

Abstract
The structural, functional, and mechanistic characterization of several types of post-translational modifications (PTMs) is well-documented. PTMs, however, may interact or interfere with one another when regulating protein function. Yet, characterization of the structural and functional signatures of their crosstalk has been hindered by the scarcity of data. To this end, we developed a unified sequence-based predictor of 23 types of PTM sites that, we believe, is a useful tool in guiding biological experiments and data interpretation. We then used experimentally determined and predicted PTM sites to investigate two particular cases of potential PTM crosstalk in eukaryotes. First, we identified proteins statistically enriched in multiple types of PTM sites and found that they show preferences toward intrinsically disordered regions as well as functional roles in transcriptional, posttranscriptional, and developmental processes. Second, we observed that target sites modified by more than one type of PTM, referred to as shared PTM sites, show even stronger preferences toward disordered regions than their single-PTM counterparts; we explain this by the need for these regions to accommodate multiple partners. Finally, we investigated the influence of single and shared PTMs on differential regulation of protein–protein interactions. We provide evidence that molecular recognition features (MoRFs) show significant preferences for PTM sites, particularly shared PTM sites, implicating PTMs in the modulation of this specific type of macromolecular recognition. We conclude that intrinsic disorder is a strong structural prerequisite for complex PTM-based regulation, particularly in context-dependent protein–protein interactions related to transcriptional and developmental processes. Availability: http://www.modpred.org
Keywords: post-translational modification, intrinsically disordered protein, molecular recognition feature, MoRF, prediction, crosstalk, steric competition, protein, protein interaction
INTRODUCTION
Protein post-translational modifications (PTMs) are important biochemical events involved in the regulation of various cellular functions.1–3 PTM-based regulation can occur through the individual effect of a modification at a single residue or through combined effects over multiple sites undergoing the same or different modifications. This modus operandi is best exemplified by the “histone code hypothesis,” according to which distinct PTMs on histones, sequentially or in combination, regulate downstream chromatin processes.4 Over the past decade, growing evidence has suggested that the concept of regulatory interplay among PTMs can be extended to other proteins,3,5–8 with specific examples including PDGFR-β,9 p300/CBP,10 RNA polymerase II (subunit RPB1),11 α-tubulin,12 Cdc25C phosphatase,13 FoxO family of transcription factors,14 and p53.15 Furthermore, several recent large-scale studies have established extensive crosstalk between different pairs of PTMs such as O-linked glycosylation-phosphorylation,16 acetylation-phosphorylation,17 acetylation-ubiquitylation18,19 and phosphorylation-ubiquitylation.20 The above studies have revealed three general modes of concerted PTM-based regulation. First, nonadjacent residues may be modified by one or more PTMs in a sequential or combinatorial manner to induce structural changes. Second, clusters of PTMs within a small region of the protein may alter local surface properties for recognition by effector molecules. Third, depending on the context, for example, tissue-type, stage of cell cycle, or external stimuli, steric competition between PTMs at the same site may result in the differential control of protein function or a different function altogether. For a given protein, it is possible that all these modes of regulation exist simultaneously (Fig. 1).
Figure 1.
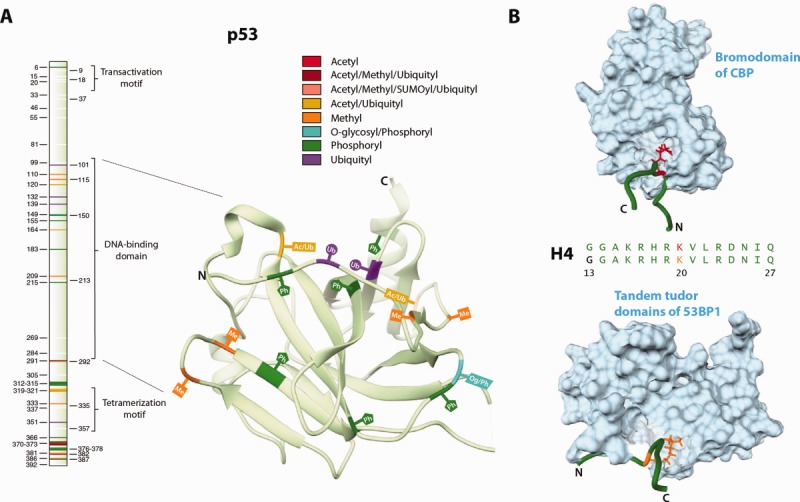
Examples of the three modes of concerted PTM regulation. (A) The primary sequence of the p53 tumor suppressor protein along with the structure of its DNA-binding domain (ID: 2YBG). The different PTM sites are highlighted and labeled in different colors. Sequential ubiquitylation of lysines in the DNA-binding domain and the C-terminus has been shown to regulate the nuclear export of p53.85 Additionally, relationships between different but overlapping clusters of N-terminal phosphorylation sites have been thought to check untimely p53 activation and enable signal integration and amplification over multiple stress pathways.86 A recent study identified 150 novel modifications and suggested that by virtue of the unusually high number of PTM sites, the combinatorial regulation of p53 is far more complex than previously thought.87 In this study, we refer to such proteins as being enriched in one or more types of PTM sites. (B) Acetylation and dimethylation of Lys20 in histone H4 results in recognition by the transcriptional coactivator CBP (ID: 2RNY, model 6) and the DNA-damage response protein 53BP1 (ID: 2LVM, model 10), respectively. Only the most informative NMR models as calculated by Olderado88 are shown here. A slight difference in structure can be seen, with the acetylated form having a larger bend than the dimethylated form. Each of these interactions results in a different functional outcome. The binding of CBP has a strong affinity in vitro89 and is speculated to increase the acetylation of H3 and H4 histones, which is generally associated with transcription activation.90,91 Unlike the recognition of the acetylation mark, the interaction between 53BP1 and dimethylated form occurs only in a specific cellular context and is important for the promotion of nonhomologous end-joining repair in response to DNA damage.92 Recently, a mass spectrometry-based study of in vivo histone acetylation dynamics reported that a sharp reduction in acetylation at Lys20 was due to increased methylation.93 For simplicity, we refer to such sites of competition (with more than one observed modification) as shared PTM sites, those sites with only one observed modification as single-PTM sites and those with no observed modifications as non-PTM sites.
PTM sites have been extensively studied with respect to their structural preferences and can broadly be divided into two groups.21–23 The first group includes PTM types with sites largely found in regions of well-defined secondary structure. Examples include acetylation,24,25 palmitoylation,23 and N-linked glycosylation,26 among others. PTMs from the second group show strong preference for intrinsically disordered regions, that is, regions without a single dominant macrostate under physiological conditions.27,28 For example, statistical associations between PTM sites and intrinsic disorder have been observed for phosphorylation,29,30 methylation,31 and ubiquitylation.32 Understanding the structure surrounding PTM sites has also provided key mechanistic insights into the effects of PTMs on protein folding and binding. Modified residues can induce orthosteric and/or allosteric effects that result in shifts toward novel low-energy conformations and their (de)stabilization.3,33–35
PTMs can also occur at interaction interfaces and can influence protein-protein binding.23,36 In this context, molecular recognition features (MoRFs) are an interesting class of interaction sites. MoRFs are short structured or loosely structured fragments within disordered regions that are important for high-specificity/low-affinity interactions in signal transduction, cell regulation, and many other functions.37,38 The observation of PTM sites in MoRFs has led to the speculation that PTMs may enable selective binding of these regions to one or more partners in a dynamic- and context-specific manner.39
Despite the progress in understanding the structural aspects of PTMs and their resulting consequences, the focus has largely been on characterizing the different types of modifications individually. However, with recent advances in the rapid and high-throughput identification of some PTMs, large-scale studies that integrate information on different types of modification sites have become realistic. For example, two recent studies have used conservation of individual sites40 or coevolution of site-pairs41 to infer global functional relationships between PTM sites. At the local level, Woodsmith et al.42 observed that more than 80% of PTM integration (PTMi) spots overlap with disordered regions. Most recently, short conserved sequence motifs containing any two PTM sites have been identified and used to assign joint functional roles to such pairings of sites.43
Interestingly, to date, there have been no large-scale studies on the characterization of properties surrounding sites shared by multiple PTMs. This can largely be attributed to a paucity of data for such sites. Even with the latest methods of detection, a full repertoire of sites has not been established for any PTM type.44 Furthermore, most methods have been applied only to a handful of PTMs such as phosphorylation, glycosylation, acetylation, and ubiquitylation, thus limiting studies on shared modification sites. A complementary approach involves the use of computational methods for predicting PTM sites. Many PTM site prediction methods have been developed;45,46 however, to the best of our knowledge, no unified tool exists for a simultaneous prediction of sites for more than a few types of modifications. Zhou et al. suggested a unified user interface to connect several independently developed predictors for different PTM types.47 This approach introduces a problem of interpreting heterogeneous prediction results, for example scores drawn from different distributions.
