

Abstract
FK506-binding protein 51 (FKBP51) is a negative regulator of glucocorticoid receptor-α (GRα), although the mechanism is unknown. We show here that FKBP51 is also a chaperone to peroxisome proliferator–activated receptor-γ (PPARγ), which is essential for activity, and uncover the mechanism underlying this differential regulation. In COS-7 cells, FKBP51 overexpression reduced GRα activity at a glucocorticoid response element-luciferase reporter, while increasing PPARγ activity at a peroxisome proliferator response element reporter. Conversely, FKBP51-deficient (knockout) (51KO) mouse embryonic fibroblasts (MEFs) showed elevated GRα but reduced PPARγ activities compared with those in wild-type MEFs. Phosphorylation is known to exert a similar pattern of reciprocal modulation of GRα and PPARγ. Knockdown of FKBP51 in 3T3-L1 preadipocytes increased phosphorylation of PPARγ at serine 112, a phospho-residue that inhibits activity. In 51KO cells, elevated phosphorylation of GRα at serines 220 and 234, phospho-residues that promote activity, was observed. Because FKBP51 is an essential chaperone to the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase, Akt signaling was investigated. Elevated Akt activation and increased activation of p38 kinase, a downstream target of Akt that phosphorylates GRα and PPARγ, were seen in 51KO MEFs, causing activation and inhibition, respectively. Inactivation of p38 with PD169316 reversed the effects of FKBP51 deficiency on GRα and PPARγ activities and reduced PPARγ phosphorylation. Last, loss of FKBP51 caused a shift of PPARγ from cytoplasm to nucleus, as previously shown for GRα. A model is proposed in which FKBP51 loss reciprocally regulates GRα and PPARγ via 2 complementary mechanisms: activation of Akt-p38–mediated phosphorylation and redistribution of the receptors to the nucleus for direct targeting by p38.
The molecular chaperone, FK506-binding protein (FKBP) 51, is an FKBP immunophilin that contains several tetratricopeptide repeat (TPR) motifs used in protein interactions (1, 2). The TPR domains form the basis for interactions with steroid receptors via the single TPR-binding domain of heat shock protein 90 (Hsp90) (for reviews, see Refs. 3 and 4). The TPR site of Hsp90 can also accommodate other chaperones, including FKBP52 and protein phosphatase-5 (PP5), each of which exerts distinct and sometimes opposing effects on steroid receptor actions. Using the glucocorticoid receptor (GRα) as an example, we and others have found that FKBP52 tends to be a positive regulator of GRα that promotes translocation of GRα to the nucleus, increases GRα hormone-binding affinity, and increases transcriptional activity at select genes, both in vitro and in vivo (5–9). In contrast, PP5 inhibits GRα activity (10), and we recently showed that this occurs through its intrinsic phosphatase activity, causing dephosphorylation of the receptor (11). Like PP5, FKBP51 is also a negative regulator of GRα, and the evidence so far suggests that it does this by sequestering GRα to the cytoplasm and by reducing its intrinsic hormone-binding affinity (12–14). However, our recent work with cells derived from FKBP51 knockout (51KO) mice has shown very high levels of GRα activity, suggesting that FKBP51 must be suppressing GRα through additional mechanisms. Thus, the recent discovery by Wang and colleagues (15) and later by Hausch and colleagues (16) that FKBP51 serves as a necessary chaperone to the Akt-specific phosphatase, PH domain leucine-rich repeat protein phosphatase (PHLPP), led us to speculate that FKBP51-mediated phosphorylation events may also account for its effects on steroid receptors.
Control of GRα by phosphorylation has been known for many years, with most phosphorylation sites located in the N-terminal domain (17, 18). Three of these sites, serines 212, 220, and 234 in the mouse (serines 203, 211, and 226 in humans), are of particular relevance because all 3 are phosphorylated in response to hormone binding and contribute positively or negatively to GRα transcriptional activity (19). Several kinases have been implicated in the targeting of these sites. Cyclin-dependent kinases target serines 212 and 220 (20, 21), leading either to inhibition if serine 212 is the target (22, 23) or activation of GRα if serine 220 is phosphorylated (22, 24). The p38 MAPK is known to phosphorylate serines 220 and 234, both of which stimulate GRα transcription activity (20, 25, 26). Recently, it has been demonstrated that activation of Akt leads to phosphorylation and activation of p38 kinase, a process that is mediated by apoptosis signal-regulating kinase-1 (27–30). For this reason, we have further speculated that phosphorylation control of GRα by FKBP51 might be mediated by the Akt-p38 pathway.
Although all steroid receptors are chaperoned by the Hsp90-TPR complex, some members of the nuclear receptor family are not known to be regulated in this fashion. In particular, the thyroid and retinoic acid receptors have been conclusively shown not to interact with Hsp90 (31, 32). Until recently, many of the so-called orphan nuclear receptors were also thought to not bind chaperones. In 2003, association of Hsp90 with the peroxisome proliferator–activated receptors (PPARα, PPARβ, and PPARγ) was demonstrated (33, 34). Since then, we have shown that the TPR cochaperone PP5 can also enter into complexes with PPARγ (11). The PPARγ-PP5 association occurred under basal conditions but was further stimulated by the binding of the thiazolidinedione agonist rosiglitazone (Rosi) to PPARγ, resulting in dephosphorylation of PPARγ at serine 112. Because phosphorylation of serine 112 is inhibitory for PPARγ (35, 36), cells deficient in PP5 were resistant to adipogenic differentiation and lipid accumulation due to compromised PPARγ action. Only a few kinases that directly target serine 112 of PPARγ have been identified. The first of these were ERK2 and c-Jun NH2-terminal kinase, members of the MAPK pathway that are typically activated by growth factor signals that suppress PPARγ activity (35, 36). More recently, negative control of PPARγ by p38 kinase has been described (37).
With these concepts in mind, we have investigated the possible contribution of FKBP51 to reciprocal regulation of GRα and PPARγ. We show that FKBP51 can indeed interact with PPARγ and that it negatively regulates GRα, while simultaneously stimulating PPARγ activity. Importantly, we provide evidence that FKBP51 achieves this duality by suppressing activation of the Akt-p38 pathway, leading to reduced phosphorylation of GRα and PPARγ at specific serine residues. Thus, FKBP51 is now identified as an important fulcrum point in differential regulation of these receptors by phosphorylation.
Materials and Methods
Materials
Dexamethasone (Dex), HEPES, Tris, EDTA, PBS, protease inhibitor cocktail, PD169316, insulin, sodium molybdate, dextran-coated charcoal, isobutylmethylxanthine and sodium chloride were all obtained from Sigma-Aldrich. Rosi was obtained from Alexis Biochemicals. Iron-supplemented bovine calf serum was from HyClone Laboratories Inc. Dialyzed fetal bovine serum was from Denville Scientific. Halt phosphatase inhibitor cocktail was from Thermo Scientific. The Immobilon-FL polyvinylidene fluoride membrane was obtained from Millipore Corporation. Puromycin and DMEM were from Fisher Scientific (Pittsburgh, PA). Lipofectamine 2000 transfection reagent and Opti-MEM were obtained from Invitrogen. DRAQ5 was obtained from Cell Signaling.
