

Abstract
Thioredoxin superfamily proteins introduce disulfide bonds into substrates, catalyze the removal of disulfides, and operate in electron relays. These functions rely on one or more dithiol/disulfide exchange reactions. The flavoenzyme quiescin sulfhydryl oxidase (QSOX), a catalyst of disulfide bond formation with an interdomain electron transfer step in its catalytic cycle, provides a unique opportunity for exploring the structural environment of enzymatic dithiol/disulfide exchange. Wild-type Rattus norvegicus QSOX1 (RnQSOX1) was crystallized in a conformation that juxtaposes the two redox-active di-cysteine motifs in the enzyme, presenting the entire electron-transfer pathway and proton-transfer participants in their native configurations. As such a state cannot generally be enriched and stabilized for analysis, RnQSOX1 gives unprecedented insight into the functional group environments of the four cysteines involved in dithiol/disulfide exchange and provides the framework for analysis of the energetics of electron transfer in the presence of the bound flavin adenine dinucleotide cofactor. Hybrid quantum mechanics/molecular mechanics (QM/MM) free energy simulations based on the X-ray crystal structure suggest that formation of the interdomain disulfide intermediate is highly favorable and secures the flexible enzyme in a state from which further electron transfer via the flavin can occur.
Keywords: enzyme mechanism, flavin adenine dinucleotide, thioredoxin fold, cis-proline, X-ray crystallography, quantum mechanics/molecular mechanics
Introduction
Cysteine amino acids in proteins are crosslinked, unlinked, or participate in electron transfer pathways by engaging in dithiol/disulfide exchange reactions. Catalyzed dithiol/disulfide exchange is essential to numerous biological pathways, including re-reduction of ribonucleotide reductase and other cytosolic proteins via the thioredoxin/thioredoxin reductase system,1 production of virulence factors in the bacterial periplasm,2 and oxidative protein folding in the eukaryotic secretory pathway.3 The most common motif for redox-active disulfides is two cysteines separated by two intervening amino acids, that is, the CXXC motif (or CAXXCB motif, to distinguish the two participating cysteines), as found in enzymes with the thioredoxin (Trx) fold.4 The identities of the “X” residues in the CXXC motif, as well as other amino acids, influence the oxidation-reduction potential of the di-cysteine motif, but how reaction rates are encoded in amino acid sequence is far from obvious.5 The structures of numerous Trx proteins have been determined, including a handful in complex with their substrates.6–8 In complementary studies, the dithiol/disulfide exchange process has been investigated theoretically using small-molecule model species.9 For the most part, however, the functional-group and structural features that promote enzymatic dithiol/disulfide exchange reactions have yet to be elucidated.
One challenge in achieving such an analysis is that the interaction of the two di-cysteine motifs in a dithiol/disulfide exchange event is typically transient. Nucleophilic attack by the dithiol CA cysteine on the disulfide results in a “mixed” disulfide between the CA cysteines from the two motifs. An additional nucleophilic attack by the CB thiolate from the initiating CAXXCB motif then resolves the mixed disulfide and completes the electron-transfer process. Alternatively, attack by the CB from the accepting CAXXCB motif restores the initial conditions. It is possible to make the mixed disulfide resistant to resolution and capture it for observation by eliminating the CB cysteines,6–8,10,11 but at the expense of losing key players in the reaction.
Mixed disulfide intermediates have been studied for the enzyme quiescin sulfhydryl oxidase (QSOX), a catalyst of disulfide bond formation found in the Golgi apparatus of animal cells and in secreted fluids.12–15 QSOX contains two Trx domains (Trx1 and Trx2) at its amino terminus, similar to tandem Trx domains seen in many proteins of the protein disulfide isomerase (PDI) family.16 The QSOX Trx1 domain contains a CXXC motif, whereas the Trx2 domain lacks such a motif and is not redox active. QSOX undergoes an internal electron-transfer reaction between the Trx1 domain and a sulfhydryl oxidase domain of the Erv family near the carboxy terminus of the enzyme. These redox-active domains are linked flexibly to one another such that the Trx1 domain can interact alternately with external substrates and with the Erv domain during the QSOX catalytic cycle [Fig. 1(A)]. The reaction accomplished by QSOX is the transfer of two electrons from pairs of substrate thiol groups, via two enzyme disulfides and a bound flavin adenine dinucleotide (FAD) cofactor, to molecular oxygen17,18 [Fig. 1(B)]. To prevent completion of the catalytic cycle, two mutations were introduced to trap the mixed disulfide between the Trx1 and Erv domains of QSOX enzymes.11 Explicitly, the CB cysteines of the CAXXCB motifs were mutated to alanine or serine, leaving only the CA cysteine of each motif to form the interdomain disulfide. Structures of these species revealed how the QSOX oxidoreductase domain docks into a berth above the redox-active disulfide and FAD of the sulfhydryl oxidase module.
Figure 1.