In this study, we predict and analyze multiple types of PTM sites simultaneously to gain structural and functional insights into the regulation of proteins by multi-PTM interplay. Specifically, we focus on both protein-level and site-level regulation and address issues of limited data through the predictor development. Our work provides evidence that intrinsic disorder is a key structural signature of complex PTM-based crosstalk in eukaryotes and enables the regulation of protein–protein interactions in transcriptional and developmental processes.
Results
Predictor development and performance evaluation
To train a combined predictor of PTM sites, we first collected data from public databases and the literature (see Materials and Methods). In total, this data contained 278,703 PTM sites. These sites were found in 54,484 proteins from 3,219 species. After the removal of redundant sites, the training data set contained 126,036 experimentally verified PTM sites (positives) and 971,129 sites not known to be modified (negatives), as shown in Table I. Next, we trained bootstrapped ensembles of logistic regression models for each PTM type and evaluated them using cross-validation. Finally, we built a webserver and standalone tool called ModPred for the prediction of PTM sites on single and multiple sequences, respectively. A schematic summary of the models in ModPred is provided in Supporting Information, Figure S1. All data sets, installable software, and the prediction server can be accessed at our website (see Materials and Methods).
Table I.
Summary of Data Sets and the Results of Cross-Validation Tests
| Number of sites | AUC | sn at sp = 0.90 | sn at sp = 0.95 | sn at sp = 0.99 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Modification type | Residue | Positive | Negative | No PSSM | PSSM | No PSSM | PSSM | No PSSM | PSSM | No PSSM | PSSM |
| Acetylation | K | 6,848 | 149,314 | 0.688 | 0.713 | 0.277 | 0.312 | 0.168 | 0.188 | 0.046 | 0.057 |
| ADP-ribosylation | E, R | 108 | 4,681 | 0.739 | 0.753 | 0.356 | 0.369 | 0.221 | 0.236 | 0.063 | 0.062 |
| Amidation | All | 457 | 29,966* | 0.964 | 0.967 | 0.923 | 0.930 | 0.851 | 0.866 | 0.570 | 0.615 |
| C-linked glycosylation | W | 32 | 118 | 0.938 | 0.928 | 0.756 | 0.837 | 0.606 | 0.750 | 0.454 | 0.415 |
| Carboxylation | E | 112 | 1,063 | 0.920 | 0.939 | 0.795 | 0.843 | 0.641 | 0.767 | 0.359 | 0.535 |
| Disulfide linkage | C | 9,736 | 7,101 | 0.646 | 0.783 | 0.182 | 0.391 | 0.110 | 0.246 | 0.037 | 0.078 |
| Farnesylation | C | 41 | 59* | 0.857 | 0.862 | 0.533 | 0.633 | 0.319 | 0.225 | 0.174 | 0.041 |
| Geranylgeranylation | C | 30 | 43* | 0.866 | 0.919 | 0.571 | 0.687 | 0.393 | 0.596 | 0.230 | 0.534 |
| GPI-anchor amidation | N | 84 | 2,362 | 0.961 | 0.966 | 0.908 | 0.905 | 0.841 | 0.853 | 0.518 | 0.528 |
| Hydroxylation | K, P, Y | 219 | 4,209 | 0.832 | 0.907 | 0.535 | 0.732 | 0.388 | 0.586 | 0.109 | 0.253 |
| Methylation | K, R | 628 | 18,561 | 0.660 | 0.674 | 0.319 | 0.349 | 0.243 | 0.264 | 0.130 | 0.127 |
| Myristoylation | G | 99 | 119* | 0.792 | 0.852 | 0.353 | 0.514 | 0.175 | 0.354 | 0.038 | 0.008 |
| N-linked glycosylation | N | 11,286 | 78,050 | 0.790 | 0.806 | 0.215 | 0.330 | 0.066 | 0.160 | 0.018 | 0.030 |
| N-terminal acetylation | A, G, M, S, T | 1,310 | 2,002* | 0.821 | 0.836 | 0.471 | 0.503 | 0.310 | 0.331 | 0.093 | 0.106 |
| O-linked glycosylation | S, T | 1,427 | 44,048 | 0.731 | 0.749 | 0.350 | 0.376 | 0.228 | 0.253 | 0.059 | 0.082 |
| Palmitoylation | C | 245 | 1,298 | 0.856 | 0.881 | 0.625 | 0.679 | 0.467 | 0.525 | 0.192 | 0.244 |
| Phosphorylation | S, T, Y | 90,058 | 320,506 | 0.771 | 0.777 | 0.422 | 0.437 | 0.296 | 0.312 | 0.113 | 0.116 |
| Proteolytic cleavage | All | 997 | 257,783 | 0.727 | 0.759 | 0.379 | 0.420 | 0.264 | 0.291 | 0.085 | 0.102 |
| PUPylation | K | 87 | 1,077 | 0.658 | 0.786 | 0.218 | 0.436 | 0.123 | 0.256 | 0.042 | 0.059 |
| Pyrrolidone carb. acid | Q | 275 | 2,789 | 0.880 | 0.906 | 0.682 | 0.770 | 0.538 | 0.658 | 0.188 | 0.389 |
| Sulfation | Y | 121 | 667 | 0.913 | 0.930 | 0.772 | 0.832 | 0.575 | 0.629 | 0.304 | 0.268 |
| SUMOylation | K | 744 | 17,539 | 0.742 | 0.739 | 0.419 | 0.458 | 0.311 | 0.360 | 0.135 | 0.172 |
| Ubiquitylation | K | 1,092 | 27,774 | 0.583 | 0.605 | 0.164 | 0.185 | 0.089 | 0.101 | 0.020 | 0.025 |
Each row shows mean performance for one PTM type, combined over all amino acids and subdata sets (motifs and non-motifs). A breakdown of performance for each amino acid and data set is provided in Supporting Information, Table S1. The “No PSSM” column represents the basic classification model and the “PSSM” column represents the model enhanced with evolutionary features. Area under the ROC curve (AUC) is shown for each PTM as well as sensitivity (sn; true positive rate) at different levels of specificity (sp; true negative rate). ROC curves for each amino acid and data set are provided in Supporting Information, Figure S3. Values marked in bold indicate the better-performing model. The data sets marked with a “*” indicate that the negatives were obtained from proteins different to those containing the positives through a random sampling procedure (See Materials and Methods).
The classification performance of ModPred combined over all amino acids for a given PTM is shown in Table I. The model using evolutionary features performed better than the model without them (21 out of 23 PTMs; P = 2.9 × 10−6; binomial test); therefore, we used this model in all subsequent analyses. Except for the cases of specialized predictors (e.g., kinase-specific predictors of phosphorylation sites48), ModPred reaches similar accuracy as other available tools and thus provides benefits by unifying the computational and statistical framework utilized across different PTMs. Unfortunately, accurate direct comparisons with other tools are very difficult, as witnessed by the emergence of critical assessments in many fields,49 because those tools use different data sets, different evaluation protocols, or have different application objectives.
Proteins containing long regions of disorder are enriched in multiple types of PTM sites
We reasoned that the probability of concerted regulation by multiple types of PTMs of a protein would be higher if the protein is predicted to contain an unusually large number of sites for more than one type of PTM. To identify such site-enriched proteins, we first ran ModPred on two data sets: the set of all eukaryotic disordered proteins obtained from DisProt50 and the set of reference proteomes of seven eukaryotic species (see Materials and Methods). We “called” PTM sites based on thresholds corresponding to a false positive rate of 0.1. Then, for a given modification, we defined a protein to be enriched in PTM sites if it contained a significantly larger proportion of predicted sites than expected by chance. A significant P-value implied that the protein held the potential to be excessively modified.