Cell lines and culture
Mouse embryonic fibroblasts (MEFs) were isolated from wild-type (WT) and FKBP51 knockout (51KO) embryonic day 13.5 embryos, as described previously (38). The knockdown of FKBP51 in 3T3-L1 cells was achieved by viral infection of 3T3-L1 cells with a specific short hairpin RNA (shRNA) for FKBP51 (GGTGAAGATATCACTACGAAGAAAGACAG). A control cell line was made using a scramble shRNA that was not specific to any known RNA sequence. Three hours after the infection, cells were washed with fresh medium and allowed to recover; 48 hours after the infection, cells were washed, followed by selection with puromycin (5 ng/μL) for 7 days, with medium changes every 24 hours. To confirm knockdown of FKBP51, Western blot analysis was performed on the cell line. Cells were routinely cultured in DMEM containing 10% bovine calf serum with 1% penicillin-streptomycin. All cell treatments were done at or near confluence. For measurement of steroid responses, cells were grown in DMEM containing 10% dialyzed fetal bovine serum 24 hours before treatment.
Immunoadsorption of GRα and PPAR complexes
Cells were harvested in HEMG (10 mM HEPES, 3 mM EDTA, 20 mM sodium molybdate, and 10% glycerol, pH 7.4) plus the protease inhibitor mixture and set on ice for 20 minutes, followed by Dounce homogenization. Supernatants (cytosol) were collected after a 10-minute centrifugation at 20 800 × g at 4°C and then were precleared with protein A- or G-Sepharose nutating for 1 hour at 4°C. Samples were spun down, split into equal aliquots of cytosol, and immunoadsorbed overnight with FiGR antibody against total GRα, PPARγ antibody against total PPARγ, and appropriate controls (nonimmune mouse IgG) at 4°C under constant rotation. Pellets were washed 5 to 7 times with TEG (10 mM Tris, 3 mM EDTA, 10% glycerol, 50 mM NaCl, and 20 mM sodium molybdate, pH 7.4), and complexes were eluted with 6× SDS sample buffer.
Transfection and reporter assay
MEFs were grown to 90% confluence in 24-well plates and then were transiently transfected with expression vectors for FKBP51, GRα, PPARγ2, and retinoid X receptor using Lipofectamine 2000, according to the manufacturer's protocol. p-glucocorticoid response element (GRE)2EIB-luciferase (Luc) or peroxisome proliferator response element (PPRE)-Luc reporters were cotransfected, as appropriate, along with a pRL-CMV-Renilla reporter to normalize for transfection efficiency. Twenty-four hours after transfection, cells were treated with vehicle, PD169316, or hormone at the indicated concentrations for an additional 24 hours until harvest. Cell lysates were prepared, and the assay was performed using the Promega luciferase system.
Whole-cell extraction
Cells were washed and collected in 1× PBS followed by centrifugation at 1500 × g for 10 minutes. The supernatant was discarded, and the pellet was resuspended in 1× PBS. After a short spin at 20 800 × g for 5 minutes at 4°C, the pellet was rapidly frozen on a dry ice-ethanol mix and stored at −80°C overnight. The frozen pellet was then resuspended in 3 volumes of cold whole-cell extract buffer (20 mM HEPES, 25% glycerol, 0.42 M NaCl, and 0.2 mM EDTA, pH 7.4) with protease and Halt phosphatase inhibitor cocktail and incubated on ice for 10 minutes. The samples were centrifuged at 100 000 × g for 5 minutes at 4°C. Protein levels were measured spectrophotometrically by a SpectraMax Plus spectrophotometer (Molecular Devices Corp). The supernatants were used immediately for Western analysis.
Gel electrophoresis and Western blotting
Protein samples were resolved by SDS-PAGE and electrophoretically transferred to Immobilon-FL membranes. Membranes were blocked at room temperature for 1 hour in Tris-buffered saline (TBS) (10 mM Tris-HCl [pH 7.4] and 150 mM NaCl) containing 3% BSA plus phosphatase inhibitors. Incubation with the primary antibody was done overnight at 4°C. After 3 washes in TBST (TBS plus 0.1% Tween 20), membranes were incubated with infrared anti-rabbit (IRDye 800, green), anti-mouse (IRDye 680, red), or anti-goat (IRDye 800, green) secondary antibodies (LI-COR Biosciences) at 1:15 000 dilution in TBS for 2 hours at 4°C. Immunoreactivity was visualized and quantified by infrared scanning in the Odyssey system (LI-COR Biosciences). Antibodies against FKBP51 (sc-11518), FiGR (sc-12763), PPARγ (sc-7273), total Akt (sc-1619), and Hsp90 (sc-13119) were obtained from Santa Cruz Biotechnology. Phospho-GRα Ser-212, Ser-220, and Ser-234 antibodies were provided by Dr Michael Garabedian (New York University School of Medicine, New York, New York). Ser-112 phospho-PPARγ2 antibody was purchased from Abcam PLC. Ser-473 phospho-Akt (4060), Thr-180/Try-182 phospho-p38 MAPK (4511), and total p38 (8690) were purchased from Cell Signaling Technology.
Green fluorescent protein (GFP) imaging
WT and 51KO MEFs were seeded in 60-mm dishes at 300 000 to 500 000 cells/dish in DMEM containing 10% iron-supplemented calf serum prestripped of endogenous steroids and free fatty acids by 1% (w/v) dextran-coated charcoal (39, 40). Cells were transfected with a GFP-PPARγ2 construct. Fluorescent images of the living cells were obtained 24 hours posttransfection using a DMIRE2 confocal microscope (Leica). DRAQ5 for nuclear staining was added to the medium just before the imaging. Cells were scanned at low laser power to avoid photobleaching. ImageJ software was used for data analysis.
Statistical analysis
Data were analyzed with Prism 5 (GraphPad Software) using ANOVA combined with the Tukey posttest to compare pairs of group means or unpaired t tests. Values of P ≤ .05 were considered statistically significant.
Results
FKBP51 interacts with PPARγ and is essential for its activity
The recent discovery of PPARγ complexes containing Hsp90 led us to investigate cochaperone interactions with the receptor. Recently, we showed that like GRα, PPARγ heterocomplexes contained PP5. In contrast to GRα, however, PP5 was not a negative regulator of receptor activity but was needed for full activation of PPARγ by reducing inhibitory phosphorylation at serine 112 (11). Because GRα complexes can also contain FKBP51, we tested for the interaction of this cochaperone with PPARγ. Figure 1 shows the results of coimmunoadsorption experiments after expression of mouse GRα and mouse PPARγ2 in receptor-less COS-7 cells. The results show that PPARγ, like GRα, interacts with both Hsp90 and FKBP51.
Figure 1.