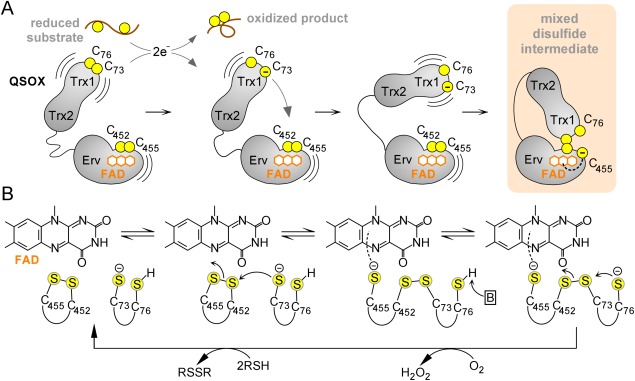
QSOX catalytic cycle. (A) Schematic diagram of initial steps in QSOX catalysis. Juxtaposed yellow balls represent disulfide-bonded cysteines, and isolated balls represent reduced, unpaired cysteines. Structural domains of QSOX are labeled, and the FAD cofactor is shown as fused orange hexagons. (B) Active-site chemistry. Only the FAD cofactor and sulfur atoms of the CXXC motifs are shown explicitly. Cysteine protonation states at various steps are surmised, and the boxed “B” represents a generic base. An interactive view is available in the electronic version of the article.
The ready formation of the interdomain disulfide by the mutant QSOX and the apparent geometrical fit between the Trx1 and Erv modules suggested that the mutant was a good representation of the intermediate state in electron transfer from the Trx1 CXXC to the FAD-proximal disulfide. However, the lack of key features present in the natural, transient intermediate, that is, the resolving CB cysteine and the charge-transfer CB cysteine, which interacts with the FAD cofactor, limited the mechanistic conclusions that could be drawn. A new crystal structure of a mammalian QSOX enzyme with these missing features restored has now been obtained. Wild-type Rattus norvegicus QSOX1 (RnQSOX1) was crystallized and found to occupy a conformation poised for interdomain electron transfer. The presence of all four redox-active cysteines in the RnQSOX1 structure allows for modeling and assessing the energetics of a dithiol/disulfide exchange reaction in a biologically relevant and representative structural context.
Results
Overall structure of RnQSOX1
The structure of RnQSOX1 spanning amino acid residues 37–547 (hereafter referred to as RnQSOX1) was determined by X-ray crystallography to 2.9 Å resolution (Table I). The structure was solved by molecular replacement (MR) using the tandem Trx and Erv modules from MmQSOX1 as separate search models. The RnQSOX1 crystals contained two molecules in the asymmetric unit, designated molecules A and B. Despite the absence of covalent constraints, both RnQSOX1 molecules were found in a “closed” conformation in which the two redox-active sites are juxtaposed (Fig. 2), similar to the configuration of the previously determined structure of the Mus musculus QSOX1 (MmQSOX1) Cys76Ala/Cys455Ser mutant constrained by a disulfide bond between the two reactive cysteines Cys73 and Cys452.11 The relative orientations of the oxidoreductase and sulfhydryl oxidase modules of the two RnQSOX1 molecules were roughly similar, differing by a rotation of about 3°, calculated as described (Materials and Methods).
Table I.
Crystallographic and Refinement Statistics
| Data collection | |
|---|---|
| Wavelength, Å | 0.872 |
| Space group | P 21 21 2 |
| Unit cell dimensions (a,b,c), Å | 96.50, 169.58, 68.37 |
| Resolution range, Å | 48.8–2.9 (3.004–2.9)* |
| Total observations | 120,214 |
| Unique observations | 25,595 (2521) |
| Completeness, % | 99.9 (99.8) |
| Mean I/σ | 10.6 (2.7) |
| Rsym | 0.149 (0.357) |
| Refinement statistics | |
|---|---|
| Rwork | 0.178 |
| Rfree | 0.243 |
| Bond length rms deviation, Å | 0.003 |
| Bond angle rms deviation, ° | 0.69 |
| Non-hydrogen atoms | 8225 |
| Macromolecular | 7897 |
| Cofactor (FAD) | 106 |
| Water | 222 |
| Ramachandran favored (%) | 97.4 |
| Ramachandran outliers (%) | 0 |
| Average B-factor | 27.8 |
| Macromolecules | 27.9 |
| Cofactor | 43.9 |
| Water | 21.3 |
Highest resolution shell is shown in parentheses.
Figure 2.
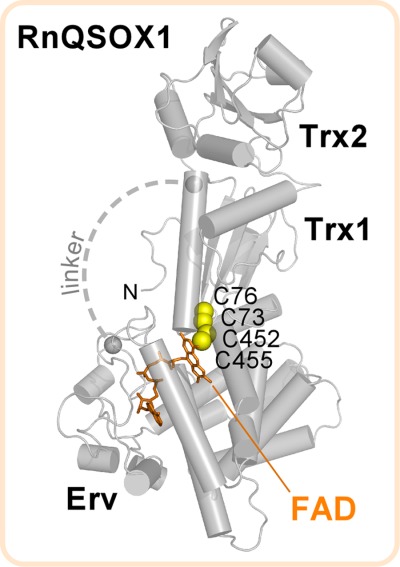
Structure of RnQSOX1. Gray spheres mark the amino acids to either side of a flexible 12-residue segment for which electron density was not observed (“linker,” approximated as a dashed line). “N” is the protein amino terminus.