Next, we investigated the structural properties of proteins enriched in sites for multiple PTM types. For all proteins, we counted the number of PTM types that showed significant P-values in our statistical enrichment test and grouped them based on whether they contained long regions of disorder (at least 30 consecutive residues) or not. On the DisProt data set, we observe that proteins containing experimentally characterized long disordered regions (LDRs) are enriched in more types of PTM sites than proteins without LDRs (1.88 vs. 0.99; P = 2.3 × 10−7; t-test). Conversely, proteins enriched in two or more types of PTM sites contain significantly larger fractions of disordered residues (0.55 vs. 0.28; P = 6.2 × 10−18; t-test). As shown in Figure 2(A), while half of all proteins containing known LDRs are enriched in at least two types of PTM sites, only a quarter of proteins without LDRs show such multi-PTM enrichment.
Figure 2.
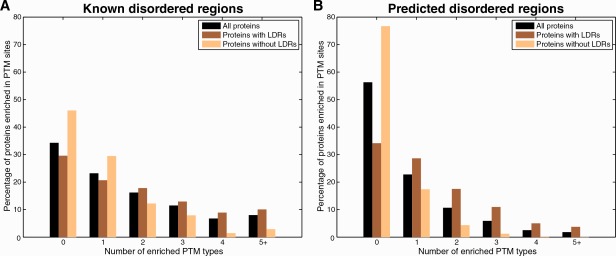
Distribution of the number of PTM types enriched in (A) the set of known disordered proteins from DisProt and (B) the proteomes of seven model organisms when all predicted sites were included. For a given protein, the number of PTM types for which a statistically significant enrichment of sites was found was recorded. This number is represented in the groupings on the x-axis. When performed over the entire data set, this analysis yielded proportions of the proteome for each PTM type enrichment count. This is represented on the y-axis.
For the data set of seven proteomes, we used predictions of structural disorder51 to address the unavailability of known disordered regions. In this case as well, we observe that proteins containing predicted LDRs are enriched in sites for a larger number of PTM types than proteins without LDRs (1.42 vs. 0.39; P < 10−64; t-test). As further support, we also find that proteins containing extremely long regions of disorder (at least 100 consecutive residues) are enriched in more types of PTM sites than LDR-containing proteins (2.27 vs. 0.88; P < 10−64; t-test). Again, proteins enriched in at least two types of PTMs show preferences for disordered regions (average disorder score of 0.43 vs. 0.11; P < 10−64; t-test). We find that 65% of proteins that are predicted to contain LDRs are enriched in sites for one or more types of PTMs [Fig. 2(B)], while 40% are enriched in sites for at least two PTM types. These fractions are considerably larger than those for the set of proteins without LDRs (23 and 7%, respectively).
Since disordered regions are likely to occur at the terminal regions of proteins, we also investigated whether these distributions are the result of a nonrandom accumulation of PTM sites at the termini. We find that when residues at the N- and C-termini (10% of protein length) are excluded, the distributions largely remain the same, indicating the absence of significant positional bias for enriched PTM sites in the primary structure (data not shown).
Functional signatures of proteins enriched in multiple types of PTM sites
To better understand the functional roles of proteins under complex PTM-based regulation, we carried out an enrichment analysis of UniProt52 keywords associated with proteins statistically enriched in predicted sites for at least two PTM types. We used a one-sided Fisher's exact test53 to ask whether the relative frequencies of occurrence of particular keywords were significantly larger in this set of proteins (main set) than in the remaining proteins (control set). We find that the top 10 keywords that correlate with enrichment of multiple PTMs are largely related to DNA-binding and transcriptional regulation processes [Fig. 3(A)]. While not represented in the top 10, keywords such as “RNA-binding,” “mRNA processing,” “mRNA splicing,” and “spliceosome” all show significant P-values (Supporting Information, Table S2). Additionally, we find significant enrichment of keywords associated with cellular differentiation and development such as “cell cycle,” “cell division,” and “Wnt signaling pathway.” Significantly depleted terms include those associated with membrane proteins and various metabolic enzymes. Interestingly, keywords related to signaling such as “transducer,”, “sensory transducer,” and “receptor” are significantly depleted for the set of proteins enriched in multiple PTMs. These terms are generally associated with proteins that exist in the membrane and participate in the initial steps of major signaling pathways. While this may be suggestive of preferential differences in upstream and downstream signaling proteins for PTM-based regulation, we hesitate to assign any biological meaning to this depletion.
Figure 3.

UniProt keyword enrichment analysis for the set of proteins enriched in sites for two or more PTM types. P-values were calculated using a one-tailed Fisher's exact test for each keyword and fold-enrichment or depletion was calculated by taking the ratios of the frequencies of keywords in the main set to the frequencies in the control set. This was repeated for three types of data sets—(A) the set of all proteins, (B) the set of proteins with LDRs, and (C) the set of proteins without LDRs. Only the top 10 keywords are shown here and additional significant keywords are reported in Supporting Information, Table S2.
Previous studies have shown that long regions of intrinsic disorder correlate with signaling, transcription, splicing, and developmental processes and anticorrelate with metabolic and transporter processes.54–56 To exclude the possibility that our results are a consequence of the presence of LDRs as opposed to the enrichment in multiple types of PTM sites, we split the main and control protein sets based on whether they contained LDRs or not. We then compared the LDR-containing proteins in the main set to the LDR-containing proteins in the control set. Similarly, we repeated the analyses only on the ordered proteins. As shown in Figure 3(B,C), the top 10 keywords in both the disordered and ordered analyses largely confirm the associations detected above (Supporting Information, Table S2). Three additional observations stand out. First, the term “alternative splicing” is significantly enriched in the disordered main set but significantly depleted in the ordered main set, which suggests a joint role of PTMs and alternative splicing in generating functional diversity of disordered proteins. On the other hand, although ordered proteins are less likely to be alternatively spliced,57 functional diversity in these proteins may still be enhanced through complex PTM-based regulation. Second, several keywords for mitochondrial localization and processes such as “tricarboxylic acid cycle,” “mitochondrion,” “pyruvate,” “respiratory chain,” “electron transport,” “ubiquinone,” and “glucose metabolism” are specifically enriched in the ordered main set. On closer inspection, the most frequently enriched PTM types in these proteins were acetylation and ubiquitylation, which have been previously observed in mitochondria.58,59 Third, molecular functions related to immunity such as “antibiotic,” “MHC I,” “defensin,” and “fungicide” are enriched in the ordered main set but not in disordered. This suggests a role for structure-based PTM regulation in immune response.
Shared PTM sites have stronger preference for disordered structures than single-PTM sites
We investigated the structural preferences of sites occupied by more than one PTM (shared PTM sites) in four ways. First, we mapped experimentally determined shared PTM sites to the DisProt data set. Second, we used ModPred's predictions to identify putative shared PTM sites and mapped them to this data set. Third, we used a data set of 648 experimentally determined shared PTM sites, obtained by combining sites for individual PTMs from our training data and mapping them to the data set consisting of seven proteomes. Finally, we mapped predicted shared PTM sites to this data set as well. For the DisProt data set, we compared the proportion of single and shared PTM sites occurring in ordered and disordered regions. In the seven proteomes, we directly compared disorder prediction scores of single-PTM sites to those of putative sites of competition.
The mapping of known shared PTM sites to the DisProt data set yielded too few sites to allow a “per-PTM” analysis. Overall, a larger percentage of shared PTM sites was found in disordered regions when compared to single-PTM sites (65.3 vs. 48.3%; P < 10−64; t-test). When we considered predicted sites, a larger proportion of shared sites was present in disordered regions than that of single-PTM sites in almost all cases [54.0 vs. 40.8%; P < 10−64; t-test; Fig. 4(A)]. Generally, disordered regions are more likely to harbor both single and shared PTM sites than ordered regions (Table II). However, we note that 72% of residues in the DisProt data set lack any structural annotation.
Figure 4.