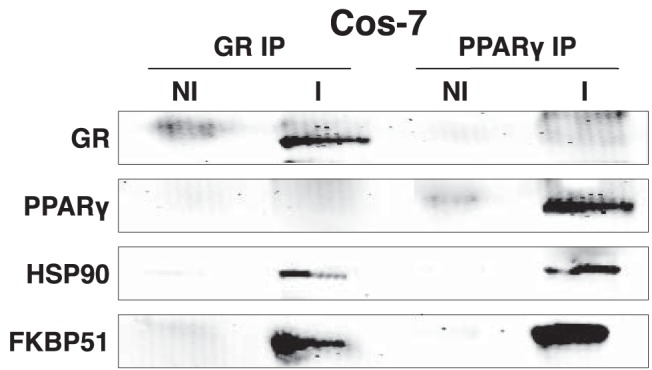
Interaction of FKBP51 with GRα and PPARγ. GRα and PPARγ transfected COS-7 cells were adsorbed to protein G-Sepharose using the FiGR monoclonal antibody to GRα (I), PPARγ monoclonal antibody (I), or nonimmune IgG (NI), followed by Western blotting for GRα, PPARγ, Hsp90, and FKBP51 proteins. A representative of 3 independent experiments is shown.
We next tested whether FKBP51 controlled PPARγ transcriptional activity. Receptor-less COS-7 cells were once again transfected with GRα and PPARγ, along with reporter genes regulated by promoters containing minimal GRE or PPRE. These cells were also transfected with increasing amounts of FKBP51 (Figure 2A). In response to FKBP51 overexpression, reduced Dex-induced GRα activity was observed, whereas PPARγ activity was increased 8-fold (Figure 2B). The relatively high level of PPARγ potentiation by FKBP51 suggested that this receptor is particularly sensitive to FKBP51 action. To confirm this hypothesis, reporter gene activities were measured in WT and 51KO MEFs (Figure 2C). As expected, Dex-induced activity was increased in the 51KO MEFs compared with that in the WT cells (Figure 2D), but no discernible increase was observed in the absence of hormone. Because these experiments were performed using serum that had been charcoal-stripped and dialyzed to remove steroids and fatty acids, all GRα was expected to be hormone free. Thus, the lack of an effect of FKBP51 loss on GRα basal activity was not surprising. In contrast, basal activity of PPARγ was greatly reduced in 51KO cells, and Rosi-induced activity was nonexistent (Figure 2D). These facts suggest that a sizable proportion of PPARγ is active under basal conditions, presumably due to binding by endogenously produced fatty acids, and that FKBP51 is essential for fatty acid–induced activity. As a last test, GRα and PPARγ activities at endogenous metabolic genes were measured in WT and 51KO MEFs (see Figure 5 in the companion article [41]). The results show elevated Dex-induced expression of pyruvate dehydrogenase kinase-4, phosphoenolpyruvate carboxykinase, and glucose 6-phosphatase, as well as a greater level of Dex-induced repression at adiponectin, CD36, and perilipin, demonstrating that loss of FKBP51 increases both the transactivation and transrepressive properties of GRα. Interestingly, Rosi-induced expression of adiponectin, CD36, perilipin, and other genes was greatly reduced.
Figure 2.
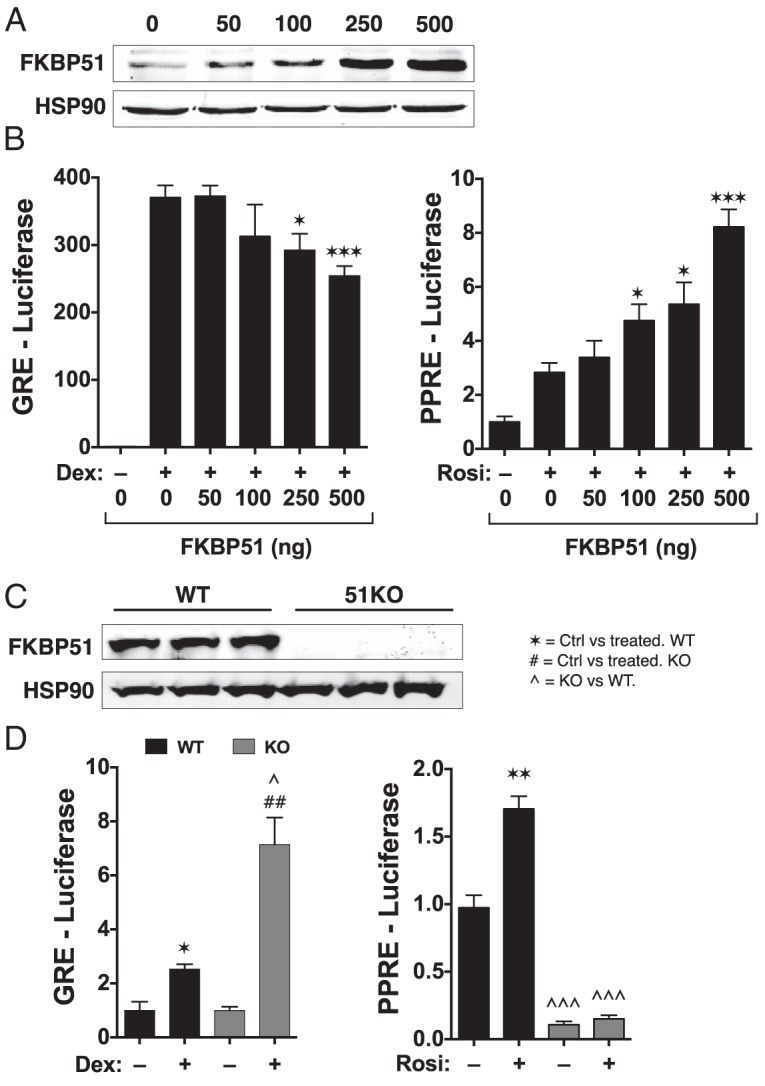
Reciprocal regulation of GRα and PPARγ activity by FKBP51. A, Receptor-less COS-7 cells were transfected with increasing concentrations of FKBP51 and assayed for FKBP51 expression by Western blotting. Hsp90 was used as a loading control. B, Cells of panel A were simultaneously transfected with expression constructs for GRα or PPARγ and analyzed for Dex-induced activity using a GRE-Luc reporter and Rosi-induced activity using PPRE-Luc (±SEM; n = 4–6). C, Western blot analysis of whole-cell extracts from WT and 51KO MEFs demonstrating a complete lack of FKBP51 in the 51KO cells. Hsp90 was used as a loading control. D, WT and 51KO cells were analyzed for GRα activity using GRE-Luc and PPARγ activity using PPRE-Luc in response to treatment with 1 μM Dex or 1 μM Rosi, respectively (±SEM; n = 3–6). All luciferase experiments used a pRL-CMV-Renilla reporter to normalize for transfection efficiency. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO.
Figure 5.