CXXC motifs and flavin cofactor
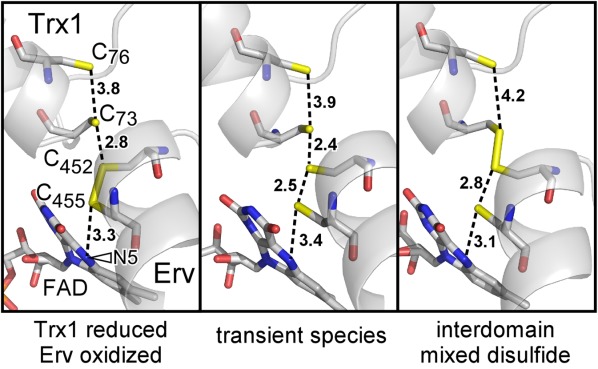
RnQSOX1 was initially modeled in the fully oxidized state, with two intradomain disulfide bonds (C73–C76 and C452–C455). However, electron density maps suggested the contribution of an interdomain disulfide (C73–C452), most notably for molecule B. This molecule was, modeled with alternate conformations, corresponding to oxidized and partially reduced states [Fig. 3(A)]. In particular, a species containing a disulfide between Cys73 and Cys452 and reduced cysteines at positions 76 and 455 was included and assigned occupancy 0.5. Although electron density corresponding to the Cys73–Cys452 interdomain disulfide was less evident in molecule A, and an interdomain disulfide was, not included in the model for this molecule, some residual electron density in the vicinity of Cys76 suggests partial reduction of molecule A as well [Fig. 3(A)].
Figure 3.
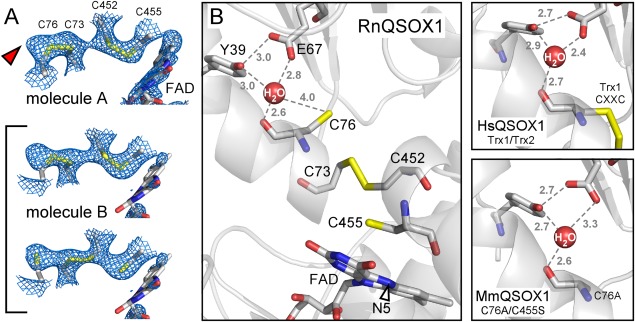
(A) Simulated annealing omit maps, contoured at 1 σ, in the vicinity of the RnQSOX1 CXXC motifs and FAD isoalloxazine. Cysteine residues and FAD are in stick representation. The alternate conformations that contribute to molecule B are bracketed. The red arrowhead indicates excess density in molecule A, which may indicate a minor population of partially reduced disulfides in this molecule as well. Electron density for the FAD isoalloxazine in molecule B is very poor, suggesting a mixture of redox states or conformational heterogeneity. (B) The putative proton transfer catalyst site of RnQSOX1, including Glu67, Tyr39, and a buried water molecule, is shown in relation to the cysteine ladder descending toward the FAD. Gray numbers are distances in Ångstrom. The N5 position of FAD is indicated by an arrowhead. Also shown is the coordination of the comparable buried water molecules in QSOX fragment and mutant structures (PDB codes 3Q6O and 3T38, respectively).
The presence of all four redox-active cysteines in the RnQSOX1 structure, captured in multiple conformations, allows a thorough analysis of the geometries and environments of important players in the electron transfer reaction. The sulfur atoms of the four cysteines that participate in the interdomain dithiol/disulfide exchange mechanism sit roughly on a line, and this line approaches the plane of the FAD isoalloxazine at an angle of about 60° [Fig. 3(B)]. In the partially reduced state, the cysteine side chains constitute a pathway for electron transfer spanning about 12 Å from Cys76 to the FAD N5 nitrogen.
The partially reduced state of RnQSOX1 shows how the resolving cysteine in a QSOX Trx1 domain might be deprotonated to enable attack on the interdomain mixed disulfide and completion of the dithiol/disulfide exchange reaction. Near the resolving Cys76 is a buried water molecule, which appears to be coordinated by the side chain hydroxyl of Tyr39, the carboxylic acid group of Glu67, and the backbone carbonyl of Cys76 itself [Fig. 3(B)]. A change in the side-chain dihedral angle of Cys76, hints of which are seen in the electron density, would place the cysteine sulfur within hydrogen bonding distance of the buried water molecule, but not within hydrogen bonding distance of the tyrosine or glutamate directly. The bound water may facilitate abstraction of a proton from Cys76 while in turn donating a proton to Glu67. Both Tyr39 and Glu67 are widely conserved in QSOX enzymes, so this mechanism is likely to apply generally to QSOXs [Fig. 3(B)].
The cis-proline
A proline residue in the cis configuration abutting the CXXC motif is a characteristic feature of Trx domains and is present in RnQSOX1 (Pro122) [Fig. 4(A)]. The carbonyl of the peptide bond before the proline (Phe121 C=O) is oriented toward the Arg453 N–H group, in the peptide bond immediately after the CA cysteine in the Erv CXXC, and via a water molecule toward the N–H of Asp454. In addition, the N–H group of Phe121 is in position to form a hydrogen bond with the carbonyl group of the residue preceding the Erv domain CA cysteine (Gly451 C=O). These interactions can be compared with other structures of mixed disulfides involving a CXXC motif, such as complexes of glutaredoxin with glutathione,19 thioredoxin and a target protein,20 and the bacterial periplasmic dithiol/disulfide oxidoreductases DsbA and DsbB.8 In each of these cases, the substrate peptide or target region is positioned antiparallel to the loop containing the cis-proline, and the N–H and C=O groups of the substrate cysteine backbone are hydrogen bonded to the two backbone amide groups immediately upstream of the proline [Fig. 4(B)]. In RnQSOX1, in contrast, the loop containing the cis-proline is locally roughly parallel to the polypeptide immediately preceding the active-site helix of the Erv domain, and the N–H and C=O groups of the Erv domain CA cysteine point away from the cis-proline. Instead, the C=O and N–H groups from the same two amides, but belonging to the two residues on either side of the cysteine (Gly and Arg), participate in hydrogen bonds with the two amides upstream of the cis-proline [Fig. 4(B)]. Though the RnQSOX1 structure supports the generality of a role for the conserved proline of thioredoxin fold domains in interacting with the polypeptide backbone in the vicinity of the substrate reactive cysteine, it shows that the details of the interaction may differ.
