

Abstract
We showed previously that the hinge region of estrogen receptor (ER) α is involved in mediating its actions. The hinge 1 (H1) ERα mutant has disrupted nuclear localization and has lost interaction with c-JUN, but retains estrogen response element (ERE)–mediated functions. The hinge 2 + nuclear export sequence (H2NES) ERα mutant does not maintain nuclear translocation with hormone and no longer activates ERE target genes but does retain a nongenomic, nonnuclear, rapid-action response. Herein, we used the human endometrial cancer Ishikawa stable cell lines (Ishikawa/vector, Ishikawa/wild-type [WT] ERα, Ishikawa/H1 ERα, or Ishikawa/H2NES ERα) to characterize the biological activities of these 2 ERα hinge region mutants. We confirmed by confocal microscopy increased cytoplasmic ERα in the H1 ERα cell line and full cytoplasmic ERα localization in the H2NES ERα cell line. Luciferase assays using the 3xERE reporter showed activation of H1 ERα and H2NES ERα by estradiol (E2) treatment, but using the endogenous pS2 reporter, luciferase activity was only seen with the H1 ERα cell line. Examining cell proliferation revealed that only the WT ERα and H1 ERα cell lines increased proliferation after treatment. Using microarrays, we found that WT ERα and H1 ERα cluster together, whereas vector and H2NES ERα are most similar and cluster independently of E2 treatment. These studies revealed that the nongenomic activities of ERα are unable to mediate proliferative changes or the transcriptional profile after treatment and demonstrate the importance of genomic action for ERα/E2-mediated responses with the nongenomic actions of ERα being complementary to elicit the full biological actions of ERα.
Estrogen receptor (ER) α belongs to the nuclear receptor superfamily with activation by estrogen (estradiol [E2]) occurring by 3 major mechanisms of action, including (1) nuclear, genomic, direct DNA binding, (2) nuclear, genomic, “tethered”-mediated protein-protein interactions, and (3) nonnuclear, nongenomic, rapid action responses (1–8). The first mechanism involves liganded ERα bound to estrogen response elements (EREs) of target genes to mediate changes in gene expression via the “classic” ERα DNA-binding responses (4, 9). The second mechanism involves recruitment and interaction of ERα to “tether” with other transcription factors, such as c-JUN and Sp1, to form a protein-protein complex that interacts directly with the AP-1 and Sp1 DNA response elements, respectively (7, 10, 11). Last, the third mechanism, involves nonnuclear ERα that mediates rapid signaling events that include calcium mobilization, nitric oxide synthesis, and activation of intracellular kinase signaling cascades (5, 12). More recently, understanding the nonclassic mechanisms of ERα has become a focused area of research. ERα serves functions outside the nucleus involving posttranslational modifications, protein-protein interactions of the ERα with G proteins and kinases (12–15).
ERα, as a member of the nuclear receptor superfamily, maintains the classic nuclear receptor domain structure. There are 4 main domain demarcations known as A/B, C, D, and E/F. Each domain can act independently, but all domains are needed for full ERα functionality. The A/B domain is the hormone-independent activation function domain (AF-1), the C-domain is where DNA binding (DNA binding domain [DBD]) occurs, the D-domain or the hinge region contains NLSs, and the E/F domain is the ligand-binding domain and the hormone-dependent activation function (AF-2) (1, 16, 17). Originally the D-domain of ERα was viewed as the flexible hinge, but studies have shown that this domain contains nuclear localization signals (NLSs) and links the C-domain to the multifunctional E/F domain in the C terminus. The interaction of ERα AF-1 and AF-2 domains is essential for effective transactivation (18–20). We previously showed that mutating the bipartite NLS in mouse ERα (R267A, K270A, K272A, R273A, and R275A; hinge 1 [H1] ERα mutant) (Figure 1A) minimally disrupts nuclear localization but blocks the c-Jun–mediated protein-protein interaction with ERα (21). In addition, amino acid residues in the hinge region of ERα mediate alterations in activity. For example, K302 and K303 of human ERα are important for ubiquitination and protect ERα from basal degradation (22). Direct acetylation of lysine residues in the hinge region between amino acids 282 and 337 of ERα regulates transactivation and hormone sensitivity by p300 (23). Within this same region, K266 of human ERα is part of an inhibitory methylation event, in which, upon E2 stimulation, ERα-K266 methylation is diminished, and allows K266 to be acetylated and promote ERα transactivation activity (24). Phosphorylation events within the hinge region of ERα show that hinge region phosphorylation patterns uniquely inform various activation mechanisms as S294 differentiates ligand-dependent from ligand-independent activation of S305 (25). These studies demonstrate not only a structural role but also a functional role for the hinge region in ERα activation.
Figure 1.