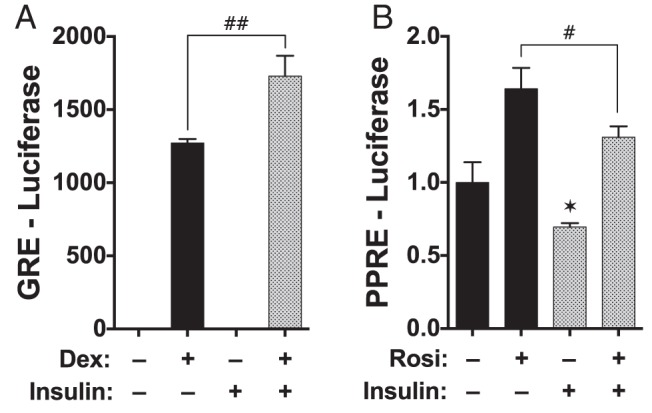
Insulin reciprocally regulates the intrinsic transcriptional activities of GRα and PPARγ. A, COS-7 cells were transiently transfected with GRα and GRE-Luc reporter in the absence or presence of insulin and Dex, as indicated. B, Similarly, COS-7 were transiently transfected with PPARγ and PPRE-Luc in the absence or presence of insulin and Rosi, as indicated. In each case, cells were transfected with pRL-CMV-Renilla to normalize for transfection efficiency. Final luciferase values were normalized to 1 for untreated cells. Results represent the means ± SEM (n = 6). *, control vs insulin; #, Dex or Rosi vs Dex or Rosi + insulin. Significant differences for all data are indicated as follows: *, P > .05; **, P > .01; the same parameters apply for #.
FKBP51 deficiency increases phosphorylation of GRα and PPARγ
In prior work, the phosphatase PP5 was found to decrease GRα but increase PPARγ activities by directly controlling the phosphorylation status of each receptor (11). Because FKBP51 exerts a similar pattern on the functions of these same receptors, we speculated that it might achieve this by indirectly controlling receptor phosphorylation. To test the phosphorylation status of PPARγ, we chose 3T3-L1 preadipocyte cells, which express detectable levels of endogenous PPARγ protein. Stable knockdown of FKBP51 in 3T3-L1 was achieved by lentivirus expression of FKBP51 shRNA. Western blot analysis demonstrated approximately 40% reduction of FKBP51 protein in 3T3-L1 FKBP51-knockdown cells (Figure 3A) and about a 2-fold increase in PPARγ phosphorylation at serine 112 (Figure 3B), suggesting that FKBP51 positively regulates PPARγ by decreasing inhibitory serine 112 phosphorylation.
Figure 3.
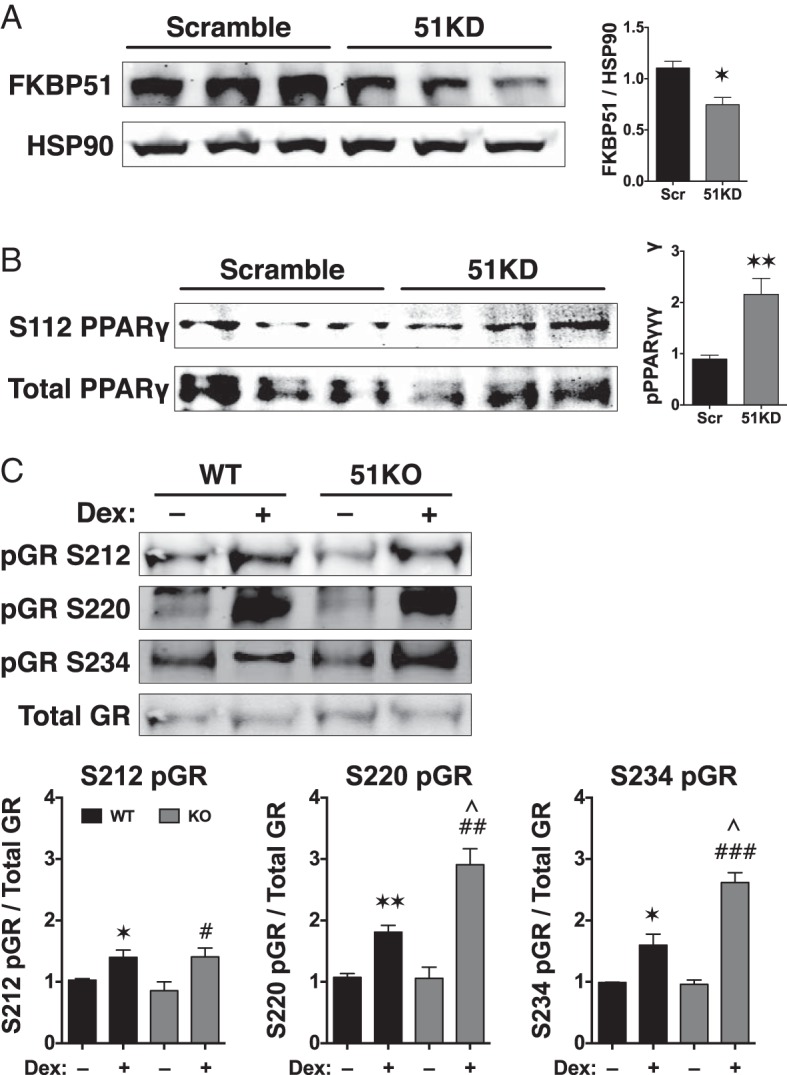
FKBP51 control of GRα and PPARγ phosphorylation. A, 3T3–L1 preadipocytes were infected with lentivirus for stable expression of shRNAs against FKBP51 (FKBP1 knockdown [51KD]) or a scrambled control (Scr). Cells were grown to confluence, total protein was isolated, and expression of FKBP51 was analyzed by Western blotting. FKBP51 expression was normalized to Hsp90 (±SEM; n = 3). B, Whole-cell extracts of scramble and 51KD 3T3–L1 preadipocytes were analyzed by Western blotting with antibodies against total PPARγ or phosphoserine 112 (S112) of PPARγ. Phospho-PPARγ signals were normalized to total PPARγ (±SEM; n = 4). C, Whole-cell extracts of WT and 51KO MEFs treated with or without Dex (100 nM) for 1 hour were analyzed by Western blotting with antibodies specific to serines 212, 220, and 234 of mouse GRα. The FiGR antibody was used to detect total GRα. Phospho-GRα signals were normalized to total GRα (±SEM; n = 4). All quantitations were performed by infrared spectrophotometry. Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO.
To date, the mechanism by which FKBP51 inhibits GRα transcriptional activity has not been uncovered. To determine whether this inhibition occurred through phosphorylation, the status of 3 Dex-inducible phosphoserines were measured in WT and 51KO cells, which express detectable levels of GRα protein. The results show Dex induction of serines 212, 220, and 234 in WT cells (Figure 3C). In 51KO cells, elevated Dex-induced phosphorylation of serines 220 and 234 was observed, whereas serine 212 remained unchanged compared with that in WT cells. Because phosphorylation of serines 220 and 234 is known to be stimulatory of GRα transcriptional activity, our data suggest that FKBP51-negative regulation of GRα occurs, at least in part, by reducing phosphorylation at these residues.