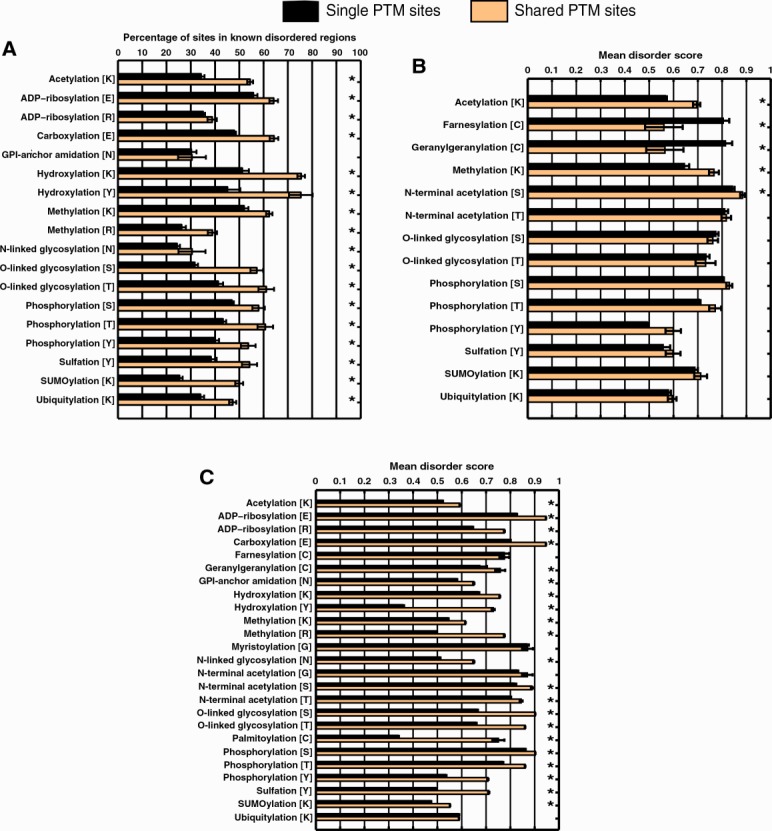
Disorder preferences of sites that are uniquely modified by one PTM when compared to those for sites modified by multiple PTMs on three data sets—(A) Predicted PTM sites mapped to known disordered regions in DisProt, (B) Known PTM sites mapped from the original training data set to predicted disordered regions in the seven proteome data set, and (C) Predicted PTM sites mapped to predicted disordered regions in the seven proteome data set. Percentage of sites in disordered regions are compared in A and mean disorder scores are compared in B and C. Error bars represent standard error derived through bootstrapping (1000 times). PTMs marked with a “*” have significantly different percentages/average disorder scores, based on two-sample t-tests, corrected for multiple testing (Benjamini–Hochberg, α = 0.05).
Table II.
Proportions of Disordered and Ordered Residues (as annotated by DisProt) Harboring Known and Predicted PTM Sites (Single and Shared)
| Known | Predicted | |||
|---|---|---|---|---|
| Types of PTM sites | Disordered (%) | Ordered (%) | Disordered (%) | Ordered (%) |
| Single | 2.81 | 1.91 | 22.91 | 9.99 |
| Shared | 0.08 | 0.00 | 4.95 | 0.06 |
Each cell contains the percentage of residues in the structural category that is either known or predicted to be a site of one or more than one modifications. Values marked in bold indicate the dominant structural category.
In the case of the seven proteomes, for most PTMs, experimentally determined shared sites have higher disorder scores than single-PTM sites for 10 out of 14 PTM types [Fig. 4(B)]. The major exceptions were farnesylation and geranylgeranylation; however, further investigation revealed that both contained only 10 shared sites, all of which were shared between the two. Since farnesylation and geranylgeranylation are considered to be specific cases of a PTM generally referred to as prenylation, it appears that these sites may not be bona fide shared PTM sites. While the general trend suggests that shared sites are more likely to be disordered than single-PTM sites (mean scores 0.74 vs. 0.72), the finding was not statistically significant (P = 0.15; t-test).
When predicted PTM sites are considered, this trend becomes more apparent with 21 out of 25 types of PTMs showing significantly increased mean disorder scores when predicted to be occupied by another PTM [Fig. 4(C)]. Overall, the mean disorder scores of shared sites are significantly higher than that of single-PTM sites (0.70 vs. 0.67; P < 10−64; t-test). The lack of significance for farnesylation, geranylgeranylation, myristoylation, and N-terminal glycine acetylation can be explained by the fact that these PTMs occur at terminal regions of proteins and are, thus, more likely to occur in disordered regions even when they are the lone modification at a site. Interestingly, the differences in scores are large when considering PTMs at order-promoting residues such as cysteine (palmitoylation) and tyrosine, further strengthening the hypothesis that sites that can be occupied by more than one PTM show distinct preferences for intrinsic disorder.
MoRFs are more likely to harbor PTM sites (particularly shared sites) than non-MoRF regions
Protein–protein interactions of intrinsically disordered proteins are commonly mediated through MoRFs. The association of PTM sites with structural disorder prompted us to ask if they preferentially occur within MoRFs and, thus, are likely to be functionally relevant in regulating protein–protein interactions. To test this, we mapped single and shared PTM sites to a data set of 897 MoRFs from 824 eukaryotic protein sequences. However, since we excluded proteins without any PTM annotation, our data set was reduced to 523 MoRFs from 502 proteins. We find that 30% of all MoRFs contained at least one experimentally verified PTM site and 12% of all MoRFs contained at least two known PTM sites. Furthermore, at least two types of PTMs were observed in 4% of all MoRFs. Since these low fractions could be a consequence of incomplete PTM annotations, we separately considered predicted sites. In this case, the data set was larger: 809 MoRFs in 787 proteins. For this data set, 70% and 45% of all MoRFs were predicted to contain at least one and two PTM sites, respectively. At least two types of PTMs were observed in 42% of the MoRF data set.
We then asked if MoRFs preferentially harbor PTM sites in general. Using a one-tailed Fisher's exact test, we tested whether the proportion of PTM sites in MoRFs was greater than that in non-MoRF regions and if this difference was greater than expected. This is indeed the case for both known and predicted PTM sites (Table III; upper half). Next, we split the PTM site counts into those of shared and single PTM sites. We then tested whether the proportion of shared PTM sites in MoRFs was significantly greater than that in non-MoRF regions. Shared sites are significantly more likely to occur in MoRFs than in non-MoRF regions (Table III; lower half). Furthermore, MoRFs contain a greater proportion of shared sites than that in other disordered regions (Known: 0.05 vs. 0.01; P = 1.8 × 10−5, Predicted: 0.18 vs. 0.14; P = 4.5 × 10−8). However, it is not clear whether PTM sites (as a whole) are more likely to be found in MoRFs than in disordered regions (Known: 0.38 vs. 0.16; P = 8.7 × 10−34, Predicted: 0.35 vs. 0.40; P = 9.6 × 10−13). This ambiguity may be attributed to the biased and incomplete annotation of known MoRFs.
Table III.
Preferences of Known and Predicted PTM Sites in MoRF Regions when Compared to Non-MoRF Regions
| Fraction of PTM sites | ||||||
|---|---|---|---|---|---|---|
| Known PTM | Predicted PTM | |||||
| Sites | MoRF | Non-MoRF | P-value | MoRF | Non-MoRF | P-value |
| PTM | 301 | 5,616 | 2.1 × 10−66 | 1,711 | 64,489 | 1.9 × 10−73 |
| Non-PTM | 758 | 53,431 | 3,242 | 215,031 | ||
| Proportion of PTM sites | 0.284 | 0.095 | 0.345 | 0.231 | ||
| Fraction of shared PTM sites | ||||||
|---|---|---|---|---|---|---|
| Known PTM | Predicted PTM | |||||
| Sites | MoRF | Non-MoRF | P-value | MoRF | Non-MoRF | P-value |
| Shared | 15 | 73 | 3.0 × 10−5 | 313 | 7,808 | 2.3 × 10−13 |
| Single-PTM | 286 | 5,543 | 1,398 | 56,681 | ||
| Proportion of shared PTM sites | 0.050 | 0.013 | 0.183 | 0.121 | ||
Comparisons of the proportions of PTM sites in MoRFs to those of non-MoRFs are provided in the upper half. In the lower half, these sites are further split into shared and single PTM sites and compared. P-values were derived from one-tailed Fisher's exact tests on 2 × 2 contingency tables of site counts. P-values marked in bold indicate that they are significant after correcting for multiple testing (Benjamini–Hochberg, α = 0.01).
Discussion
The importance of PTMs and their complex interplay in increasing proteomic diversity at low evolutionary costs is only now being recognized.1,3 However, the structural and functional aspects of this layer of regulation are not completely understood. In this study, we focused on two extreme cases of concerted PTM-based regulation using modification site information obtained through high-throughput data integration and computational prediction. We found statistical associations between the presence of intrinsic disorder and both protein-level and site-level PTM crosstalk. Our results also highlight the role of PTMs in jointly regulating molecular recognition in processes such as transcription and cell development.