Schematic illustration and intracellular localization of WT ERα, H1 ERα, and H2NES ERα in Ishikawa stable cells. A, Mutated ERα sequences for H1 ERα and H2NES ERα. B, Western blot of ERα in Ishikawa parent cell line (Ishikawa) and Ishikawa stable cell lines Ishikawa/vector, Ishikawa/WT ERα, Ishikawa/H1 ERα, and Ishikawa/H2NES ERα. C, Confocal microscopy imaging of ERα in Ishikawa stable cell lines as described in Materials and Methods. ERα is shown in green and 4′,6-diamidino-2-phenylindole is shown in blue. The insert in the lower right corner shows only ERα in green. Intracellular localization of WT and ERα mutants in the absence (vehicle, 0.01% DMSO) or presence (10 nM E2) is shown below each mutant and is based on at least 200 cells. N, nuclear; C, cytoplasm.
Studies provide evidence that estrogens exert their effects by eliciting both direct nuclear activities and extranuclear–mediated actions to mediate the diverse activities of ERα (26). These studies have begun to dissect the nonnuclear ERα activities using different molecular approaches, including ligand alteration and mutations in the receptor to alter receptor function in a targeted manner. We previously mutated NLS sequences in the hinge 2 region of ERα to preclude unliganded, predominant nuclear localization (H2) and added a nuclear export sequence (NES) to maintain ERα in the cytoplasm (H2NES ERα) (Figure 1A) to examine rapid action–mediated responses (21). The use of the estrogen dendrimer conjugate, a ligand that remains outside the cell and can only initiate extranuclear signaling demonstrated in MCF-7 cells that a subset of E2-regulated genes are responsive to estrogen dendrimer conjugate treatment (5). Further analysis revealed that these extranuclear responses are ERα regulated and involve kinase signaling to initiate changes in gene expression (5). Using a different approach, mutating the ERα Gαi-binding domain (R256A and D258A) blocks nonnuclear-mediated activity in endothelial cells and prevents E2-induced migration, endothelial nitric oxide synthase (eNOS) activation, and ERK1/2 activation in vitro (27). In an in vivo transgenic mouse model, expression of only a functional E-domain of ERα at the plasma membrane demonstrates that the nuclear activity of ERα is required for normal development and adult reproductive functions (28). Very recently, mutation of the palmitoylation site of ERα in a mouse model (C451A-ERα) revealed that abnormal reproductive function occurred due to an ovarian phenotype and that vascular actions were abrogated (29). Moreover, these studies reemphasize the complexity of E2/ERα-mediated signaling and the impact that the integration of ERα signaling has on other cellular or tissue response activities.
Previously, we established a stable cell system using a transformed/modified human endometrial cancer cell line, Ishikawa ERα(−) (30, 31) to investigate our ERα hinge region mutants, H1 ERα and H2NES ERα (21). In the current study, we explored further the activities of E2 to examine cellular localization of ERα, cell proliferation, ERE reporter activity, and endogenous gene expression profile changes. Our findings demonstrate that ERα functions predominately in the nucleus, and we conclude that intricate trafficking and intracellular communication of nonnuclear and nuclear-ERα must occur for alterations in endogenous gene expression to take place in our system.
Materials and Methods
Reagents and antibody
17β-Estradiol (E2) was purchased from Sigma-Aldrich and ICI 182 780 (ICI) was from Tocris Bioscience. Anti-ERα antibody (MC-20, sc-542) was purchased from Santa Cruz Biotechnology. Alexa Fluor 488 goat anti-rabbit IgG (A-11034) was purchased from Life Technologies.
Plasmids
The expression vector plasmid, pcDNA, was purchased from Promega. An intercontrol plasmid for the reporter assay, pRL-TK, was from Invitrogen. The luciferase (Luc) reporters, 3xERE-Luc and pS2-Luc, were described previously (43, 44).
Cell lines and tissue culture
The transformed/modified human endometrial cancer cell line, Ishikawa ERα(−) has been described previously (30, 31, 33). Ishikawa stable cell lines, Ishikawa/vector, Ishikawa/wild-type (WT) ERα, Ishikawa/H1 ERα, and Ishikawa/H2NES ERα, were generated from our research group as described previously (21). All Ishikawa stable cell lines were maintained in phenol red–free DMEM:F12 (no. 11039; Life Technologies) supplemented with 10% fetal bovine serum (FBS) and Geneticin (G418, 1.2 mg/mL, 11811–031; Life Technologies).
DNA isolation and short tandem repeat (STR) analysis
For the Ishikawa ERα(−) cell line, genomic DNA (5–10 μg) was extracted using a DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer's protocol. The concentration and qualification of DNA were measured by an ND-1000 NanoDrop spectrophotometer. STR analysis was performed at the Cancer Institute Cell Culture and Cytogenetics Facility Core at the University of Pittsburgh (Pittsburgh, Pennsylvania).
