of GR.
Although FKBP51 interacts with heat shock protein 90 (Hsp90)–containing PPARγ heterocomplexes, it appears that the principal mechanism by which FKBP51 controls GRα and PPARγ activities is through activation of the Akt-specific phosphatase PHLPP and down-regulation of the Akt-p38 MAPK pathway (13–15). This leads to elevated p38-mediated phosphorylation of each receptor in 51KO cells and reciprocal regulation of their activities. In this work, we demonstrate the significance of this mechanism to adipogenesis by showing that a specific p38 inhibitor can rescue lipid accumulation in the 51KO cells and that mutant forms of each receptor missing the targeted phosphoserines also elevated lipid content in the KO cells. These results show that elevated p38 activity targeting specific serines on each receptor is the primary reason for resistance to lipid accumulation in the 51KO cells. It should be noted that even overexpression of WT-PPARγ did not increase lipid content in the KO cells, indicating high levels of p38 kinase activity. Taken our results as a whole, we can now propose the model seen in Figure 8 in which FKBP51/Akt/p38 signaling to GRα and PPARγ is a central pathway controlling adipogenesis.
Figure 8.
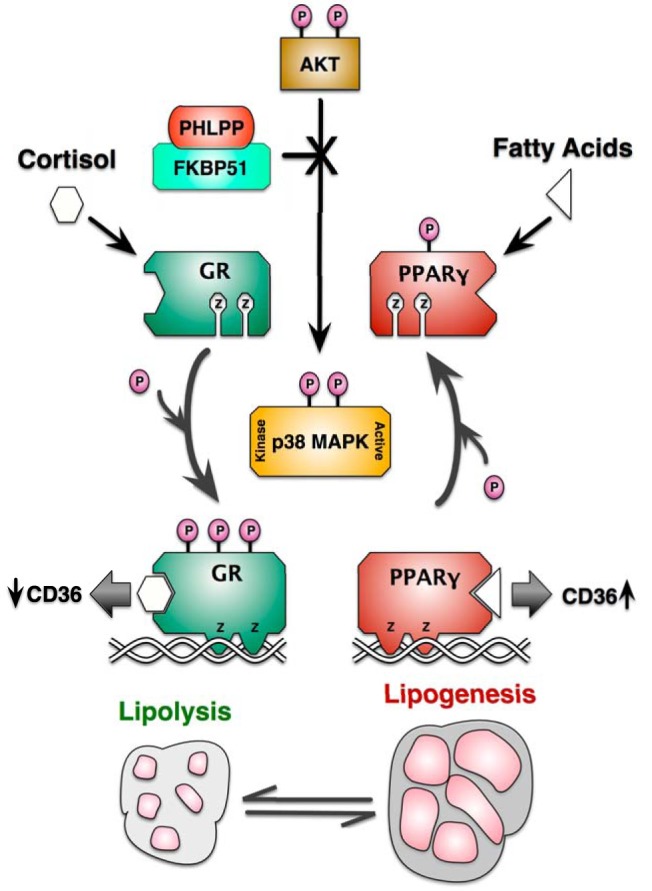
Model for FKBP51 control of lipid metabolism via Akt/p38 targeting of GRα and PPARγ. Based on this work and our companion article (13), it is proposed that FKBP51 is essential to adipogenesis and the balance between lipolysis and lipogenesis by regulating phosphorylation targeting of GRα and PPARγ by the Akt-p38 MAPK pathway. According to the model, FKBP51 activates the Akt-specific phosphatase PHLPP to inactivate downstream p38 kinase. Because p38 serves to promote the lipolytic actions of GRα, while inhibiting the lipogenic actions of PPARγ, FKBP51 can be viewed as a proadipogenic factor essential to lipid accumulation in mature adipocytes. This action is best exemplified by the ability of FKBP51 loss to decrease the transrepressive properties of GR at the CD36 gene (an essential fatty acid importer of adipocytes), while simultaneously increasing PPARγ-mediated transcription of the same gene.