Predictor development
We first developed ModPred, a PTM site predictor from amino acid sequence that is amenable to an array of applications ranging from whole proteome characterization to guiding experimental studies on a single protein. An important characteristic of ModPred is that its objective is to estimate the overall propensity of a particular amino acid to be modified, across all species and different modifying enzymes. We achieved this by training a robust model to achieve similar accuracy across the entire feature space. In comparison, models that are organism-specific and/or potentially utilize information other than sequence such as structural, modifying enzyme-specificity, or protein–protein interaction data would be expected to outperform ModPred on such organisms. ModPred can serve as a reference model to these more sophisticated solutions. However, such data are currently available for only a handful of species (and is noisy and incomplete even there), for example kinase information is available for about 12% of curated phosphorylation sites in Phospho.ELM.60 It is thus worthwhile to develop sequence-based tools that generalize well across organisms and modifying enzymes even at a cost of somewhat increased error rates. Furthermore, the use of a common statistical framework alleviates several practical problems encountered when connecting independently developed predictors, such as differences in code implementation, feature generation, software dependencies, or output score distributions. Additionally, the interpretation of scores from these different predictors is nontrivial. For each predictor, the use of default score thresholds or arbitrary user-defined cutoffs such as 0.5 results in a different false positive rate. This variation in the number of false positive predictions is particularly problematic in studies such as ours, where different types of PTM sites are analyzed at the same time. Thus, in this context, using a unified predictor is beneficial.
Multiply modified proteins tend to be intrinsically disordered
Theoretically, even a handful of sites for different PTMs on a protein are sufficient to elicit combinatorial regulation. However, without more detailed information, interpreting the presence of a few sites as evidence of PTM site interplay becomes unreasonable. Therefore, we adopted a more conservative approach (analogous to the extreme scenario of “hypermodification”61,62) and identified proteins statistically enriched in sites for multiple PTM types, as predicted by ModPred. We found that these proteins contained a greater fraction of disordered residues than proteins enriched for at most one type of PTM sites. These proteins largely participate in context-dependent processes such as transcriptional and posttranscriptional regulation. Incidentally, most known cases of concerted PTM-based regulation have been recorded in histones and transcription factors. For example, Benayoun and Veitia have proposed that a sophisticated “PTM code” is perhaps necessary for transcription factors as differently modified isoforms could result in distinct DNA- and protein-binding specificities and affinities, thus enabling them to participate in a variety of signal-dependent processes.7
We note that because of spatiotemporal differences of certain PTMs, sites shared by PTMs may not necessarily be sites of competition. Nevertheless, we also investigated the structural properties surrounding known or predicted shared PTM sites and found that they preferentially lie in intrinsically disordered regions. Remarkably, this was the case even with PTMs typically known to prefer ordered regions such as acetylation, palmitoylation, and N-linked glycosylation. We reason that since shared PTM sites need to be recognized and modified by multiple enzymes with varying substrate specificities, they benefit from the structural flexibility present in disordered regions to accommodate multiple partners. Within disordered regions, we observed that the proportion of PTM sites, particularly that of shared sites in MoRFs was greater than that for non-MoRF regions. Taken together, our results suggest that both single and shared PTM sites are important in modulating disorder-based interactions. On modification, sites in MoRFs may induce local changes in structure that result in the formation of partially or fully developed secondary structure elements, recognizable to binding partners. Alternatively, modifications in MoRFs may act as inhibitors of protein–protein interactions through transition to an unfavorable secondary structure element. Additionally, the presence of shared PTM sites in MoRFs can allow for the presentation of structurally different recognition surfaces, thus enhancing binding-partner diversity. Interestingly, while 70% of MoRFs were predicted to contain at least one PTM site, only 20% of all MoRFs were predicted to contain at least one shared PTM site, suggesting that the modification of a single site and the interaction between neighboring single-PTM sites may be more common mechanisms in MoRF interactions. We note that eukaryotic linear motifs (ELMs)63 and short linear motifs (SLiMs)64 have also been implicated in disorder-based protein–protein interactions. A natural extension of our work would be to investigate the relationship between shared sites and linear motifs.
Intrinsic disorder inversely correlates with rates of synthesis and protein half-life.30 The same study also suggested that phosphorylation may fine-tune the abundance and availability of such proteins, based on requirements in the cell at any given time. We speculate that the above mechanisms of complex PTM-based regulation provide elegant solutions to counter the limited availability of intrinsically disordered proteins. We suspect that at the protein-level, the unusually large number of PTM sites provides a framework for conformational selection, as suggested by Ma and Nussinov.35 This may be particularly effective in LDRs, which are known to harbor many binding sites that could be used sequentially to bind multiple partners.65 Additionally, adjacent and shared PTM sites may result in local changes that allow for one-to-many mechanisms of multipartner binding. For example, a site shared by two PTMs may result in the simultaneous availability of three distinct recognition surfaces for a particular period of time and it is possible that all binding scenarios occur to varying degrees. In fact, recent arguments support the notion of the simultaneous existence of multiple “mod-forms” (specific patterns of modifications of a protein) with distributions that vary according to the cellular context.66
Related work
To the best of our knowledge, there have been only four large-scale studies that integrate PTM data from multiple sources to characterize relationships between PTMs. Beltrao et al. inferred conservation of individual PTM sites within protein domains to define PTM “hotspots” and assigned functional roles to small groups of PTM sites.40 Minguez et al. used coevolution of sites for 13 PTMs and considered them in pairs to infer functional relationships between them.41 Woodsmith et al. used only four types of PTM sites to comprehensively characterize protein-level and region-level regulation in protein complexes.42 Peng et al. identified conserved sequence motifs suggestive of crosstalk between pairs of PTM sites close to each other.43 Our work differs from previous studies in three ways. First, by taking advantage of our predictor, the scale of the analysis, and the coverage of PTMs in this study are far greater than in any previous work. Second, unlike these studies, our primary objective was to gain broad structural and functional insights into PTM-based crosstalk. Last, due to the complexities of translating static PTM site data to dynamic regulatory phenomena, we only concentrate on two extreme cases of concerted PTM-based control. During the revision of this manuscript, Huang et al. reported that proteins containing multiple types of PTM sites possess more disordered regions and are involved in chromatin and DNA-related processes.67 While our study differs from this work in the use of statistical enrichment of PTM sites in a protein, our findings are in general agreement with its results.
Limitations
The use of a predictive approach gives rise to potential sources of bias. First, redundant sequences in the data sets are likely to skew the observed results as predictors would, in effect, “call” the same PTM sites and disordered regions more than once. We accounted for this by running CD-HIT68 on our data sets to filter out redundant sequences at 40% sequence identity and repeating all our analyses. This reduction did not change our original observations (data not shown). Second, the effects of false positive predictions of PTM sites on the study are a concern. When we called PTM sites based on even more stringent thresholds (corresponding to a false positive rate of 0.01), the observed trends did not change in general (data not shown). However, due to low sensitivities of the predictor at this threshold (Table I), it is likely that a lot of true PTM sites are missed. Third, the use of intrinsic disorder as a feature in ModPred may potentially lead to biased inferences on its relationship with concerted PTM regulation, particularly if its contribution to each prediction is high. To address this, we trained models without features related to intrinsic disorder, performed 10-fold cross-validation and compared the resulting performance to the original ModPred model. We found that the removal of these features had negligible effects on predictor performance and that the resulting prediction scores were highly correlated with those of the original model (data not shown). Last, not all PTMs occur in all proteins in all species. For example, sulfation occurs only on proteins that pass through the Golgi apparatus and canonical O-linked GlcNAcylation is not known to occur in yeast. ModPred does not explicitly take these factors into consideration when making predictions. Therefore, we repeated all analyses while limiting our PTMs to lysine acetylation, methylation, N-linked glycosylation, N-terminal acetylation, phosphorylation, SUMOylation, and ubiquitylation. Again, we found that the trends did not change (data not shown). In general, while the particular numbers obtained in this study through prediction may differ from the actual (but unknown) values, we believe that our results provide confident assessments of all trends. We note that, unlike intrinsic disorder, PTM-based regulation is frequently observed in all domains of life and using only eukaryotic proteins may be an additional source of bias. However, because of their underrepresentation in the training data, archaeal, bacterial, and viral proteins are prone to false positive predictions by ModPred. The potential severity of this bias led us to limit our data set to eukaryotic species.
Materials and methods
Data collection
Training data
Experimentally verified PTM sites were mainly collected from Swiss-Prot52 (Release 2011_08), Protein Data Bank69 (January 2012), and Human Protein Reference Database70 (Release 9). Sites annotated with terms such as “by similarity,” “probable,” “potential,” and “partial” were excluded. This data set was supplemented by high-throughput data from PHOSIDA,71 Phospho.ELM60 (Release 9.0), PhosphoSitePlus,72 and sites that we manually extracted from the literature.