Figure 4.
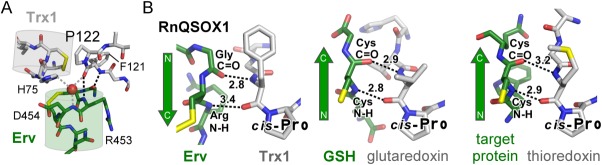
Interactions between the RnQSOX1 cis-proline and the target of nucleophilic attack. (A) The two RnQSOX1 CXXC motifs, at the amino-termini of their respective helices (cylinders), are shown in relation to cis-proline P122. Dashed lines indicate potential hydrogen bonds. Hydrogen bonds in black are shown in a different orientation in panel B. (B) Hydrogen bonding from the amino acid residue immediately upstream of the cis-proline to the substrate peptide is compared among RnQSOX1, a glutathione/glutaredoxin complex (PDB code 3FZ9), and a complex between thioredoxin and a target protein (PDB code 2IWT). Numbers indicate distances in Ångstrom.
Stabilization of the mixed disulfide
The observation of a mixed disulfide intermediate in the RnQSOX1 crystals indicated that the conformation observed is competent for electron transfer and suggested that the mixed disulfide is more stable than other partially reduced states of the system. To explore the thermodynamics of the RnQSOX1 electron relay more thoroughly, we conducted free energy simulations to extract thermodynamic parameters. On the basis of the fully oxidized RnQSOX1 variant (Cys73–Cys76 and Cys452–Cys455), an in silico model was prepared. The Cys73–Cys76 disulfide was broken, and Cys73 was left in the deprotonated state, while Cys76 was protonated. The Cys452–Cys455 disulfide was preserved. Unbiased hybrid QM/MM molecular dynamics (MD) simulations starting with this model [Fig. 5(A)] resulted in spontaneous formation of a Cys73–Cys452 interdomain disulfide bridge as a result of nucleophilic attack by Cys73 on the remaining Cys452–Cys455 disulfide.
Figure 5.
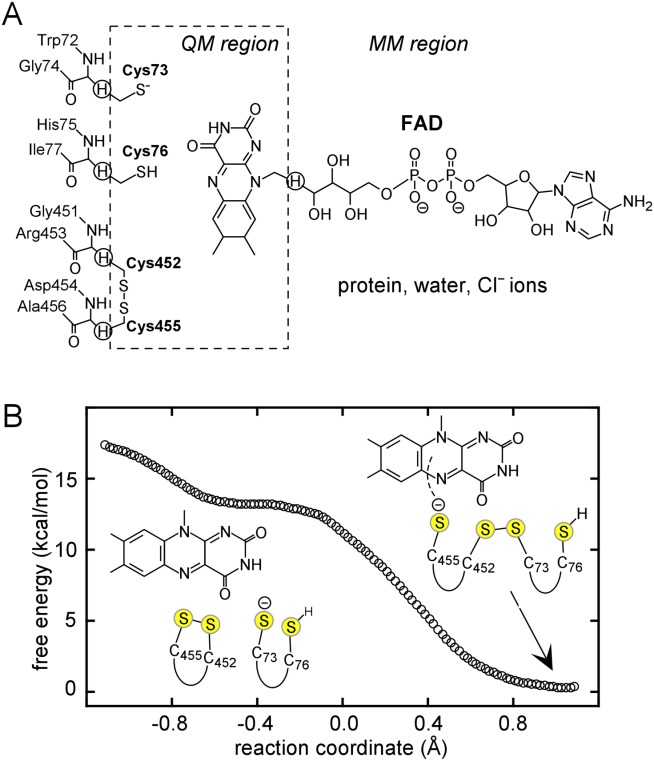
(A) QM/MM partitioning scheme. Link atoms are marked by circled hydrogens. (B) Free energy profile for formation of the interdomain disulfide in RnQSOX1. The reaction coordinate (x-axis) is the antisymmetric stretch coordinate, defined as the difference between S–S bond distances: R(S452–S455)–R(S73–S452).
Potential of mean force (PMF) simulations were performed to obtain the free energy reaction profile for the formation of the interdomain disulfide in RnQSOX1. Mixed disulfide formation between Cys73 and Cys452 lacks a free energy barrier and has a reaction free energy of ΔGr = −13.3 kcal/mol [Fig. 5(B)]. In the starting structure, the average distances are 2.19 (±0.08) between Cys452 Sγ and Cys455 Sγ Å and 2.80 (±0.08) Å between Cys73 Sγ and Cys452 Sγ (Table II). As Cys73 attacks Cys452, the mixed disulfide bond is gradually formed between these two cysteines, while an electron transfers to Cys455. Representative snapshots from the reaction trajectory are shown (Fig. 6). At the stationary point of the PMF, the average distance between Cys452 Sγ and Cys455 Sγ is 2.94 (±0.10) Å, and the distance between Cys73 Sγ and Cys452 Sγ is 2.14 (±0.06) Å. The gradual structural changes during the trajectory support the notion of an SN2 reaction. Additional 100 ps sampling of the mixed disulfide structure without any restraints resulted in an average Cys452 Sγ − Cys455 Sγ distance of 3.49 (±0.39) Å and Cys73 Sγ − Cys452 Sγ distance of 2.08 (±0.05). These distances are in good agreement with the crystallographic structure of the mixed disulfide form of molecule B (Table II).