Our studies also serve to explain the long-known observation that FKBP51 is a prominent marker of mature adipocytes (17). In our hands, the transition from preadipocyte to mature adipocyte in 3T3-L1 cells led to an approximately 3.5-fold increase in FKBP51 protein on day 8 of differentiation. The earlier study by Yeh et al (17) identified FKBP51 as a marker that was only transiently expressed during the proliferative phase of differentiation (days 2–4), with FKBP51 protein levels returning to normal by day 8. Although this discrepancy might be due to any number of factors (such as differences in the adipogenic cocktails used), it is probable that FKBP51 expression in our cells is indeed higher at the earlier proliferative phase time points. However, it does not appear that the main function of FKBP51 is to control proliferation, as 51KO cells showed no obvious alteration in cell density by phase-contrast imaging of differentiated cells at day 8 and had the same growth rates in response to insulin as WT cells. Because FKBP51 has intrinsic peptidyl-prolyl cis-trans isomerase (PPIase) activity, Yeh et al (18) also investigated the effects of PPIase inhibitors on adipogenesis potential in 3T3-L1 cells. Interestingly, rapamycin was found to completely block adipogenic conversion, whereas FK506 did not. Because both compounds inhibit the PPIase function of FKBP51, it was concluded that FKBP51 was not an essential factor in adipogenesis. Yet, we now know that the PPIase functions of FKBP51 and the closely related FKBP52 are completely irrelevant to their effects on steroid receptor action (26). Instead, it appears that FKBP51 provides a more general chaperone or scaffolding function that is key to regulating not only the steroid receptor and Akt-p38 kinase pathways but also adipogenesis. Because lipolysis at adipose cells is also controlled by catecholamines, such as norepinephrine, it is possible that FKBP51 may control this pathway as well. At present, however, no precedents exist for FKBP51 regulation of β-adrenergic receptor signaling, protein kinase A activity, or cross-talk between p38 kinase and protein kinase A. Further studies will be needed to address this possibility.
Differentiated 3T3-L1 cells also exhibited increased expression of PP5 protein, another nuclear receptor cochaperone that has very similar effects on both adipogenesis and the actions of GRα and PPARγ (7). In the case of PP5, we showed that it controlled dephosphorylation of GRα and PPARγ at the same serine residues, leading to negative regulation of the former, but activation of the latter. PP5-deficient cells were also resistant to adipogenic conversion principally because of a lack of PPARγ activity and, to a lesser degree, elevated GR-mediated lipolysis. The question then arises whether PP5 and FKBP51 are redundant regulators of adipogenesis mediated by GRα and PPARγ or whether they act independently. Although we have not yet investigated this question, it seems reasonable to predict that they are independent regulators. In the case of PP5, the major known mechanism for its activation is either binding to Hsp90 via its TPR domain or binding of the TPR domain by certain polyunsaturated fatty acids, such as arachidonic acid (27–29). It is likely, therefore, that the major signal for PP5 control of GRα and PPARγ in adipocytes is a dietary signal, such as elevated fatty acids after meals. FKBP51 is not known to bind fatty acids nor does it have intrinsic phosphorylation activity. Instead, it appears to be central to control of Akt signaling and thus is most likely to be controlled by insulin or other growth factors. Although no evidence as yet exists for growth factor–induced targeting of FKBP51, an intriguing possibility is that it acts as an autologous inhibitor of GR. FKBP51 has been known as a negative regulator of GR activity for many years, but it is also one of the most highly induced GR target genes in most cells (30, 31). It has been speculated that it must serve as an intracellular, negative feedback mechanism to attenuate overstimulation of GR. To date, however, no cellular or physiological models have been found to implicate this mechanism. Our studies suggest that GR-induced FKBP51 expression might serve to decrease GR activity by inhibiting Akt-p38 kinase activation of the receptor, perhaps to attenuate glucocorticoid-induced lipid loss from adipocytes. Of course, our model also predicts that GR-induced FKBP51 leads to activation of PPARγ to promote lipid retention.
Acknowledgments
We are especially indebted to Dr Michael J. Garabedian (New York University School of Medicine, New York, New York) for the gift of the 3AGR mutant and to Dr Mitch Lazar (University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania) for the gift of the S112A mutant PPARγ.
This work was supported in part by Department of Physiology and Pharmacology (University of Toledo College of Medicine) funds awarded to E.R.S. and by National Institutes of Health (Grants DK70127 [to E.R.S.], DK73402 [to W.S.], and DK054254 and DK083850 [to S.M.N.]). L.A.S. was supported in part by a Center for Diabetes and Endocrine Research Hiss Foundation Fellowship. T.D.H. was supported by National Institutes of Health PRIDE Grant HL106365.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CoA
- coenzyme A
- Cyp40
- cyclophilin-40
- Dex
- dexamethasone
- FAS
- fatty acid synthase
- FBS
- fetal bovine serum
- FKBP
- FK506-binding protein
- GR
- glucocorticoid receptor
- GRα
- glucocorticoid receptor-α
- Hsp90
- heat shock protein 90
- IBMX
- isobutylmethylxanthine
- 51KD
- FKBP51 knockdown
- 51KO
- FKBP51 knockout
- MEF
- mouse embryonic fibroblast
- PHLPP
- PH domain leucine-rich repeat protein phosphatase
- PP5
- protein phosphatase-5
- PPARγ
- peroxisome proliferator–activated receptor-γ
- PPIase
- peptidyl-prolyl cis-trans isomerase
- shRNA
- short hairpin RNA
- TBS
- Tris-buffered saline
- UCP
- uncoupling protein
- WT
- wild-type.