Eukaryotic reference proteomes
Reference proteomes of seven model organisms were downloaded from Swiss-Prot (Release 2013_08). These included Saccharomyces cerevisiae (6621 proteins), Caenorhabditis elegans (3430), Arabidopsis thaliana (12,187), Drosophila melanogaster (3169), Rattus norvegicus (7858), Mus musculus (16,618), and Homo sapiens (20,260). In total, 70,143 proteins were used for all analyses.
DisProt data set
A set of experimentally verified disordered proteins were downloaded from DisProt (Release 6.02)50 and its annotations were used to identify disordered and ordered regions. Archaeal, bacterial, and viral proteins were excluded, as several PTMs considered in this study are not known to occur in these organisms. Only proteins with a minimum length of 30 amino acids were considered. The final set contained 493 proteins from 55 eukaryotic species, with 877 annotated disordered regions and 58 annotated ordered regions.
MoRF data set
A data set consisting of 4839 MoRFs extracted from the Protein Data Bank (PDB) was obtained using a method similar to that described in Hsu et al.39 (structured partner >40 residues). After removing duplicate MoRFs and those that map to ambiguous regions of protein sequence, 1769 MoRFs remained. Since some of these MoRFs overlap with each other, MoRFs from the same protein sequence (as per UniProt IDs) were merged together. After merging, only MoRFs between lengths 5 and 25 were included. Next, overlapping MoRFs from 100% identical sequences (such as those from orthologous or paralagous sequences) were merged together. Finally, MoRFs from archaeal, bacterial, and viral proteins were excluded. The final set used in this study consisted of 897 MoRFs from 824 protein sequences.
Predictor construction
Data preparation and redundancy removal
All sites in our compiled data set that were annotated as PTM sites were defined to be positive training examples and all other occurrences of the corresponding residues were defined to be negative examples. More specifically, for each PTM, a set of proteins that contained positive examples for that particular PTM was also used to define negative examples. In the case of amidation (motif), due to the small number of negatives present in our original data set, we randomly sampled residues from our overall PTM data set (excluding plant proteins) and added them to our training data. Farnesylation, geranylgeranylation, myristoylation, and N-terminal acetylation are PTMs that are known to occur at specific termini and/or positions in a protein. In these cases, negative examples from the same protein could not be used and were solely obtained through the above random-sampling procedure.
Each positive and negative site was associated with a 25-residue fragment centered at the residue of interest (for the sites near termini, the fragments were asymmetric). To use a nonredundant training set and achieve good generalization, we removed all residues associated with fragments that were more than 40% identical to other fragments in the data set.29,32 In cases where a fragment containing a negative site was 40% identical to a fragment containing a positive site, the one with the negative class label was removed because its class designation was less reliable.
Additional constraints were applied to the phosphorylation data sets due to their large sizes and the variation in quality in different data sources. First, only data from Swiss-Prot, HPRD, and PDB were used as the addition of high-throughput data offered little improvement in performance (data not shown). Second, for every positive site in a protein, we limited the number of negative sites sampled from it to five.
Feature extraction
Three types of features were generated for model training and evaluation. We distinguish (1) sequence-based features, (2) features based on physicochemical and other predicted properties, and (3) evolutionary features.
The first type included amino acid relative frequencies as well as beta entropies73 (β∈ {1, 1.25, 1.50, 1.75}) calculated using concentric windows centered at the positive and negative sites. We also calculated the net and total charge by counting the number of positively charged residues (K and R) and negatively charged residues (D and E) within these windows. Additionally, we calculated the proportions of aromatic residues (F, Y, and W) and the charge-hydrophobicity ratios74 within these windows. For this set of features, windows of sizes 3, 7, 11, and 21 were used. We then added binary features indicating the presence (one) or absence (zero) of the 20 amino acids within three positions N-terminal and C- terminal to the central residue.
The second set of features included physicochemical properties and structural properties, calculated or predicted for each residue and then averaged over windows of sizes 1, 7, 11, and 21. These consisted of VL2 intrinsic disorder,75 VSL2B intrinsic disorder,51 flexibility,76 hydrophobic moment,77 B-factor,78 amino acid volumes, and secondary structure (in-house predictor). Apart from the mean, the standard deviation and the maximum values in these windows were also included as features. In total, 418 features were obtained for the basic model derived from these two classes of features.
Finally, the third set of features was derived from position-specific scoring matrices (PSSMs) and was designed to incorporate evolutionary constraints around sites. First, PSSMs were constructed for full-length protein sequences by running PSI-BLAST (v.2.2.18; E-value threshold: 0.0001; number of passes: 3) against the NCBI nonredundant database (June 2013).79 Then, each of the columns of a PSSM was treated as a sequence of numbers and the features were constructed by averaging the values around the residue of interest using window sizes of 1, 3, 11, and 21. For our features, we excluded the last column because its values differ, depending on which version of PSI-BLAST is used. In this manner, 164 additional evolutionary features were added and the final number of features used to train the “with PSSM” model was 582.
Training
Logistic regression classifiers are linear classifiers that use the logistic function, applied to a linear combination of features, to calculate class posterior probabilities. They usually perform well on high-dimensional biological data sets and are robust to noise. To ensure stability in training and enhance performance, we Z-score normalized original data sets and performed principal component analysis (PCA) on these data with the retained variance set to 95%. The value of 95% was selected with the goal of eliminating nearly colinear features and no parameter optimization was attempted. In addition, normalization and transform matrices for PCA were calculated on the training partition only and then applied to the test data.
We adopted a bagging approach.80 In each bootstrap iteration during training, positive and negative examples were sampled separately to ensure an equal number of examples from the positive and negative classes. We had initially constructed random forest models consisting of 100 regression trees.81 However, we found that, for this problem, while random forests resulted in slightly better performance accuracies than the ensembles of logistic regression models, the actual prediction scores could not be interpreted meaningfully (i.e., the majority of scores on test data was limited to a relatively narrow part of the 0–1 interval; data not shown). Therefore, we chose logistic regression ensembles over random forests to achieve more stable and interpretable prediction scores.
For all PTMs, we trained an ensemble of 30 logistic regression models for each modified residue type separately. For example, in the case of methylation, separate models were built for lysine and arginine. ADP-ribosylation, amidation, hydroxylation, and proteolytic cleavage were exceptions to this rule as training data was insufficient for a per-residue split. Furthermore, special treatment was provided for PTMs for which a sequence motif had been known, as we observed from two-sample logos82 that motifs alone are not predictive of these modifications (Supporting Information, Fig. S2). In those situations, we adopted a novel approach by separately constructing models on positive versus negative motif-containing sequences and positive versus negative non-motif sequences. We defined motifs based on rules in the literature and PROSITE.83 The importance of such training can be seen in the case of N-linked glycosylation. Here, most positive sites contain an N[!P][ST][!P] motif, whereas most negative sites do not. Training a single classifier on such data may result in a model that predicts positively on all motif sequences but still have relatively low precision due to its inability to correctly classify negative motif-containing sequences. This arises from the facts that the data set for N-linked glycosylation is highly imbalanced and that the number of motif-containing negatives is comparable to the positives.
Evaluation
To evaluate the performance of the ensemble models, 10-fold cross-validation was performed on most data sets. The first exception was made for phosphorylation, where twofold cross-validation was adopted due to the sufficiently large data set size for stable accuracy estimation. Furthermore, in cases where the number of positive instances was less than 100, a leave-one-out approach was adopted. To avoid intraprotein biases, partitions for cross-validation were defined at the protein level rather than the site (residue) level. Predictions were made by passing each data point from the test partition into each member of an ensemble model for a given PTM. Scores were then averaged across the 30 models to obtain scores for each residue. To assign classes (modified or not modified), a threshold score was set and any residue with a score above this threshold was defined as a PTM site. Any residue with a score lower than this threshold was defined as a non-PTM site.
We varied score thresholds between zero and one in small increments and calculated sensitivity (sn; true positive rate) and specificity (sp; true negative rate) at each threshold as follows:
Here, TP = number of true positives, that is instances where a positive example is predicted to be a positive; TN = number of true negatives, that is instances where a negative example is predicted to be a negative; FP = number of false positives, that is instances where a negative example is predicted to be a positive; FN = number of false negatives, that is instances where a positive example is predicted to be a negative.