Table II.
Comparison of Computed and Crystallographic Geometrical Parameters for RnQSOX1
| Conf. 1 | Conf. 2 | Free MD | X-ray molecule A | X-ray molecule B1 | X-ray molecule B2 | |
|---|---|---|---|---|---|---|
| SγC73 − SγC76 (Å) | 3.64 ± 0.43 | 3.81 ± 0.40 | 3.48 ± 0.38 | 2.03 | 2.02 | 3.57 |
| SγC452 − SγC455 (Å) | 2.19 ± 0.08 | 2.94 ± 0.10 | 3.49 ± 0.39 | 2.03 | 2.03 | 4.16 |
| SγC73 − SγC452 (Å) | 2.80 ± 0.08 | 2.14 ± 0.06 | 2.08 ± 0.05 | 3.68 | 3.70 | 2.03 |
| SγC73 − SγC452 − SγC455 (°) | 156.5 ± 5.5 | 152.6 ± 6.5 | 151.4 ± 6.6 | 143.4 | 125.0 | 119.5 |
| χ1C73 (°) | 170.2 ± 8.5 | 174.8 ± 7.5 | 173.9 ± 7.6 | 155.1 | 165.2 | −174.2 |
| χ1C76 (°) | −52.3 ± 8.8 | −55.1 ± 8.0 | −53.5 ± 8.0 | −51.0 | −43.20 | −89.5 |
| χ1C452 (°) | −171.1 ± 6.1 | −161.0 ± 6.9 | −159.5 ± 7.6 | −174.2 | 179.2 | −58.5 |
| χ1C455 (°) | −54.1 ± 6.0 | −59.5 ± 6.2 | −62.7 ± 7.5 | −52.5 | −52.1 | −55.8 |
| SγC455 − N5FAD (Å) | 3.59 ± 0.23 | 3.32 ± 0.12 | 3.24 ± 0.11 | 3.92 | 3.95 | 3.82 |
The computed geometrical parameters were obtained as an average over 520 MD configurations each. Values in columns “Conf. 1” and “Conf. 2” were obtained from trajectories based on PMF simulations in the reactant and product wells, with intra- and interdomain disulfide bridges, correspondingly. Values in column “Free MD” were obtained from free MD simulations without restraints, which correspond to the interdomain disulfide conformation. For the geometrical parameters obtained from the X-ray crystal structures, molecule A and the first modeled conformation of molecule B (B1) contain the disulfide bonds C73–C76 and C452–C455. The second modeled conformation of molecule B (B2) contains the interdomain disulfide C73–C452.
Figure 6.
Snapshots from the free energy simulations, including the starting configuration, a transient species during formation of the interdomain disulfide, and the interdomain disulfide bridged state. Nonbonded sulfur–sulfur distances and distances between Cys455 and the FAD N5 position (arrowhead) are indicated in Ångstrom.
The QSOX1 active-site architecture appears well-suited for charge relay from Trx1 to Erv. To initiate electron transfer along the ladder consisting of cysteines 76, 73, 452, and 455 [Fig. 3(B)], a negative charge located on Cys73 of Trx1 is passed via an SN2 reaction to Cys455 of Erv, in proximity to the FAD. Subsequently, a second electron is moved from Cys76 to Cys452 along the same ladder toward the FAD electron sink via an additional proposed SN2 reaction. Based on the current simulations of the first of these steps, the negative charge migration from Cys73 to Cys455 and the cofactor is expected to be spontaneous and thermodynamically highly favorable. During the course of this step, the average distance between Cys455 Sγ and the FAD N5 position changes from 3.59 (±0.23) Å to 3.32 (±0.12) Å based on our PMF simulations (Table II). The decreased distance between Cys455 and FAD likely prepares the system for the next step in the charge relay, from Cys455 to FAD.
Discussion
In solution, QSOX enzymes populate conformations in which the redox-active sites are distant from one another, as indicated by measurements of energy transfer between fluorescent groups on the Trx1 and Erv domains.11 Nevertheless, the two molecules in the RnQSOX1 crystal asymmetric unit were found with the redox-active sites poised for interaction. These observations are consistent with this “closed” state being a local minimum on the configurational energy landscape. Trapped in proximity by the crystal lattice, the RnQSOX1 redox-active sites may then have been affected by the intense synchrotron radiation required to obtain X-ray diffraction data of even moderate resolution. RnQSOX1 was prepared for crystallization in aerated solutions free of reductants, and was thus presumably crystallized in the oxidized state. Possibly, photoelectrons released during data collection reacted with a fraction of the CXXC disulfides or FAD isoalloxazines in the crystal, and the electrons introduced into the system were subsequently distributed according to the oxidation/reduction potentials of the linked redox centers. In contrast to other oxidoreductases that have been trapped in intermediate states by mutagenesis,8,19,20 wild-type RnQSOX1 fortuitously formed the mixed disulfide intermediate state during the experiment.
It has been suggested that the reduction of FAD to FADH2 may be rate limiting in disulfide formation in QSOX.21 It would, therefore, seem necessary to hold QSOX in the closed state for sufficient time for the electron relay and concomitant proton transfer to occur. The current crystallographic and computational study suggests that, though QSOX may fluctuate between open and closed states, once the proper closed state is formed with Cys73 oriented for an SN2 reaction, the interdomain disulfide bridge is formed with a substantial free energy gain and without significant barriers. The active-site architecture of QSOX allows the negative charge to migrate closer to the FAD cofactor, making the process of electron transfer highly favorable in this case. Formation of the interdomain disulfide may lock the enzyme in the closed state for long enough to permit the subsequent chemistry to take place.