Confocal microscopy
Ishikawa stable cell lines were plated on Lab-Tek 2-well chamber slides (NUNC) in DMEM:F12 + 10% charcoal/dextran-stripped FBS (sFBS) (Thermo Scientific) for 48 hours and treated with 10 nM E2 for 4 hours. The cells were then fixed with 4% paraformaldehyde for 1 hour. Cells were washed with PBS, permeabilized with 0.5% Triton X-100 for 30 minutes, washed with PBS, and blocked with serum blocking solution (Zymed solution) for 60 minutes. The primary ERα antibody was incubated at 1:250 overnight at 4°C. Cells were washed with PBS and then were incubated with antirabbit Alexa Fluor 488 at 1:1000 for 2 hours. Cells were washed and cover-slipped with ProLong Gold antifade reagent with DAPI (P-36931; Life Technologies). The cellular localization of ERα was visualized by confocal microscopy (Zeiss LSM-510 microscope equipped with an argon-krypton laser) using a ×40 1.2 objective lens.
Transient transfection and luciferase assay
Cells were seeded in 24-well plates in DMEM:F12 + 10% sFBS overnight. A total of 0.3 μg of DNA, including 0.2 μg of reporter (3xERE-Luc or pS2-Luc) and 0.1 μg of pRL-TK plasmids, was transfected overnight using the Effectene transfection reagent (Qiagen) according to the manufacturer's protocol. Cells were starved with 10% sFBS for 8 hours and then were treated as described in the figure legends. Luciferase assays were performed using the Dual-Luciferase Reporter Activity System (Promega) according to the manufacturer's protocol. Data are from 3 independent experiments.
Protein extraction and Western blot analysis
Whole cell lysates were prepared by using BD TransFactor Extraction Kit (BD Biosciences). Western blot analysis has been described previously (Burns et al, 2011) (21). Briefly, the samples were loaded on a 10% SDS-PAGE gel after heating and separated by electrophoresis. The proteins were electrotransferred onto nitrocellulose membrane and blocked in PBS with 5% nonfat milk for 2 hours. The blots were incubated with primary antibody (ERα) at 4°C overnight, rinsed with PBS + Tween (0.1%) and then incubated with the antirabbit horseradish peroxidase conjugated secondary antibody at room temperature for 1 hour. The immunoreactive products were detected by the ECL system (Amersham Pharmacia). β-actin was used as a loading control.
Proliferation assay
For proliferation assays, Ishikawa/vector, Ishikawa/WT ERα, Ishikawa/H1 ERα, and Ishikawa/H2NES ERα cell lines were plated in 6-well plates in triplicate at 15 000 cells/well. Cells were starved for 6 days before treatment in DMEM:F12 + 5% sFBS + G418 (1.2 mg/ml). Medium was changed every 2 days. Vehicle (0.01% dimethylsulfoxide [DMSO]) and E2 (10 nM) were added on day 7, and medium was changed per day for 5 days. Cells were trypsinized, and each well was counted 3 times by trypan blue exclusion using a hemocytometer to obtain cell number per well.
RNA extraction and real-time PCR
Total RNA was extracted by using TRIzol Reagent (Life Technologies) according to the manufacturer's protocol. First-strand cDNA synthesis was performed using SuperScript RT according to the manufacturer's protocol (Life Technologies). The mRNA levels of genes were measured using SYBR Green assays (Life Technologies). The names of genes and sequences of primers used in the real-time PCRs are shown in Table 1.
Table 1.
Primer Sequences
| Gene Name | Accession No. | Forward Primer | Reverse Primer |
|---|---|---|---|
| CA12: carbonic anhydrase XII | NM_206925 | 5′-CCCTCGGACATGCACATCC-3′ | 5′-GCACTGTAGCGAGACTGGA-3′ |
| VIM: vimentin | NM_003380 | 5′-GGAAGCCGAAAACACCCTG-3′ | 5′-GAGACGCATTGTCAACATCCT-3′ |
| WDR69: dynein assembly factor with WD repeat domains 1 | NM_178821 | 5′-GCCAGAACAGCAATCACACG-3′ | 5′-TGCACGTCCGATCATAGCTTC-3′ |
| LRRC54: tsukushi, small leucine rich proteoglycan | NM_001258210 | 5′-CTTCTCCCGCCTTCGCTAC-3′ | 5′-GTTGTGGCTAAGGTTCACGTC-3′ |
| FGF19: fibroblast growth factor 19 | NM_005117 | 5′-CGGAGGAAGACTGTGCTTTCG-3′ | 5′-CTCGGATCGGTACACATTGTAG-3′ |
| ITGB3: platelet glycoprotein IIIa | NM_000212 | 5′-CATGAAGGATGATCTGTGGAGC-3′ | 5′-AATCCGCAGGTTACTGGTGAG-3′ |
| GSG1L: GSG1-like | NM_144675 | 5′-GTCACCACGCTCAACTCCTAC-3′ | 5′-CCGGAACTCAATGACCGTCTT-3′ |
| CYP1A1: cytochrome P450 1A1 | NM_000499 | 5′-TCGGCCACGGAGTTTCTTC-3′ | 5′-TCTTGAGGCCCTGATTACCCA-3′ |
| HES2: hes family bHLH transcription factor 2 | NM_019089 | 5′-CCAACTGCTCGAAGCTAGAGA-3′ | 5′-GCACGGTCATTTCCAGGAC-3′ |
| KCNF1: potassium voltage-gated channel subfamily F member 1 | NM_002236 | 5′-GCCAGCGACGACATAGAGATA-3′ | 5′-GACTGAGGAGGTCCCCGTA-3′ |
| TSCOT: solute carrier family 46, member 2 | NM_033051 | 5′-CTGGCACGGTTGGCACATA-3′ | 5′-CCCACTGCATACTGTTGGTCC-3′ |
| SMTNL2: smoothelin-like 2 | NM_001114974 | 5′-GTGAGCGCCTCATCGAAGT-3′ | 5′-AGCGACTGGACGTAGGTGA-3′ |
| GAPDH: glyceraldehyde-3-phosphate dehydrogenase | NM_002046 | 5′-ATGGGGAAGGTGAAGGTCG-3′ | 5′-GGGGTCATTGATGGCAACAATA-3′ |
Cycle time values were obtained using the ABI PRISM 7900 sequence detection system and analysis software (Applied Biosystems). Each sample was quantified against its glyceraldehyde-3-phosphate dehydrogenase transcript content and then normalized with respect to the control group. Quantification was performed according to the mathematical model described by Pfaffl (34) and as described previously (35). The experiments were repeated 3 times, and results are presented as fold increase ± SD.