Loss of FKBP51 activates the Akt-p38 pathway to regulate GRα and PPARγ
Because FKBP51 has no intrinsic kinase or phosphatase activity, it must be indirectly controlling GRα and PPARγ phosphorylation. A long-standing clue to how FKBP51 might achieve this is the targeting of serines 220 and 234 by p38 kinase (20, 25, 26). More recently, another clue emerged when it was shown that FKBP51 acts as an essential chaperone to the Akt-specific phosphatase PHLPP (15). Last, Akt signaling that leads to phosphorylation and activation of p38 kinase has recently been established (27–30). We therefore reasoned that FKBP51 control of the Akt-p38 pathway is responsible for differential regulation of GRα and PPARγ. This theory was tested by measuring Akt phosphorylation at serine 473, the residue specifically targeted by PHLPP (15), in WT and 51KO cells subjected to insulin treatment (Figure 4A). The results show insulin induction of Akt phosphorylation in both WT and 51KO cells, as expected. However, 51KO cells had higher levels of Akt phosphorylation under both basal and insulin treatment conditions. To establish the Akt-p38 connection, levels of phospho-p38 were also measured in response to insulin (Figure 4B). In WT cells, insulin treatment did indeed increase p38 activation and once again phospho-p38 levels were higher in the 51KO cells under basal and insulin treatment conditions. As a second approach, we exploited the ability of Dex to rapidly activate p38 (42), which was increased in WT cells and which was further increased by Dex in 51KO cells (Figure 4C). Because GRα is known to inhibit Akt activation in muscle models of insulin resistance (43), we also tested whether Dex might blunt the Akt-p38 pathway by reducing Akt phosphorylation. Results in Figure 4D show that this is not the case in either WT or 51KO MEFs. Thus, the effects of FKBP51 loss on activation of the Akt-p38 pathway are not attenuated by Dex activation of GRα. Indeed, the higher levels of Dex-activated p38 seen in the 51KO cells is probably a factor contributing to the elevated GRα transcriptional activities seen in Figure 2.
Figure 4.

FKBP51 regulates Akt phosphorylation at serine 473 and p38 MAPK activation. A, WT and 51KO MEFs treated with or without insulin (INS) for 30 minutes were analyzed by Western blotting with Akt antibodies specific to phosphoserine 473 or total Akt. B, WT and 51KO MEFs treated with or without insulin for 30 minutes were analyzed by Western blotting with a p38 antibody with dual specificity against phosphothreonine 180/phosphotyrosine 182 or an antibody against total p38 MAPK. C, WT and 51KO MEFs treated with or without Dex (100 nM) for 1 hour were analyzed by Western blotting with the p38 antibodies (see panel B). D, WT and 51KO MEFs treated with or without Dex for 1 hour were analyzed by Western blotting with Akt antibodies (see panel A). All quantitations were performed by infrared spectrophotometry and phospho-Akt levels were normalized to total Akt while phospho-p38 levels were normalized to total p38. Hsp90 was used as a loading control. All results represent means ± SEM (n = 4). Statistical differences are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols. *, WT vs WT; #, 51KO vs 51KO; ∧, WT vs 51KO.
Because insulin caused activation of the Akt-p38 pathway, we reasoned that insulin treatment should reciprocally regulate GRα and PPARγ activities. This was tested by measuring the intrinsic transcriptional activities of each receptor in receptor-less COS-7 cells transfected with luciferase reporter constructs regulated by minimal response elements (Figure 5). The results show elevated GRα but reduced PPARγ activities in response to insulin.
To directly test the requirement of p38 activity in FKBP51 control of GRα and PPARγ, we used the p38-specific inhibitor PD169316 (37). As a first test, COS-7 cells were transfected with GRα or PPARγ and their respective reporter genes, followed by treatment with cognate ligand in the presence or absence of PD169316 (Figure 6A). The results show inhibition of GRα activity by PD169316 but stimulation of PPARγ. We next determined whether PD169316 could rescue GRα or PPARγ activities in the 51KO MEFs (Figure 6B). In 51KO cells, treatment with PD169316 decreased GRα activity at the GRE-Luc reporter to the levels seen in WT cells. In contrast, PD169316 increased PPARγ activity in 51KO cells to apparent WT levels. The rescue of PPARγ activity in the 51KO cells correlated with a decrease in receptor phosphorylation at serine 112 (Figure 6C).
Figure 6.

p38 MAPK inhibition decreases GRα and increases PPARγ activity by reducing its phosphorylation. A, Analysis of GRα and PPARγ in PD169316 (PD)-treated COS-7 cells. COS-7 cells were transiently transfected with GRα and GRE-Luc reporter in the absence or presence of PD169316 and Dex, as indicated. Similarly, COS-7 were transiently transfected with PPARγ and PPRE-Luc in the absence or presence of PD169316 and Rosi, as indicated. In each case, luciferase activities were normalized to 1 for untreated cells. Results represent the means ±SEM (n = 6). *, Ctrl vs PD169316; #, Dex or Rosi vs Dex or Rosi + PD169316. B, Reversion of GRα and PPARγ activities in PD169316-treated 51KO MEFs. WT and 51KO cells were transiently transfected with GRα and GRE-Luc reporter or PPARγ and PPRE-Luc reporter, in the absence or presence of PD169316 and cognate ligand, as indicated. In each case, luciferase activities were normalized to 1 for untreated cells. Results represent the means ± SEM (n = 6). *, Ctrl vs Dex or Rosi; #, Dex or Rosi vs Dex or Rosi + PD169316. C, Inhibition of p38MAPK decreases PPARγ phosphorylation in 51KO cells. To increase PPARγ expression and activation, WT and 51KO cells were induced to differentiate into adipocytes, as described in companion paper (41). At day 5 postinduction, the extent of PPARγ phosphorylation was determined by Western blotting with antibodies against total PPARγ or phosphoserine 112 (S112) PPARγ in the absence or presence of PD169316, as indicated. Phospho-PPARγ signals were normalized to total PPARγ. Hsp90 was used as loading control. Results represent the means ± SEM (n = 3). *, WT control vs WT PD169316; #, 51KO control vs 51KO PD169316; ∧, WT control vs 51KO control. Significant differences for all data are indicated as follows: *, P > .05; **, P > .01; ***, P > .001; the same parameters apply for # and ∧ symbols.