The importance of interdomain mixed disulfides and charge-transfer intermediates between CB cysteines and oxidizing cofactors was emphasized recently in a thermodynamics and kinetics study of trypanosome QSOX and a comparison with the bacterial team of disulfide catalysts DsbA and DsbB.22,23 In both QSOX and DsbA/DsbB, a challenging step in the catalytic cycle is reoxidation of a highly oxidizing Trx domain. According to the redox potentials of the two CXXC disulfides in trypanosome QSOX, and presumably other QSOX orthologs, a two-electron transfer between the reduced Trx1 CXXC and the oxidized Erv domain CXXC would be a thermodynamically uphill event.22 However, transfer of the first electron from the Trx-fold domain CXXC motif to the vicinity of the FAD may facilitate the transfer of the second electron and completion of disulfide formation in the Trx domain. A similar scenario exists in DsbB, through its ubiquinone cofactor.8,23 In another related example, various single-cysteine mutants of the vitamin K epoxide reductase (VKOR), a transmembrane enzyme that generates disulfide bonds by reducing vitamin K epoxide, spontaneously formed a mixed disulfide between two successive di-cysteine motifs, despite the presence of the cysteine that could in principle resolve the back-reaction and eliminate the mixed disulfide.24 This cysteine, Cys133 in Synechococcus sp. VKOR, abuts the ubiquinone, and stabilization of the cysteine thiolate in the presence of the quinone ring may protect the mixed disulfide from attack. The current study shows that a mixed disulfide between successive di-cysteine motifs can be detected even in the presence of the CB cysteine thiol that naturally resolves the mixed disulfide in the forward direction. This observation suggests that, in contrast to the facile formation of the mixed disulfide intermediate once the participants are aligned, resolution of the mixed disulfide may present a significant kinetic barrier.
A feature of the QSOX structure that was not noted previously, but which may contribute to the resolution of the mixed disulfide and completion of the catalytic cycle, is the conserved environment presenting a buried water molecule to the CB cysteine of the Trx1 CAXXCB motif. The structures of the oxidoreductase fragment of human QSOX1 and the MmQSOX1 Cys76Ala/Cys455Ser mutant [Fig. 3(B)] both contained a water molecule in this position,11 but the distance between the water and the CB sulfur in the former and the absence of the CB cysteine in the latter obscured the potential role of this water as a proton acceptor from the resolving cysteine. Other Trx-fold proteins, such as thioredoxin itself, contain buried acidic residues, which have been proposed to serve as proton transfer catalysts to the cysteine that resolves the mixed disulfide.25,26 An ultrahigh resolution structure of Acetobacter aceti thioredoxin revealed a water molecule hydrogen bonded to a buried aspartate and to the carbonyl of the CB cysteine, 5.6 Å from the CB cysteine sulfur.27 For comparison, the corresponding water molecule is 5.3 Å from the sulfur atom of Cys76 in the oxidized state of RnQSOX1. In the reduced state of RnQSOX1 molecule B, the Cys76 sulfur was modeled 4.0 Å from the water molecule, and a further turn toward the trans X1 dihedral angle rotamer would bring the cysteine sulfur even closer to the bound water. In PDI family proteins, glutamate is also the dominant amino acid at the position corresponding to Glu67 in RnQSOX1. We propose that QSOX shares a general scheme with PDI family proteins in which a buried water molecule functions as a bridge for proton transfer to the buried glutamate. Proton transfer would be a prerequisite for formation of the Cys73–Cys76 disulfide bond and relay of the second electron down the cysteine ladder in QSOX [Fig. 3(B)].
It has been noted previously that Trx-like CXXC motifs are often interspersed with redox-active disulfides in other structural contexts in biological disulfide relays.28 However, Trx-fold proteins also undergo direct dithiol/disulfide exchange with other Trx-fold proteins.29,30 Therefore, the interaction between two CXXC motifs at the amino termini of helices with the aid of the neighboring cis-proline as observed in RnQSOX1 may be relevant to other biological dithiol/disulfide exchange events.
Materials and Methods
Vector construction, protein expression, and protein purification
The region of the rat QSOX1 gene encoding amino acids 36 to 550 codon-optimized for expression in Escherichia coli (Genscript), was introduced into the pET-15b vector (Novagen) between the Nde I and Bam HI restriction sites. The recombinant vector was transformed into E. coli strain BL21 (DE3) plysS (Novagen). Cells were grown in LB containing 100 mg/L ampicillin and 30 mg/L chloramphenicol to OD600 nm 0.5–0.6 at 37°C. Isopropyl-1-thio-β-d-galactopyranoside was added to a final concentration of 0.5 mM, and the cultures were grown for a further 36 h at 15°C. Cells were harvested and suspended in 20 mM sodium phosphate buffer, 500 mM NaCl, 20 mM imidazole, pH 7.4, sonicated on ice, and centrifuged for 1 h at 40,000g. The supernatant was applied to a Ni-NTA column, and protein was eluted in 20 mM sodium phosphate buffer, 500 mM NaCl, and a gradient of imidazole (20–500 mM), pH 7.4. The eluted enzyme was exchanged into 20 mM sodium phosphate buffer, pH 7.4, 100 mM NaCl, 20 mM imidazole using a PD-10 desalting column (GE Healthcare). Thrombin (10 units/mg protein) was added, and the cleavage reaction was incubated overnight at room temperature. PMSF was added to 1 mM to inhibit the thrombin, and the protein was reapplied to a Ni-NTA column and collected in the flow-through. Further purification was performed by size exclusion chromatography on a Superdex 75 16/60 column in 100 mM NaCl, 10 mM Tris buffer, pH 8.0.