Microarray methodology
Gene expression analysis was conducted using Agilent Whole Human Genome 4x44 multiplex format oligo arrays (014850; Agilent Technologies) following the Agilent 1-color microarray-based gene expression analysis protocol. Starting with 500 ng of total RNA, Cy3-labeled cRNA was produced according to the manufacturer's protocol. For each sample, 1.6 μg of Cy3-labeled cRNAs was fragmented and hybridized for 17 hours in a rotating hybridization oven. Slides were washed and then scanned with an Agilent scanner. Data were obtained using Agilent Feature Extraction software (version 9.5), with the 1-color defaults for all parameters. The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. Feature extraction data files were then imported into Partek Genomics Suite 6.6 beta, for which an intensity of >80 was set. The resulting probes were then analyzed for differentially expressed genes by ANOVA. The cutoff for genes was set at −1.5- to 1.5-fold with a false discovery rate of P < .05. Contrasts were set at Ishikawa/vector 4 hours E2 vs. Ishikawa/vector vehicle, Ishikawa/WT ERα 4 hours E2 vs Ishikawa/WT ERα vehicle, Ishikawa/H1 ERα 4 hours E2 vs Ishikawa/H1 ERα vehicle, and Ishikawa/H2NES ERα 4 hours E2 vs. Ishikawa/H2NES ER α vehicle. Genes that changed in all groups were filtered from analysis. Hierarchical clustering and Venn diagrams were derived from this filtered data set.
Data access
Microarray expression data has been submitted to the NCBI Gene Expression Omunibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession number GSE56946.
Statistical analysis
One-way ANOVA with a Tukey's posttest was performed using GraphPad Prism (version 6.00 for Windows; GraphPad Software). Two-way ANOVA with a Tukey's or Bonferroni's posttest was also performed using GraphPad Prism.
Results
STR analysis of Ishikawa ER(−) cells and nuclear localization of H1 and H2NES ERα mutants using Ishikawa stable cells
STR profiling is the accepted international standard for genetic analysis of cell lines for authentication (36). To verify the ERα(−) Ishikawa cell line, we compared the STR profile with the original Ishikawa ERα(+) cells (36, 37). STR analysis showed that >80% of the standard 9 STR loci matched between these 2 cell lines (Supplemental Table 1). These data suggested that this transformed Ishikawa ERα(−) cell line is related to the original Ishikawa ERα(+) cells. Ishikawa ERα(−) cells stably expressing ERα hinge region mutants were described previously (21) (Figure 1A). To further characterize the hinge region mutants, we examined their function in ERα(−) Ishikawa cells stably expressing ERα mutants (vector, WT ERα, H1 ERα, and H2NES ERα). Western blotting for ERα in Figure 1B confirms the presence of ERα in the stable cell lines. Confocal microscopy was used to demonstrate ERα localization by visualizing ERα and mutant ERα proteins by ERα antibody in these cell lines (Figure 1C). ERα protein was not detected in the empty vector–transfected cells. WT ERα is predominately nuclear in the absence or presence of ligand, E2 (10 nM). H1 ERα demonstrated a disruption of nuclear localization in the absence of ligand; however, in the presence of E2, it was translocated to the nucleus. The H2NES ERα is not localized to the nucleus with or without E2. We confirmed that H1 ERα and H2NES ERα cellular localization in this stable cell system is comparable to that in our previous study with transiently transfected HeLa cells (21).
Functional analysis and cell proliferation analysis of H1 and H2NES mutant ERα using Ishikawa stable cells
Our previous study with HeLa cells demonstrated that H2NES ERα is shuttled and can be trapped in the nucleus after leptomycin B treatment (21); therefore, in these experiments, we used two different ERE-driven promoters, a strong artificial ERE (3xERE-Luc) and a natural 1xERE (pS2-Luc), to examine reporter activation in the Ishikawa stable cell lines. Activation by E2 was seen with WT ERα, H1 ERα, and H2NES ERα using the 3xERE-Luc reporter, but this activation was not seen using the pS2-Luc reporter in the H2NES ERα cell line (Figure 2A). ICI was used as a pure ERα antagonist to show the specificity of reporter activity.