References
- 1. Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361 [DOI] [PubMed] [Google Scholar]
- 2. Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123:993–999 [DOI] [PubMed] [Google Scholar]
- 3. Ahmed W, Ziouzenkova O, Brown J, et al. PPARs and their metabolic modulation: new mechanisms for transcriptional regulation? J Intern Med. 2007;262:184–198 [DOI] [PubMed] [Google Scholar]
- 4. Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61 [DOI] [PubMed] [Google Scholar]
- 5. Xu C, He J, Jiang H, et al. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol Endocrinol. 2009;23:1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campbell JE, Fediuc S, Hawke TJ, Riddell MC. Endurance exercise training increases adipose tissue glucocorticoid exposure: adaptations that facilitate lipolysis. Metabolism. 2009;58:651–660 [DOI] [PubMed] [Google Scholar]
- 7. Hinds TD, Jr, Stechschulte LA, Cash HA, et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ). J Biol Chem. 2011;286:42911–42922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science. 1996;274:2100–2103 [DOI] [PubMed] [Google Scholar]
- 9. Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272:5128–5132 [DOI] [PubMed] [Google Scholar]
- 10. Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aouadi M, Laurent K, Prot M, Le Marchand-Brustel Y, Binétruy B, Bost F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes. 2006;55:281–289 [DOI] [PubMed] [Google Scholar]
- 12. Aouadi M, Jager J, Laurent K, et al. p38MAP kinase activity is required for human primary adipocyte differentiation. FEBS Lett. 2007;581:5591–5596 [DOI] [PubMed] [Google Scholar]
- 13. Stechschulte LA, Hinds TD, Jr, Ghanem SS, Shou W, Najjar SM, Sanchez ER. FKBP51 reciprocally regulates GRα and PPARγ activation via the Akt-p38 pathway. Mol Endocrinol. 2014;28:1254–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pei H, Li L, Fridley BL, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fabian AK, Marz A, Neimanis S, Biondi RM, Kozany C, Hausch F. InterAKTions with FKBPs—mutational and pharmacological exploration. PLoS One. 2013;8:e57508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller AL, Webb MS, Copik AJ, et al. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583 [DOI] [PubMed] [Google Scholar]
- 17. Yeh WC, Li TK, Bierer BE, McKnight SL. Identification and characterization of an immunophilin expressed during the clonal expansion phase of adipocyte differentiation. Proc Natl Acad Sci USA. 1995;92:11081–11085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yeh WC, Bierer BE, McKnight SL. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc Natl Acad Sci USA. 1995;92:11086–11090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toneatto J, Guber S, Charo NL, et al. Dynamic mitochondrial-nuclear redistribution of the immunophilin FKBP51 is regulated by the PKA signaling pathway to control gene expression during adipocyte differentiation. J Cell Sci. 2013;126(Pt 23):5357–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yong W, Yang Z, Periyasamy S, et al. Essential role for co-chaperone Fkbp52 but not Fkbp51 in androgen receptor-mediated signaling and physiology. J Biol Chem. 2007;282:5026–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809 [DOI] [PubMed] [Google Scholar]
- 22. Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem. 1997;272:10811–10816 [DOI] [PubMed] [Google Scholar]
- 23. Camp HS, Tafuri SR, Leff T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-γ1 and negatively regulates its transcriptional activity. Endocrinology. 1999;140:392–397 [DOI] [PubMed] [Google Scholar]
- 24. Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277:26573–26580 [DOI] [PubMed] [Google Scholar]
- 25. Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riggs DL, Cox MB, Tardif HL, Hessling M, Buchner J, Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol. 2007;27:8658–8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen MS, Silverstein AM, Pratt WB, Chinkers M. The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem. 1996;271:32315–32320 [DOI] [PubMed] [Google Scholar]
- 28. Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M. Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry. 2001;40:10485–10490 [DOI] [PubMed] [Google Scholar]
- 29. Ramsey AJ, Chinkers M. Identification of potential physiological activators of protein phosphatase 5. Biochemistry. 2002;41:5625–5632 [DOI] [PubMed] [Google Scholar]
- 30. Vermeer H, Hendriks-Stegeman BI, van der Burg B, van Buul-Offers SC, Jansen M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: a potential marker for glucocorticoid sensitivity, potency, and bioavailability. J Clin Endocrinol Metab. 2003;88:277–284 [DOI] [PubMed] [Google Scholar]
- 31. Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Periyasamy S, Warrier M, Tillekeratne MP, Shou W, Sanchez ER. The immunophilin ligands cyclosporin a and FK506 suppress prostate cancer cell growth by androgen receptor-dependent and -independent mechanisms. Endocrinology. 2007;148:4716–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]