Crystallization, data collection, and structure refinement
Crystals of RnQSOX1 in space group P21212 were grown by hanging drop vapor diffusion over well solution containing 0.1 M sodium citrate buffer, pH 6.0, 0.25 M NaCl, 27% w/v polyethylene glycol 3350. Crystals were transferred to 0.1 M sodium citrate buffer pH 6.0, 0.25 M NaCl, 25% w/v polyethylene glycol 3350, 27% glycerol, and flash frozen for data collection. Data were collected on the ID23-2 beamline at the European synchrotron radiation facility and processed and scaled using DENZO and SCALEPACK.31 The structure was solved by MR with Phaser,32 using as search models two fragments of mouse QSOX1 (Protein Data Bank (PDB) code 3T58), spanning residues 36–278 and 289–547, respectively. Rebuilding was done using Coot,33 and structure models were refined initially with CNS and then with Phenix,34,35 maintaining the same free set of reflections. Iterative build omit maps were inspected at the last stage of refinement.36 Data collection and refinement statistics are reported in Table I. Structure superposition and comparison of domain orientations was done using lsqkab in the CCP4 package.37 Chains A and B of RnQSOX1 were first aligned by a least-squares fit of Cα atoms in their ψErv/Erv domains, and then the rotation matrix for a least-squares fit of Cα atoms between the Trx1/Trx2 domains in the two chains was calculated. The trace of this matrix provided the rotation angle θ according to trace = 1 + 2cos θ.
Model construction for simulations
The fully disulfide bonded version of the RnQSOX1 crystal structure, at a stage close to final refinement, was used to construct the initial configuration for simulations. The X-ray crystal structure model contained 498 amino acid residues in monomer A, 91 waters, and the cofactor FAD. Amino acid residues missing from the model (25–36, 279–290, and 548–550) were added using Discovery Studio 3.5 (Accelrys), prior to commencing simulations. Residues 25–36 and 548–550 were added in an extended conformation and subjected to geometry minimization and MD simulations, whereas the remainder of the enzyme was kept fixed. During these steps, distance restraints were used between Leu25 and Ser36 (ca. 8–9 Å) to hasten folding of the N-terminus into a well-packed conformation during the minimization and dynamics. The modeling of the missing loop residues 279–290 was done using the LOOPER algorithm.38 In addition, the Cys73–Cys76 disulfide bond was reduced, resulting in a neutral Cys76 and a negatively charged Cys73, while the Cys452–Cys455 disulfide bridge was kept intact.
The protonation states of all ionizable residues were set corresponding to pH 7. The hydrogen bonding patterns of the ionizable residues with the surrounding environment were visually inspected to verify that the protonation states were reasonable. The coordinates of hydrogen atoms of the protein and water were determined using the HBUILD facility in the program CHARMM.39,40 The possible protonation states of His residues were determined by examination of hydrogen bonding interactions. Peripheral/surface His residues were assumed to be positively charged. The resulting positively charged enzyme (+12) has dimensions of about 95 × 76 × 82 Å3. To this system 12 chlorine ions were added in random positions outside the protein to obtain a net-neutral system.41 Subsequently, the protein, cofactor, crystal waters, and counter ions were embedded in a water box as detailed below.
Hybrid QM/MM potential energy surface
The hydride transfer reaction in QSOX1 was described using a hybrid QM/MM potential energy surface (PES).42
| (1) |
The system was partitioned into a QM region consisting of 45 atoms and a MM region containing the rest of the system. The QM subsystem includes 28 atoms from the FAD cofactor and 17 atoms from the cysteine residues in the protein (Cys73, Cys76, Cys452, and Cys455). Additionally, five hydrogen link atoms were introduced along the covalent bonds crossing the boundary between the QM and the MM regions, to satisfy the valence requirements of the QM fragments [Fig. 5(A)]. The QM region was treated by the self-consistent-charge tight-binding density functional (SCC-DFTB) Hamiltonian.43 The all-atom CHARMM22/27 force field44,45 with grid-based energy correction maps46 for peptide dihedral angles was used to treat the entire protein, the cofactor, and the ions. The water molecules were represented by the three-point charge TIP3P model.47 QM/MM interactions were treated by electrostatic embedding, wherein the MM partial atomic charges are included in the one-electron Hamiltonian.
MD simulations
MD simulations were conducted under periodic boundary conditions, with Ewald summation for electrostatic interactions.48 The solute was soaked in a pre-equilibrated 90 × 90 × 90 Å3 cubic box of 24,875 water molecules, with its longest axis lying along the space diagonal of the box to ensure that all protein atoms are at least 10 Å away from the edges of the box. The final model contained 73,769 atoms. For van der Waals and electrostatic interactions, a 13.0 Å group-based cutoff was used. The Ewald method was used for reciprocal space summations between MM sites as well as for the QM/MM interactions using a 90 × 90 × 90 FFT grid. The κ value was set to 0.340 Å−1.