Figure 2.

Functional analysis and cell proliferation analysis of WT ERα, H1 ERα, and H2NES ERα Ishikawa stable cell lines. A, luciferase activity. Ishikawa stable cell lines were transfected with pRL-TK and 3xERE-Luc or pS2-Luc. Cells were treated with vehicle (veh, 0.01% DMSO), E2 (10 nM), or E2 + ICI (1 μM) for 18 hours. Relative luciferase activity to vehicle is shown. Transfection efficiency was normalized by Renilla luciferase. Data shown are representative of 3 experiments. *, P < .05 (one-way ANOVA with a Tukey's multicomparison posttest). B, Cell proliferation analysis. Ishikawa stable cell lines were treated for 5 days with vehicle (0.01% DMSO) or E2 (10 nM), and cells were counted on days 3, 4, and 5 to examine cell proliferation. n = 3 biological replicates. *, P < .05 (two-way ANOVA with Bonferroni's posttest).
To characterize how these mutations in ERα affected cell growth, we performed cell proliferation assays using each cell line with and without E2 treatment (Figure 2B). In the vector Ishikawa cell line, no additional increase in cell growth was observed with E2 treatment. In the WT ERα Ishikawa cell line, we observed a significant increase in cell growth with E2 treatment relative to that of vehicle-treated cells. Similarly, the H1 ERα Ishikawa cell line also exhibited increased cell growth with E2 treatment. The H2NES ERα Ishikawa cell line did not show increased cell growth after E2 treatment. In addition, we tested the ERα DBD mutated receptor, AA ERα (10) and found no increased cell growth after E2 treatment (data not shown). These data suggest that for increased cell proliferation with E2 treatment, DNA binding activity of ERα is required.
Hierarchical clustering
To determine whether there are endogenous genes regulated by the nongenomic activities of ERα alone, we performed microarray analysis on vector, WT ERα, H1 ERα, and H2NES ERα Ishikawa cell lines after E2 (10 nM) treatment for 4 hours. After an intensity cutoff of >80, approximately 28 000 probes remained. The data were analyzed by examining the fold change from vehicle to 4 hours of E2 treatment (vector E2 4 hours vs vector vehicle, WT ERα E2 4 hours vs WT ERα vehicle, H1 ERα E2 4 hours vs H1 ERα vehicle, and H2NES ERα 4 hours vs H2NES ERα vehicle). Hierarchical clustering demonstrates 2 main branches (Figure 3A); the large branch clusters all vehicle-treated cell lines together with the vector- and H2NES ERα 4 hour–treated lines. Within that branch, there are two separate clusters, one having all vehicle-treated cell lines and one containing the E2-treated vector and H2NES ERα cell lines. The smaller branch contains WT ERα and H1 ERα cell lines that were treated with E2 for 4 hours. The WT ERα- and H1 ERα E2-treated cell lines cluster closely together, suggesting similar gene expression changes. Microarray analysis was also done at 24 hours; however, minimal to no significant change in gene expression after E2 treatment was observed in WT, H1, and H2NES ERα cell lines.
Figure 3.

Hierarchical clustering analysis of Ishikawa stable cell lines. A, Cluster analysis. Data analysis was performed in Partek Genomics Suite 6.6 beta with −1.5- to 1.5-fold changes to compare Ishikawa vector, WT ERα, H1 ERα, and H2NES ERα cell lines. Green, vector; orange, WT ERα; yellow, H1 ERα; blue, H2NES ERα; purple, vehicle; pink, 10 nM E2 4 hours. B, Venn diagram. Data shows overlap between Ishikawa/WT ERα, H1 ERα, and H2NES ERα stable cell lines.
Venn diagram analysis for fold changes (±1.5-fold cutoff) in gene expression and overlapping with other cell lines revealed that the most overlap occurred with WT ERα and H1 ERα (77 genes) (Figure 3B). Very minimal overlap occurred with H2NES ERα and WT ERα (6 genes) or H2NES ERα and H1 ERα (12 genes) cell lines. At 4 hours, the genes that were elevated in the WT ERα cells were not elevated in the H2NES ERα cells (Supplemental Table 2). Our data suggest that changes in gene expression that occur at 4 hours after E2 treatments are ERE dependent because they are only seen with the 2 cell lines with nuclear localization and DBD activity.