Increased nuclear retention of PPARγ in 51KO cells
The PHLPP/Akt mechanism and the results of Figures 3 to 6 support a model of FKBP51 action that is independent of its interaction with nuclear receptors. What then is the role of FKBP51 within GRα and PPARγ heterocomplexes? In prior work, we demonstrated that most hormone-free GRα is localized to the cytoplasm as heterocomplexes principally containing FKBP51 and that hormone binding initiates an exchange of FKBP52 for FKBP51 that correlates with nuclear translocation (13). We have also shown that a sizable fraction of hormone-free GRα shifts to the nucleus in 51KO MEFs (7). Because the active form of p38 kinase is uniformly localized to the nucleus (44), we propose that a major role of receptor-bound FKBP51 is to sequester the receptor away from nuclear p38 activation. To test this model with respect to PPARγ, steady-state intracellular localization in live cells was determined by transfecting WT and 51KO cells with PPARγ linked to GFP (Figure 7). Moreover, to ensure as much as possible that this analysis reflected ligand-free PPARγ, the cells were grown in media stripped of free fatty acids by dextran-coated charcoal (39, 40). As reported by others, PPARγ in WT cells was found predominantly in the nucleus; however, a clearly visible portion (∼35%) is localized to the cytoplasm. Interestingly, in 51KO cells, PPARγ distribution became almost exclusively nuclear (∼90%). Thus, in 51KO cells, both GRα and PPARγ, even in the absence of hormone, are more likely to serve as targets for the active, nuclear form of p38.
Figure 7.
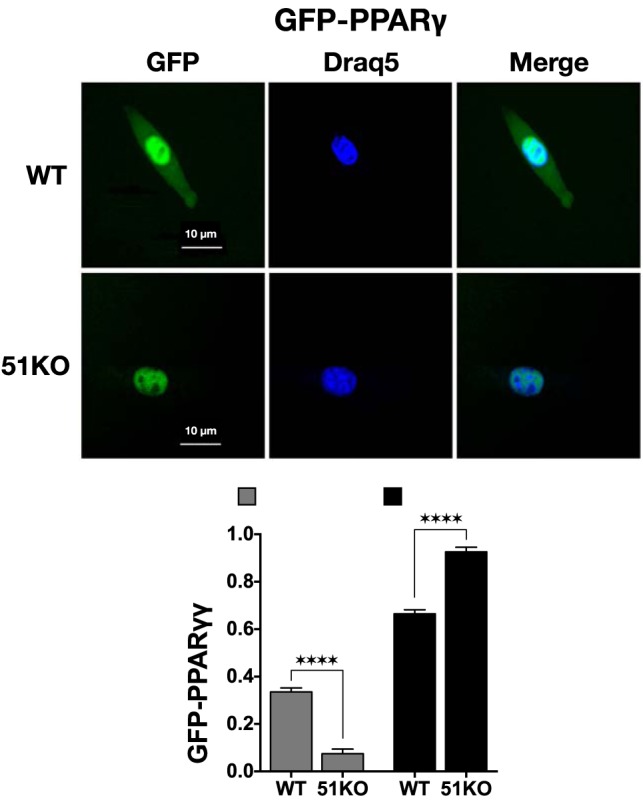
FKPB51 regulates steady-state intracellular localization of PPARγ to the cytoplasm. WT and 51KO MEFs were transfected with PPARγ-GFP in media containing charcoal-stripped serum to remove free fatty acids. DRAQ5 was used to label nuclear DNA. GFP and DRAQ5 were visualized 24 hours after transfection using a Leica DMIRE2 confocal microscope. Images shown are representative of 3 independent experiments where a minimum of 50 cells per condition were inspected. The fluorescence microscopy images were used with ImageJ to measure cellular localization of PPARγ-GFP. All results represent means ± SEM (n = 9). ****, P > .0001.
Discussion
In this work, we show the interaction of the FKBP51 cochaperone with the PPARγ-Hsp90 heterocomplex and demonstrate that it is a required for full transcriptional activity of the receptor. We contrast this role of FKBP51 with its ability to interact with GRα complexes, but cause repression, rather than stimulation, of transcriptional activity. A model depicting the essential features of this regulatory process is seen in Figure 8. According to the model, reciprocal regulation of GRα and PPARγ by FKBP51 is attributed to 2 complementary mechanisms. In the first, FKBP51 serves to inhibit Akt kinase activation at serine 473 by acting as a chaperone or scaffold protein for the Akt-specific phosphatase PHLPP, as previously reported by the laboratories of Wang and colleagues (15) and Hausch and colleagues (16). Suppression of Akt kinase by FKBP51, in turn, leads to repression of p38 kinase activation, a kinase known to activate GRα at serines 220 and 234 but to repress PPARγ at serine 112. In the second, FKBP51 also controls intracellular localization of each receptor, promoting the cytoplasmic localization of each. The latter feature is essential because the activated form of p38 kinase is known to reside exclusively within the nucleus (44). Thus, FKBP51 simultaneously represses the Akt-p38 phosphorylation cascade that regulates each receptor, while at the same time sequestering at least a fraction of each receptor away from the active form of p38.
Figure 8.

Model for FKBP51 regulation of GRα and PPARγ. FKBP51 reciprocally modulates GRα and PPARγ by 2 distinct pathways. In the first, FKBP51 serves as a scaffold for recruitment of PHLPP phosphatase to Akt (15, 16), leading to Akt inactivation by dephosphorylation at serine 473. In turn, this reduces activation of p38 MAPK, leading to reduced stimulatory phosphorylation of GRα at serines 220 and 234, while simultaneously reducing the inhibitory phosphorylation of PPARγ at serine 112. In the second, FKBP51 serves to sequester both GRα and PPARγ to the cytoplasm, away from the active state of p38 MAPK in the nucleus. The net effect of both mechanisms is negative regulation of GRα, but positive regulation of PPARγ by FKBP51. Ask1, apoptosis signal-regulating kinase-1.
Because FKBP51 functions as a scaffold for PHLPP and Akt, we wondered whether FKBP51 might recruit these proteins to GRα or PPARγ heterocomplexes. However, efforts to detect PHLPP or Akt interaction with either receptor through immunoaffinity purification in WT cells proved unsuccessful (data not shown). This result reinforces the fact that PHLPP interaction is mediated by the TPR domain of FKBP51 (15), the same domain required for recruitment to nuclear receptor complexes via binding to Hsp90 (2). Thus, FKBP51 appears to be targeting 2 distinct clients (PHLPP and Hsp90) in its role as regulator of nuclear receptor function. It is interesting to note that FKBP51 is a prominent GRα-induced gene (45, 46). Thus, it is also likely that FKBP51 repression of the Akt/p38 axis serves as an autoregulatory loop for down-regulation of GRα activity.