The receiver operating characteristic (ROC) curve was obtained by plotting these true positive rates against the false positive rates (fpr = 1 − sp) at the various threshold values. Areas under the curve (AUCs) were calculated as the main performance measure. We note that in this problem one cannot accurately estimate the precision-recall curve because the ratio of positive versus negative sites in nature is unknown. To assess whether models with evolutionary features performed better than those without them, we counted the number of PTMs where this was observed to be the case. We then used a binomial test to check if this observed count was significant, under the null hypothesis that the model with PSSMs would perform better than the model without PSSMs half the time.
Implementation
ModPred was implemented in MATLAB and compiled to run as a standalone application on different platforms. The Common Gateway Interface program for the webserver runs this executable and was written in Python. The output of ModPred is a score between zero and one, with higher scores indicating residues more likely to be modified. For each PTM, three strict confidence levels are provided for easy interpretation of results (low, with the decision threshold set to 0.5; medium, with the decision threshold set to the value corresponding to the false positive rate of 0.1; and high, with the value corresponding to the false positive rate of 0.01). The code and data are available at http://www.modpred.org.
Structural and functional analysis
Selection of PTMs
While ModPred could be used to predict sites for up to 23 PTMs, disulfide linkage and proteolytic cleavage were excluded as they do not fit the conventional definition of PTMs. Additionally, PUPylation was excluded as it is exclusively a prokaryotic PTM and amidation was excluded because it can act on any amino acid and predictions usually show a distribution of high scores around the actual amidation site, thus potentially biasing any statistical enrichment tests.
Definition of LDRs in proteins
The VSL2B predictor was run on every protein in the data set. A protein was considered to contain a long region of disorder if it contained at least 30 consecutive residues with prediction scores of 0.78 or greater. This cutoff corresponded to an fpr of 0.05 when we tested VSL2B on a data set of known LDR-containing proteins derived from DisProt.
Identification of PTM site-enriched proteins
For each PTM in this study, we applied a binomial test to assign a P-value P to each protein, as follows:
where P represents the probability that, at least k out of m modifiable residues in a protein are strongly predicted to be modified by chance. A low value suggests that an unusually high number of strong predictions cannot be explained by random chance. Here, k is derived by counting the number of modifiable residues with ModPred scores above a threshold t, for a given PTM, p is a value such that, under the null model, a randomly selected modifiable residue from any protein has a 100·p % chance of being a strongly predicted PTM site. In this study, for each PTM, we used values of t corresponding to a false positive rate of 0.1 to derive k. Therefore, p was set to 0.1 in all cases.
The above method addresses two major issues when trying to identify proteins enriched in PTM sites. First, the number of strongly predicted PTM sites in a given protein will be proportional to its length or the number of modifiable residues. As can be seen above, the calculation of P takes both k and m into account. Second, through the selection of a low value for p, this method takes into account the occurrence of false positive predictions. Finally, after P-values were calculated for all PTMs over all proteins in a data set, we used the Benjamini–Hochberg84 method to correct for multiple testing (α = 0.01). For subsequent analyses, we designated each protein as being enriched in sites for zero, one, two, three PTM types, so on and so forth.
Since LDRs were defined to be 30 residues or more, short proteins (of length less than 50 residues) were excluded for this enrichment analysis. Furthermore, specific to this analysis, PTMs such as farnesylation, geranylgeranylation, myristoylation, and N-terminal acetylation were excluded, as their specificity to either terminus would result in the underestimation of statistical enrichment.
UniProt keyword analysis
For each data set, we obtained keywords corresponding to each protein from the UniProt database. We then extracted only those proteins that were enriched in at least two types of PTM sites. To identify keywords enriched and depleted in the set of proteins enriched in multiple types of PTM sites, we used a one-tailed Fisher's exact test to calculate P-values for each keyword. This test asks whether the proportion of proteins with a keyword in the main set is significantly greater than or less than that in the control set. We compared this set to the set of all proteins enriched for less than two types of PTM sites. Additionally, we made comparisons when considering only the LDR-containing proteins in both these sets and only the ordered proteins in both these sets. P-values were Benjamini–Hochberg corrected and an association was considered significant if its P-value was less than 0.05 (with at least 10 occurrences of the keyword in the main set). Fold-enrichment of a keyword was calculated by dividing its frequency in the main set (set of proteins enriched in multiple types of PTM sites) by its frequency in the control set. Fold-depletion was calculated by taking the reciprocal of this value.
Definition of shared PTM sites
Known PTM sites from the training data set were mapped to the DisProt data set and shared sites were defined as those where one or more PTMs have been experimentally identified. In the case of predicted sites, scores for all of the competing PTMs had to be equal to or exceed thresholds corresponding to an fpr of 0.1. The same was done for the model organism data set.
MoRF analysis
The number of single-PTM sites, shared sites, and non-PTM sites occurring in and outside MoRF regions were counted. When considering non-PTM sites in a given protein, only residues relevant to its modifications were counted. For example, if a protein contained sites for only acetylation and phosphorylation, only the remaining lysine, serine, threonine, and tyrosine residues would be counted as non-PTM sites. Additionally, if a protein was not known (or predicted) to contain any PTM sites, it was excluded from the counting process.
Two 2 × 2 contingency tables were set up to perform comparisons of the proportions of different types of sites in these regions. First, the fraction of PTM sites in MoRFs was compared to that in non-MoRF regions. Second, among these sites, the fraction of shared sites was also compared to that in non-MoRF regions. One-tailed Fisher's exact tests with Benjamini–Hochberg correction (α = 0.05) were used to assign P-values to these comparisons. This was done for both known and predicted PTM sites.
Acknowledgments
The authors thank Jose Lugo-Martinez for meaningful discussions, Vladimir Vacic for providing style sheets for the predictor's website, and anonymous reviewers for their comments that helped improve the quality of the manuscript.
Glossary
- AUC
area under the curve
- CGI
common gateway interface
- ELM
eukaryotic linear motif
- LDR
long disordered region
- MoRF
molecular recognition feature
- PSSM
position-specific scoring matrix
- PTM
post-translational modification
- ROC
receiver operating characteristic
- SLiM
short linear motif
- α
significance level threshold for statistical tests.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Walsh C. Posttranslational modification of proteins: expanding nature's inventory. Greenwood Village: Roberts and Company Publishers; 2006. [Google Scholar]
- 2.Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–672. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 3.Nussinov R, Tsai C-J, Xin F, Radivojac P. Allosteric post-translational modification codes. Trends Biochem Sci. 2012;37:447–455. doi: 10.1016/j.tibs.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Yang X-J. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24:1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- 6.Sims RJ, III, Reinberg D. Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol. 2008;9:815–820. doi: 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- 7.Benayoun BA, Veitia RA. A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 2009;19:189–197. doi: 10.1016/j.tcb.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Lothrop AP, Torres MP, Fuchs SM. Deciphering post-translational modification codes. FEBS Lett. 2013;587:1247–1257. doi: 10.1016/j.febslet.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heldin C-H, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 10.Gamble MJ, Freedman LP. A coactivator code for transcription. Trends Biochem Sci. 2002;27:165–167. doi: 10.1016/s0968-0004(02)02076-5. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y-X, Hirose Y, Zhou XZ, Lu KP, Manley JL. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4:938–948. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 13.Hutchins JR, Clarke PR. Many fingers on the mitotic trigger: post-translational regulation of the Cdc25C phosphatase. Cell Cycle. 2004;3:41–45. [PubMed] [Google Scholar]
- 14.Calnan D, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 15.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1 doi: 10.1101/cshperspect.a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci USA. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Noort V, Seebacher J, Bader S, Mohammed S, Vonkova I, Betts MJ, Kühner S, Kumar R, Maier T, O'Flaherty M, V Rybin, A Schmeisky, E Yus, J Stulke, Serrano L, RB Russell, AJR Heck, P Bork, AC Gavin. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol. 2012;8:571. doi: 10.1038/msb.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danielsen JM, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, Jensen LJ, Mailand N, Nielsen ML. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.003590. M110.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villén J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods. 2013;10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pang CNI, Hayen A, Wilkins MR. Surface accessibility of protein post-translational modifications. J Proteome Res. 2007;6:1833–1845. doi: 10.1021/pr060674u. [DOI] [PubMed] [Google Scholar]
- 22.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Xu D. Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp Biocomput. 2012;17:94–103. [PMC free article] [PubMed] [Google Scholar]
- 24.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 25.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, NV Grishin, M White, XJ Yang, Y Zhao. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Petrescu A-J, Milac A-L, Petrescu SM, Dwek RA, Wormald MR. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology. 2004;14:103–114. doi: 10.1093/glycob/cwh008. [DOI] [PubMed] [Google Scholar]
- 27.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW. J Ausio, MS Nissen, R Reeves, C Kang, CR Kissinger, RW Bailey, MD Griswold, W Chiu, EC Garner, Z Obradovic. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 28.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daily KM, Radivojac P, Dunker AK. 2005. pp. 475–481. Intrinsic disorder and protein modifications: building an SVM predictor for methylation. In: IEEE Symp Comp Int in Bioinfo Comp Biol, CIBCB'05. IEEE, La Jolla, California.