Gene expression data and microarray confirmation
A number of genes were chosen for microarray confirmation by real-time PCR (Figure 4). Genes that suggested a larger fold change (carbonic anhydrase XII [CA12], fibroblast growth factor 19 [FGF19], and potassium voltage-gated channel subfamily F member 1 [KCNF1]) and a smaller fold change (tsukushi, small leucine rich proteoglycan [LRRC54], cytochrome P450 1A1 [CYP11A1], solute carrier family 46, member 2 [TSCOT], hes family bHLH transcription factor 2 [HES2], and smoothelin-like 2 [SMTNL2]) activation were selected. Cell lines containing vector, WT ERα, H1 ERα, and H2NES ERα were used and treated for 4 or 24 hours with E2 (10 nM). The Ishikawa vector cell line was used as the negative control. The H1 ERα cells retained DNA-binding activity with gene activation comparable to that of WT ERα cells, whereas there are no changes in the gene expression of these genes with the AA or the H2NES ERα mutant cell lines. ERE-mediated response genes at 4 and 24 hours are shown in Figure 4. Microarray data analysis at 4 or 24 hours (data not shown) did not reveal any reproducible alterations in gene expression changes with the H2NES ERα mutant cell line. In addition, the H1 ERα cell line showed activity comparable to that of the WT ERα cell line. Interestingly, there was a group of genes that were repressed relative to vector cells in WT ERα and H1 ERα cell lines in an E2-independent manner (Supplemental Figure 1). Partial reduction was seen with the H2NES ERα cell line. Taken together, our results show that for E2-mediated changes in gene expression, DNA binding activity and sustained nuclear localization of ERα are required for changes in endogenous gene expression.
Figure 4.

Gene expression analysis from Ishikawa stable cell lines. Total RNA was isolated from Ishikawa cells stably expressing vector, WT ERα, H1 ERα, or H2NES ERα. Cells were treated with vehicle (0.01% DMSO) or E2 (10 nM) for 4 or 24 hours. CA12, LRRC54, FGF19, CYP1A1, TSCOT, KCN1, HES2, and SMTNL2 transcripts were quantified by real time-PCR as described under Materials and Methods and were calculated relative to vehicle. n = 3 biological replicates. *, P < .05 (two-way ANOVA with a Tukey's posttest).
Discussion
Estrogens have been shown to activate nonnuclear signaling as well as the classic nuclear signaling for the various actions mediated by ERα. Evidence demonstrates that the use of the estrogen dendrimer conjugates, membrane-initiated ERα signaling, mediates ∼25% of the genes activated by E2 alone in MCF-7 cells, which contain endogenous ERα (5). To determine whether activation of nonnuclear ERα activity alone would lead to changes in endogenous gene expression or whether the initiation by E2 occurs at the membrane and still requires ERα in the nucleus, we used ERα mutants we created previously (21). We demonstrate that these effects, in our Ishikawa stable cell system, are most likely extranuclear initiated, but do require ERα translocation to the nucleus for expression of genes (ie, LRRC54). With the H2NES ERα, we find no changes in gene expression at 4 or 24 hours after E2 treatments in WT ERα cells. Our results suggest that cooperative signaling occurs with nongenomic (extranuclear-initiated) and genomic (nuclear-initiated) signaling to obtain the overall alterations in gene expression and cell responses.
Our data are supported by other studies suggesting tissue/cell-specific changes with ERα mutants to study nonnuclear-mediated activities. Mutation of the palmitoylation site of ERα (C451A; blocking nonnuclear initiated signaling) leads to tissue-specific changes in a mouse model (29). ERα C451A in vivo in the uterus is similar to that in a WT uterus, but the female mice are infertile owing to ovarian defects. Rapid action vascular responses such as rapid vasodilation, acceleration of endothelial repair, and eNOS synthase phosphorylation were abrogated in the ERα C451A mice (29). Blocking the Gαi binding domain of ERα prevented E2-induced endothelial cell migration and eNOS activation, but this mutation maintained ERE-mediated activity in vitro (27). The estrogen dendrimer conjugate stimulates endothelial cell proliferation and migration via ERα direct ERα Gαi interaction and eNOS activation in mice (38). Treatment of an ERE-luciferase reporter mouse with estrogen dendrimer conjugate showed stimulation of carotid artery reendothelization and attenuated development of neointimal hyperplasia after injury, but, in contrast, endometrial carcinoma cell growth, uterine enlargement, and MCF-7 cell xenograft growth were not stimulated by estrogen dendrimer conjugate (38). We similarly find that the Ishikawa cell line with H2NES ERα does not proliferate with E2 treatment, suggesting that the rapid action responses alone do not mediate cell proliferation in uterine cells.
We know from our previous work that H2NES ERα cycles in and out of the nucleus (21). In the current study, we do see luciferase activity with H2NES ERα stable line, but with an endogenous reporter (pS2; 1xERE), we do not see increased luciferase activity with H2NES ERα. We hypothesize that the strength of the ERE allows for this activity and is a potential explanation for why we do not see activity with the 1xERE and why this ERE-mediated activation is promoter specific. The H2NES ERα binds to DNA (21); therefore, we postulate that ERα must remain in the nucleus long enough to allow for chromatin remodeling to permit the binding of ERα to the endogenous EREs contained in the genome for the initiation of endogenous gene expression chances.