The major mechanism by which FKBP51 serves to repress not only GRα but also progesterone and mineralocorticoid receptors (MRs) has remained elusive (47). We and others have shown that most GRα and MR complexes in most cells are found in the cytoplasm in the absence of hormone, with FKBP51 as the dominant TPR protein, and that hormone binding generates a transient state in which GRα and MR translocate to the nucleus with FKBP52 as the dominant TPR (8, 13, 48). Supporting this is the observation that hormone-free progesterone receptor complexes reside in the nucleus principally bound by FKBP52 (7). Thus, FKBP51 appears to promote cytoplasmic localization of GRα. Yet, in 51KO cells only a fraction of GRα was found to redistribute to the nucleus (7). Because cellular localization of nuclear receptors is dynamic, even in the presence of hormone (49, 50), a small kinetic redistribution of receptors could have a significant impact on activity by making receptors accessible to regulatory factors, such as nuclear kinases. However, based on our static measurements alone, the change in receptor localization observed in 51KO cells did not seem to fully account for the highly elevated GRα activity seen in these cells. We and others have also measured reduced hormone-binding affinity of approximately 3-fold for GRα bound with FKBP51 compared with that for FKBP52 (14). Yet, here too this result cannot fully explain the approximately 20-fold increase in GRα activity seen at some genes in 51KO cells (see pyruvate dehydrogenase kinase-44; Figure 5 in companion article [41]), especially considering that saturating concentrations of Dex (100 nM) were used in those experiments. Thus, the FKBP51-mediated phosphorylation mechanism uncovered here appears to be a major missing element in the prominent role played by FKBP51, not only on GRα activity, but now also on the actions of PPARγ.
Here we have shown insulin activation of the Akt-p38 axis. However, because p38 and Ask1 are known to be activated by cellular stresses, such as heat shock, arsenite, and hydrogen peroxide (44, 51, 52), our discovery may have also uncovered a mechanism by which heat shock and other forms of stress cause potentiation of GRα and progesterone receptor transcriptional activities (53, 54). Although p38 has yet to be directly implicated in heat shock potentiation of GRα, it is responsible for stress-induced phosphorylation of heat shock transcription factor-1 (55), whose activity contributes to stimulation of GRα under stress conditions (56). It is now likely that stress-activated p38 may be targeting both GRα and heat shock transcription factor-1.
Although strictly a molecular investigation, this work implies that FKBP51 should be an important regulator of the cellular and physiological processes controlled by GRα and PPARγ. In our companion work (41), we test this hypothesis by investigating a cellular response shared by each receptor: lipid metabolism in adipocytes.
Acknowledgments
We thank Dr Michael J. Garabedian (New York University School of Medicine, New York, New York) for the gift of phospho-specific antibodies to GRα, Dr Guillermo Vazquez (University of Toledo College of Medicine, Toledo, Ohio) for the gift of phospho-Akt antibody, and Dr Andrea Nestor-Kalinoski (University of Toledo College of Medicine) for the gift of DRAQ5.
This work was supported in part by Department of Physiology and Pharmacology (University of Toledo College of Medicine) funds awarded to E.R.S. and by National Institutes of Health (Grants DK70127 [to E.R.S.], DK73402 [to W.S.], and DK054254 and DK083850 [to S.M.N.]). L.A.S. was supported in part by the Center for Diabetes and Endocrine Research Hiss Foundation Fellowship. T.D.H. was supported by National Institutes of Health PRIDE Grant HL106365.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Dex
- dexamethasone
- FKBP
- FK506-binding protein
- GFP
- green fluorescent protein
- GRα
- glucocorticoid receptor-α
- GRE
- glucocorticoid response element
- Hsp90
- heat shock protein-90
- 51KO
- FKBP51 knockout
- Luc
- luciferase
- MEF
- mouse embryonic fibroblast
- MR
- mineralocorticoid receptor
- PHLPP
- PH domain leucine-rich repeat protein phosphatase
- PP5
- protein phosphatase-5
- PPAR
- peroxisome proliferator-activated receptor
- PPRE
- peroxisome proliferator response element
- Rosi
- rosiglitazone
- shRNA
- short hairpin RNA
- TBS
- Tris-buffered saline
- TPR
- tetratricopeptide repeat
- WT
- wild-type.
References
- 1. Smith DF, Baggenstoss BA, Marion TN, Rimerman RA. Two FKBP-related proteins are associated with progesterone receptor complexes. J Biol Chem. 1993;268:18365–18371 [PubMed] [Google Scholar]
- 2. Nair SC, Rimerman RA, Toran EJ, et al. Molecular cloning of human FKBP51 and comparisons of immunophilin interactions with Hsp90 and progesterone receptor. Mol Cell Biol. 1997;17:594–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360 [DOI] [PubMed] [Google Scholar]
- 4. Smith DF, Toft DO. Minireview: The intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22:2229–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Riggs DL, Roberts PJ, Chirillo SC, et al. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riggs DL, Cox MB, Tardif HL, Hessling M, Buchner J, Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol. 2007;27:8658–8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banerjee A, Periyasamy S, Wolf IM, et al. Control of glucocorticoid and progesterone receptor subcellular localization by the ligand-binding domain is mediated by distinct interactions with tetratricopeptide repeat proteins. Biochemistry. 2008;47:10471–10480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wolf IM, Periyasamy S, Hinds T, Jr, Yong W, Shou W, Sanchez ER. Targeted ablation reveals a novel role of FKBP52 in gene-specific regulation of glucocorticoid receptor transcriptional activity. J Steroid Biochem Mol Biol. 2009;113:36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warrier M, Hinds TD, Jr, Ledford KJ, et al. Susceptibility to diet-induced hepatic steatosis and glucocorticoid resistance in FK506-binding protein 52-deficient mice. Endocrinology. 2010;151:3225–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zuo Z, Urban G, Scammell JG, et al. Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry. 1999;38:8849–8857 [DOI] [PubMed] [Google Scholar]
- 11. Hinds TD, Jr, Stechschulte LA, Cash HA, et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ). J Biol Chem. 2011;286:42911–42922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141:4107–4113 [DOI] [PubMed] [Google Scholar]
- 13. Davies TH, Ning YM, Sánchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277:4597–4600 [DOI] [PubMed] [Google Scholar]
- 14. Davies TH, Ning YM, Sánchez ER. Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry. 2005;44:2030–2038 [DOI] [PubMed] [Google Scholar]
- 15. Pei H, Li L, Fridley BL, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fabian AK, Marz A, Neimanis S, Biondi RM, Kozany C, Hausch F. InterAKTions with FKBPs—mutational and pharmacological exploration. PLoS One. 2013;8:e57508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bodwell JE, Webster JC, Jewell CM, Cidlowski JA, Hu JM, Munck A. Glucocorticoid receptor phosphorylation: overview, function and cell cycle-dependence. J Steroid Biochem Mol Biol. 1998;65:91–99 [DOI] [PubMed] [Google Scholar]