- 32.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 34.Xin F, Radivojac P. Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics. 2012;28:2905–2913. doi: 10.1093/bioinformatics/bts541. [DOI] [PubMed] [Google Scholar]
- 35.Ma B, Nussinov R. Regulating highly dynamic unstructured proteins and their coding mRNAs. Genome Biol. 2009;10:204. doi: 10.1186/gb-2009-10-1-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in protein-protein binding: effect on stability and function. Structure. 2011;19:1807–1815. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 38.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu WL, Oldfield CJ, Xue B, Meng J, Huang F, Romero P, Uversky VN, Dunker AK. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ. Systematic functional prioritization of protein posttranslational modifications. Cell. 2012;150:413–425. doi: 10.1016/j.cell.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minguez P, Parca L, Diella F, Mende DR, Kumar R, Helmer-Citterich M, Gavin A-C, van Noort V, Bork P. Deciphering a global network of functionally associated post-translational modifications. Mol Syst Biol. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woodsmith J, Kamburov A, Stelzl U. Dual coordination of post translational modifications in human protein networks. PLoS Comput Biol. 2013;9:e1002933. doi: 10.1371/journal.pcbi.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng M, Scholten A, Heck AJ, van Breukelen B. Identification of enriched PTM crosstalk motifs from large-scale experimental data sets. J Proteome Res. 2014;13:249–259. doi: 10.1021/pr4005579. [DOI] [PubMed] [Google Scholar]
- 44.Olsen JV, Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 2013;12:3444–3452. doi: 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisenhaber B, Eisenhaber F. Prediction of posttranslational modification of proteins from their amino acid sequence. Methods Mol Biol. 2010;609:365–384. doi: 10.1007/978-1-60327-241-4_21. [DOI] [PubMed] [Google Scholar]
- 46.Xue Y, Liu Z, Cao J, Ren J. Computational prediction of post-translational modification sites in proteins. In: Yang N-S, editor. Systems and computational biology: molecular and cellular experimental systems. InTech; 2011. pp. 105–124. [Google Scholar]
- 47.Zhou F, Xue Y, Yao X, Xu Y. A general user interface for prediction servers of proteins' post-translational modification sites. Nat Protoc. 2006;1:1318–1321. doi: 10.1038/nprot.2006.209. [DOI] [PubMed] [Google Scholar]
- 48.Gao J, Thelen JJ, Dunker AK, Xu D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol Cell Proteomics. 2010;9:2586–2600. doi: 10.1074/mcp.M110.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costello J, Stolovitzky G. Seeking the wisdom of crowds through challenge-based competitions in biomedical research. Clin Pharmacol Ther. 2013;93:396–398. doi: 10.1038/clpt.2013.36. [DOI] [PubMed] [Google Scholar]
- 50.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Z Obradovic, AK Dunker. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fisher RA. On the interpretation of χ2 from contingency tables, and the calculation of P. J R Stat Soc. 1922;85:87–94. [Google Scholar]
- 54.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korneta I, Bujnicki JM. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput Biol. 2012;8:e1002641. doi: 10.1371/journal.pcbi.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, AK Dunker. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirschey MD, Shimazu T, Huang JY, Verdin E. Acetylation of mitochondrial proteins. Methods Enzymol. 2009;457:137–147. doi: 10.1016/S0076-6879(09)05008-3. [DOI] [PubMed] [Google Scholar]
- 59.Neutzner A, Benard G, Youle RJ, Karbowski M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci. 2008;1147:242–253. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- 60.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci USA. 2007;104:2086–2091. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Querfurth C, Diernfellner AC, Gin E, Malzahn E, Höfer T, Brunner M. Circadian conformational change of the Neurospora clock protein FREQUENCY triggered by clustered hyperphosphorylation of a basic domain. Mol Cell. 2011;43:713–722. doi: 10.1016/j.molcel.2011.06.033. [DOI] [PubMed] [Google Scholar]
- 63.Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Travé G, Gibson TJ. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 64.Davey NE, Edwards RJ, Shields DC. Computational identification and analysis of protein short linear motifs. Front Biosci. 2010;15:801–825. doi: 10.2741/3647. [DOI] [PubMed] [Google Scholar]
- 65.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prabakaran S, Lippens G, Steen H, Gunawardena J. Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip Rev Syst Biol and Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang Q, Chang J, Cheung MK, Nong W, Li L, Lee M-t, Kwan HS. Human proteins with target sites of multiple post-translational modification types are more prone to be involved in disease. J Proteome Res. doi: 10.1021/pr401019d. (in press) doi: 10.1021/pr401019d. [DOI] [PubMed] [Google Scholar]
- 68.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 69.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prasad TK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, L Balakrishnan, A Marimuthu, S Banerjee, DS Somanathan, A Sebastian, S Rani, S Ray, CJH Kishore, S Kanth, M Ahmed, MK Kashyap, R Mohmood, YL Ramachandra, V Krishna, BA Rahiman, S Mohan, P Ranganathan, S Ramabadran, R Chaerkady, A Pandey. Human protein reference database—2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daróczy Z. Generalized information functions. Inform Control. 1970;16:36–51. [Google Scholar]
- 74.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 75.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52:573–584. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 76.Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19:141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 77.Eisenberg D, Weiss RM, Terwilliger TC. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci USA. 1984;81:140–144. doi: 10.1073/pnas.81.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Radivojac P, Obradovic Z, Smith DK, Zhu G, Vucetic S, Brown CJ, Lawson JD, Dunker AK. Protein flexibility and intrinsic disorder. Protein Sci. 2004;13:71–80. doi: 10.1110/ps.03128904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Breiman L. Bagging predictors. Mach Learn. 1996;24:123–140. [Google Scholar]
- 81.Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 82.Vacic V, Iakoucheva LM, Radivojac P. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006;22:1536–1537. doi: 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
- 83.Sigrist CJ, De Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, Bougueleret L, Xenarios I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41:D344–D347. doi: 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 85.Nie L, Sasaki M, Maki CG. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J Biol Chem. 2007;282:14616–14625. doi: 10.1074/jbc.M610515200. [DOI] [PubMed] [Google Scholar]
- 86.Saito Si, Yamaguchi H, Higashimoto Y, Chao C, Xu Y, Fornace AJ, Appella E, Anderson CW. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J Biol Chem. 2003;278:37536–37544. doi: 10.1074/jbc.M305135200. [DOI] [PubMed] [Google Scholar]
- 87.DeHart CJ, Chahal JS, Flint S, Perlman DH. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol Cell Proteomics. 2014;13:1–17. doi: 10.1074/mcp.M113.030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kelley LA, Sutcliffe MJ. OLDERADO: On-line database of ensemble representatives and domains. Protein Sci. 1997;6:2628–2630. doi: 10.1002/pro.5560061215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeng L, Zhang Q, Gerona-Navarro G, Moshkina N, Zhou M-M. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure. 2008;16:643–652. doi: 10.1016/j.str.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng Z, Chen C-J, Chamberlin M, Lu F, Blobel GA, Speicher D, Cirillo LA, Zaret KS, Lieberman PM. The CBP bromodomain and nucleosome targeting are required for Zta-directed nucleosome acetylation and transcription activation. Mol Cell Biol. 2003;23:2633–2644. doi: 10.1128/MCB.23.8.2633-2644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Das C, Roy S, Namjoshi S, Malarkey CS, Jones DN, Kutateladze TG, Churchill ME, Tyler JK. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc Natl Acad Sci USA. 2014;111:E1072–E1081. doi: 10.1073/pnas.1319122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 93.Zheng Y, Thomas PM, Kelleher NL. Measurement of acetylation turnover at distinct lysines in human histones identifies long-lived acetylation sites. Nat Commun. 2013;4:2203. doi: 10.1038/ncomms3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information