The hinge region of ERα is important for nuclear localization and interaction via the tethered mechanism (10, 21, 39, 40). The H1 ERα mutant loses the ability to form tethered protein-protein interactions with c-Jun but maintains ERE-mediated activations. We also examined the H1 ERα mutant properties in Ishikawa cells and hypothesized that removing the “tethered”-mediated function between ERα and Jun would alter endogenous gene regulation but instead find that blocking the tethered-mediated functionality of ERα does not alter the ability of E2 to activate cell proliferation or endogenous gene expression changes. Coupled with the use of the AA ERα known to block ERE DNA binding activity (41, 42), we find many gene targets in the Ishikawa WT ERα or H1 ERα cell lines that are ERE-mediated gene changes occurring at 4 and 24 hours after E2 treatment.
Our data suggest that with the complex nonnuclear to nuclear cellular cross talk events ERα must be able to localize to the nucleus to initiate changes in gene expression. It is believed that, upon ligand binding to nonnuclear ERα, a variety of signaling events are prompted that can themselves modify cellular behavior or influence cellular responses (15); however, our study design only allowed for nonnuclear activity, and we found no changes in gene expression initiated from the nongenomic ERα signaling cascade in Ishikawa cells at the time points analyzed. Shown in Figure 5 is a simplistic schematic diagram of the proposed ERα activity from our findings, indicating that ERα must be in the nucleus for a period of time to initiate changes in gene transcription and ultimately alter the growth response from E2 treatment. Ultimately, many cell types and animal models, which are currently in development, will be needed to examine what nonnuclear action(s) ERα/E2 mediates without nuclear genomic ERα and evaluate whether tissue selectivity is relevant to the different types of ERα action.
Figure 5.
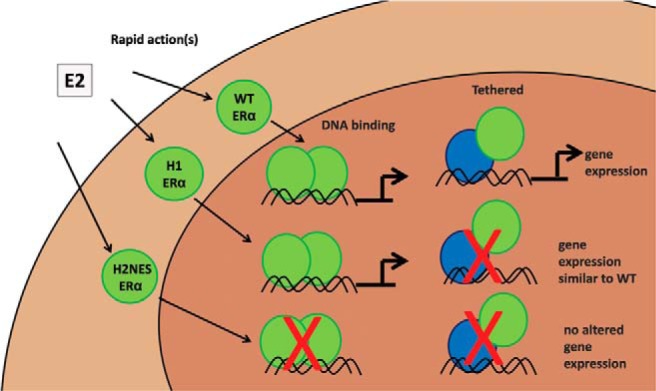
Schematic representation of altered changes in gene expression from WT ERα, H1 ERα, and H2NES ERα mutants.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr Kevin Gerrish and Laura Wharey in the National Institute of Environmental Health Sciences (NIEHS) Microarray Core and Dr C. Jeff Tucker of the NIEHS Fluorescence Microscopy and Imaging Center for assistance, Dr Steffi Oesterreich at the University of Pittsburgh Cancer Institute Cell Culture and Cytogenetics Facility for STR analysis of Ishikawa cell lines, and Drs Tatsuya Sueyoshi and Robert Oakley for critical review of this article.
This work was supported by the National Institutes of Health (Grant Z01ES70065 to K.S.K.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- DBD
- DNA-binding domain
- DMSO
- dimethylsulfoxide
- E2
- 17β-estradiol
- eNOS
- endothelial nitric oxide synthase
- ER
- estrogen receptor
- ERE
- estrogen response element
- FBS
- fetal bovine serum
- H1
- hinge 1
- H2
- hinge 2
- ICI
- ICI 182 780
- Luc
- luciferase
- NES
- nuclear export sequence
- NLS
- nuclear localization sequence
- sFBS
- charcoal/dextran-stripped FBS
- STR
- short tandem repeat
- WT
- wild-type.