- 18. Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann NY Acad Sci. 2004;1024:86–101 [DOI] [PubMed] [Google Scholar]
- 19. Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3947–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kino T, Ichijo T, Amin ND, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568 [DOI] [PubMed] [Google Scholar]
- 22. Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277:26573–26580 [DOI] [PubMed] [Google Scholar]
- 23. Wang Z, Chen W, Kono E, Dang T, Garabedian MJ. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol Endocrinol. 2007;21:625–634 [DOI] [PubMed] [Google Scholar]
- 24. Bodwell JE, Hu JM, Orti E, Munck A. Hormone-induced hyperphosphorylation of specific phosphorylated sites in the mouse glucocorticoid receptor. J Steroid Biochem Mol Biol. 1995;52:135–140 [DOI] [PubMed] [Google Scholar]
- 25. Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–2392 [DOI] [PubMed] [Google Scholar]
- 26. Miller AL, Webb MS, Copik AJ, et al. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583 [DOI] [PubMed] [Google Scholar]
- 27. Fortin J, Patenaude A, Deschesnes RG, Côté MF, Petitclerc E, C-Gaudreault R. ASK1–P38 pathway is important for anoikis induced by microtubule-targeting aryl chloroethylureas. J Pharm Pharm Sci. 2010;13:175–190 [DOI] [PubMed] [Google Scholar]
- 28. Hyun MS, Hur JM, Mun YJ, Kim D, Woo WH. BBR induces apoptosis in HepG2 cell through an Akt-ASK1-ROS-p38MAPKs-linked cascade. J Cell Biochem. 2010;109:329–338 [DOI] [PubMed] [Google Scholar]
- 29. Pan J, Chang Q, Wang X, et al. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem Res Toxicol. 2010;23:568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klawitter J, Klawitter J, Agardi E, et al. Association of DJ-1/PTEN/AKT- and ASK1/p38-mediated cell signalling with ischaemic cardiomyopathy. Cardiovasc Res. 2013;97:66–76 [DOI] [PubMed] [Google Scholar]
- 31. Dalman FC, Koenig RJ, Perdew GH, Massa E, Pratt WB. In contrast to the glucocorticoid receptor, the thyroid hormone receptor is translated in the DNA binding state and is not associated with hsp90. J Biol Chem. 1990;265:3615–3618 [PubMed] [Google Scholar]
- 32. Dalman FC, Sturzenbecker LJ, Levin AA, et al. Retinoic acid receptor belongs to a subclass of nuclear receptors that do not form “docking” complexes with hsp90. Biochemistry. 1991;30:5605–5608 [DOI] [PubMed] [Google Scholar]
- 33. Sumanasekera WK, Tien ES, Turpey R, Vanden Heuvel JP, Perdew GH. Evidence that peroxisome proliferator-activated receptor α is complexed with the 90-kDa heat shock protein and the hepatitis virus B X-associated protein 2. J Biol Chem. 2003;278:4467–4473 [DOI] [PubMed] [Google Scholar]
- 34. Sumanasekera WK, Tien ES, Davis JW, 2nd, Turpey R, Perdew GH, Vanden Heuvel JP. Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-α (PPARα) and PPARβ activity. Biochemistry. 2003;42:10726–10735 [DOI] [PubMed] [Google Scholar]
- 35. Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science. 1996;274:2100–2103 [DOI] [PubMed] [Google Scholar]
- 36. Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272:5128–5132 [DOI] [PubMed] [Google Scholar]
- 37. Aouadi M, Laurent K, Prot M, Le Marchand-Brustel Y, Binétruy B, Bost F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes. 2006;55:281–289 [DOI] [PubMed] [Google Scholar]
- 38. Yong W, Yang Z, Periyasamy S, et al. Essential role for co-chaperone Fkbp52 but not Fkbp51 in androgen receptor-mediated signaling and physiology. J Biol Chem. 2007;282:5026–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen RF. Removal of fatty acids from serum albumin by charcoal treatment. J Biol Chem. 1967;242:173–181 [PubMed] [Google Scholar]
- 40. Viscardi RM, Ullsperger S, McKenna MC. Carbon stripping extracts serum free fatty acids: implications for media supplementation of cultured type II pneumocytes. Lab Invest. 1991;65:250–257 [PubMed] [Google Scholar]
- 41. Stechschulte LA, Hinds TD, Jr, Khuder S, Shou W, Najjar SM, Sanchez ER. FKBP51 controls cellular adipogenesis through p38 kinase-mediated phosphorylation of GRα and PPARγ. Mol Endocrinol. 2014;28:1265–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kewalramani G, Puthanveetil P, Kim MS, Wang F, et al. Acute dexamethasone-induced increase in cardiac lipoprotein lipase requires activation of both Akt and stress kinases. Am J Physiol Endocrinol Metab. 2008;295:E137–E147 [DOI] [PubMed] [Google Scholar]
- 43. Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61 [DOI] [PubMed] [Google Scholar]
- 44. Gong X, Ming X, Deng P, Jiang Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J Cell Biochem. 2010;110:1420–1429 [DOI] [PubMed] [Google Scholar]
- 45. Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leclerc N, Luppen CA, Ho VV, et al. Gene expression profiling of glucocorticoid-inhibited osteoblasts. J Mol Endocrinol. 2004;33:175–193 [DOI] [PubMed] [Google Scholar]
- 47. Hubler TR, Denny WB, Valentine DL, Cheung-Flynn J, Smith DF, Scammell JG. The FK506-binding immunophilin FKBP51 is transcriptionally regulated by progestin and attenuates progestin responsiveness. Endocrinology. 2003;144:2380–2387 [DOI] [PubMed] [Google Scholar]
- 48. Galigniana MD, Erlejman AG, Monte M, Gomez-Sanchez C, Piwien-Pilipuk G. The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol. 2010;30:1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Htun H, Barsony J, Renyi I, Gould DL, Hager GL. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc Natl Acad Sci USA. 1996;93:4845–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DeFranco DB, Ramakrishnan C, Tang Y. Molecular chaperones and subcellular trafficking of steroid receptors. J Steroid Biochem Mol Biol. 1998;65:51–58 [DOI] [PubMed] [Google Scholar]
- 51. Ludwig S, Hoffmeyer A, Goebeler M, et al. The stress inducer arsenite activates mitogen-activated protein kinases extracellular signal-regulated kinases 1 and 2 via a MAPK kinase 6/p38-dependent pathway. J Biol Chem. 1998;273:1917–1922 [DOI] [PubMed] [Google Scholar]
- 52. Morita K, Saitoh M, Tobiume K, et al. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001;20:6028–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Edwards DP, Estes PA, Fadok VA, et al. Heat shock alters the composition of heteromeric steroid receptor complexes and enhances receptor activity in vivo. Biochemistry. 1992;31:2482–2491 [DOI] [PubMed] [Google Scholar]
- 54. Sanchez ER, Hu JL, Zhong S, Shen P, Greene MJ, Housley PR. Potentiation of glucocorticoid receptor-mediated gene expression by heat and chemical shock. Mol Endocrinol. 1994;8:408–421 [DOI] [PubMed] [Google Scholar]
- 55. Kim J, Nueda A, Meng YH, Dynan WS, Mivechi NF. Analysis of the phosphorylation of human heat shock transcription factor-1 by MAP kinase family members. J Cell Biochem. 1997;67:43–54 [DOI] [PubMed] [Google Scholar]
- 56. Jones TJ, Li D, Wolf IM, Wadekar SA, Periyasamy S, Sánchez ER. Enhancement of glucocorticoid receptor-mediated gene expression by constitutively active heat shock factor 1. Mol Endocrinol. 2004;18:509–520 [DOI] [PubMed] [Google Scholar]