References
- 1. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor α to estrogen-responsive elements. J Biol Chem. 2010;285:2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hewitt SC, O'Brien JE, Jameson JL, Kissling GE, Korach KS. Selective disruption of ERα DNA-binding activity alters uterine responsiveness to estradiol. Mol Endocrinol. 2009;23:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McDevitt MA, Glidewell-Kenney C, Jimenez MA, et al. New insights into the classical and non-classical actions of estrogen: evidence from estrogen receptor knock-out and knock-in mice. Mol Cell Endocrinol. 2008;290:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol Metab. 2005;16:347–353. [DOI] [PubMed] [Google Scholar]
- 7. Kushner PJ, Agard DA, Greene GL, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. [DOI] [PubMed] [Google Scholar]
- 8. Webb P, Nguyen P, Valentine C, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13:1672–1685. [DOI] [PubMed] [Google Scholar]
- 9. Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr Rev. 1994;15:391–407. [DOI] [PubMed] [Google Scholar]
- 10. Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J Biol Chem. 2001;276:13615–13621. [DOI] [PubMed] [Google Scholar]
- 11. Safe S. Transcriptional activation of genes by 17β-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. [DOI] [PubMed] [Google Scholar]
- 12. Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–741. [DOI] [PubMed] [Google Scholar]
- 13. Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov. 2004;3:27–41. [DOI] [PubMed] [Google Scholar]
- 14. Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;23:665–686. [DOI] [PubMed] [Google Scholar]
- 15. Banerjee S, Chambliss KL, Mineo C, Shaul PW. Recent insights into non-nuclear actions of estrogen receptor α. Steroids. 2014;81:64–69. [DOI] [PubMed] [Google Scholar]
- 16. Mader S, Chambon P, White JH. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res. 1993;21:1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids. 1999;64:310–319. [DOI] [PubMed] [Google Scholar]
- 18. Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, Michalides R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci. 2010;123(Pt 8):1253–1261. [DOI] [PubMed] [Google Scholar]
- 19. Métivier R, Penot G, Flouriot G, Pakdel F. Synergism between ERα transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 α-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol. 2001;15:1953–1970. [DOI] [PubMed] [Google Scholar]
- 20. Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M. Differences in the abilities of estrogen receptors to integrate activation functions are critical for subtype-specific transcriptional responses. Mol Endocrinol. 2002;16:1810–1827. [DOI] [PubMed] [Google Scholar]
- 21. Burns KA, Li Y, Arao Y, Petrovich RM, Korach KS. Selective mutations in estrogen receptor α D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. J Biol Chem. 2011;286:12640–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berry NB, Fan M, Nephew KP. Estrogen receptor-α hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol Endocrinol. 2008;22:1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang C, Fu M, Angeletti RH, et al. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem. 2001;276:18375–18383. [DOI] [PubMed] [Google Scholar]
- 24. Zhang X, Tanaka K, Yan J, et al. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc Natl Acad Sci USA. 2013;110:17284–17289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Held JM, Britton DJ, Scott GK, et al. Ligand binding promotes CDK-dependent phosphorylation of ER-α on hinge serine 294 but inhibits ligand-independent phosphorylation of serine 305. Mol Cancer Res. 2012;10:1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Genomic collaboration of estrogen receptor α and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol. 2011;31:226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu Q, Chambliss K, Lee WR, Yuhanna IS, Mineo C, Shaul PW. Point mutations in the ERα Gαi binding domain segregate nonnuclear from nuclear receptor function. Mol Endocrinol. 2013;27:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pedram A, Razandi M, Kim JK, et al. Developmental phenotype of a membrane only estrogen receptor α (MOER) mouse. J Biol Chem. 2009;284:3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111:E283–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ignar-Trowbridge DM, Teng CT, Ross KA, Parker MG, Korach KS, McLachlan JA. Peptide growth factors elicit estrogen receptor-dependent transcriptional activation of an estrogen-responsive element. Mol Endocrinol. 1993;7:992–998. [DOI] [PubMed] [Google Scholar]
- 31. Yamada H, Sasaki M, Honda T, et al. Suppression of endometrial carcinoma cell tumorigenicity by human chromosome 18. Genes Chromosomes Cancer. 1995;13:18–24. [DOI] [PubMed] [Google Scholar]
- 32. Hall JM, McDonnell DP. Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol Interv. 2005;5:343–357. [DOI] [PubMed] [Google Scholar]
- 33. Mueller SO, Hall JM, Swope DL, Pedersen LC, Korach KS. Molecular determinants of the stereoselectivity of agonist activity of estrogen receptors (ER) α and β. J Biol Chem. 2003;278:12255–12262. [DOI] [PubMed] [Google Scholar]
- 34. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winuthayanon W, Piyachaturawat P, Suksamrarn A, et al. Diarylheptanoid phytoestrogens isolated from the medicinal plant Curcuma comosa: biologic actions in vitro and in vivo indicate estrogen receptor-dependent mechanisms. Environ Health Perspect. 2009;117:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korch C, Spillman MA, Jackson TA, et al. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol Oncol. 2012;127:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishida M. The Ishikawa cells from birth to the present. Hum Cell. 2002;15:104–117. [DOI] [PubMed] [Google Scholar]
- 38. Chambliss KL, Wu Q, Oltmann S, et al. Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teyssier C, Belguise K, Galtier F, Chalbos D. Characterization of the physical interaction between estrogen receptor α and JUN proteins. J Biol Chem. 2001;276:36361–36369. [DOI] [PubMed] [Google Scholar]
- 40. Francis MK, Phinney DG, Ryder K. Analysis of the hormone-dependent regulation of a JunD-estrogen receptor chimera. J Biol Chem. 1995;270:11502–11513. [DOI] [PubMed] [Google Scholar]
- 41. Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER) α deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002;16:2188–2201. [DOI] [PubMed] [Google Scholar]
- 42. Li Y, Burns KA, Arao Y, Luh CJ, Korach KS. Differential estrogenic actions of endocrine-disrupting chemicals bisphenol A, bisphenol AF, and zearalenone through estrogen receptor α and β in vitro. Environ Health Perspect. 2012;120:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Masiakowski P, Breathnach R, Bloch J, et al. Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res. 1982;10:7895–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tremblay GB, Tremblay A, Copeland NG, et al. 1997 Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol 11:353–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.