All water molecules were relaxed using the adopted-basis set Newton–Raphson (ABNR) minimization method (100 steps), while the crystal water oxygens were harmonically restrained to their original positions. This procedure was followed by a 100 ps MD equilibration of the water molecules, which were then minimized again (30 steps ABNR). Subsequently, all atoms were subjected to minimization in a stepwise fashion, while removing restraints, to eliminate close contacts in the initial protein–cofactor–solvent system. Finally, the entire system was minimized (30 steps ABNR) without any restraints.
The isothermal–isobaric ensemble number of particles, pressure, temperature (NPT) was used at 1 atm and 298 K using the extended system pressure/temperature algorithm of Andersen with an effective mass of 500 amu and the Hoover thermostat with an effective mass of 1000 kcal/mol·ps2. The SHAKE algorithm was applied to constrain all MM bonds involving hydrogen atoms. The system was gradually heated up from 48 to 298 K during five sessions of 5 ps for a total of 25 ps, and thereafter equilibrated at the target temperature (298 K) over the course of 1 ns using the QM(SCC-DFTB)/MM potential.48
Potential of mean force
The classical–mechanical potential of mean force (CM-PMF) was determined using the umbrella sampling technique to sample the high-energy regions of the PES.49 The reaction coordinate (ζ) was defined as the difference between the lengths of the (breaking) C452 Sγ − C455 Sγ and (forming) C73 Sγ − C452 Sγ disulfide bonds.
| (2) |
A total of eight discrete regions along the reaction coordinate (windows) were defined with spacing of 0.20 Å. Each simulation was performed with the addition of a biasing potential to the reaction coordinate (roughly the negative of the computed PMF), and a harmonic restraint centered in each window for the reaction coordinate. The harmonic force constants, k, ranged from 20.0 to 75.0 kcal/mol/Å2 [Eharm= k(ζ − ζref)2]. Each window was equilibrated for 25 ps, followed by 125 ps production simulations that collected the probability densities of configurations (ρ) along the reaction coordinate (ζ), and sorted them into bins of width 0.02 Å. The PMF curve was obtained using a multidimensional version of the weighted histogram analysis method.50,51 To assure convergence of the PMF, the simulations were run until the difference between sequential PMF profiles was less than ±1 kcal/mol.
Coordinates
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 4P2L.
Acknowledgments
The authors declare no conflict of interest.
References
- 1.Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 2.Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7:215–225. doi: 10.1038/nrmicro2087. [DOI] [PubMed] [Google Scholar]
- 3.Bulleid N, Ellgaard L. Multiple ways to make disulfides. Trends Biochem Sci. 2011;36:485–492. doi: 10.1016/j.tibs.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Martin JL. Thioredoxin—a fold for all reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 5.Chivers PT, Prehoda KE, Raines RT. The CXXC motif: a rheostat in the active site. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 6.Wang P-F, Veine DM, Ahn SH. Williams CH., Jr A stable mixed disulfide between thioredoxin reductase and its substrate thioredoxin: preparation and characterization. Biochemistry. 1996;35:4812–4819. doi: 10.1021/bi9526793. [DOI] [PubMed] [Google Scholar]
- 7.Nordstrand K, Åslund F, Holmgren A, Otting G, Berndt KD. NMR structure of Escherichia coli glutaredoxin 3-glutathione mixed disulfide complex: implications for the enzyme mechanism. J Mol Biol. 1999;286:541–552. doi: 10.1006/jmbi.1998.2444. [DOI] [PubMed] [Google Scholar]
- 8.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 9.Fernandes PA, Ramos MJ. Theoretical insights into the mechanism for thiol/disulfide exchange. Chem Eur J. 2004;10:257–266. doi: 10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alon A, Grossman I, Gat Y, Kodali VK, DiMaio F, Mehlman T, Haran G, Baker D, Thorpe C, Fass D. The dynamic disulphide relay of quiescin sulfhydryl oxidase. Nature. 2012;488:414–418. doi: 10.1038/nature11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrowski MC, Kistler WS. Properties of a flavoprotein sulfhydryl oxidase from rat seminal vesicle secretion. Biochemistry. 1980;19:2639–2645. doi: 10.1021/bi00553a016. [DOI] [PubMed] [Google Scholar]
- 13.Coppock D, Kopman C, Gudas J, Cina-Poppe DA. Regulation of the quiescence-induced genes: quiescin Q6, decorin, and ribosomal protein S29. Biochem Biophys Res Commun. 2000;269:604–610. doi: 10.1006/bbrc.2000.2324. [DOI] [PubMed] [Google Scholar]
- 14.Chakravarthi S, Jessop CE, Willer M, Stirling CJ, Bulleid NJ. Intracellular catalysis of disulfide bond formation by the human sulfhydryl oxidase, QSOX1. Biochem J. 2007;404:403–411. doi: 10.1042/BJ20061510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilani T, Alon A, Grossman I, Horowitz B, Kartvelishvily E, Cohen SR, Fass D. A secreted disulfide catalyst controls extracellular matrix composition and function. Science. 2013;341:74–76. doi: 10.1126/science.1238279. [DOI] [PubMed] [Google Scholar]
- 16.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Hoober KL, Thorpe C. Egg white sulfhydryl oxidase: kinetic mechanism of the catalysis of disulfide bond formation. Biochemistry. 1999;38:3211–3217. doi: 10.1021/bi9820816. [DOI] [PubMed] [Google Scholar]
- 18.Heckler EJ, Alon A, Fass D, Thorpe C. Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry. 2008;47:4955–4963. doi: 10.1021/bi702522q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couturier J, Koh CS, Zaffagnini M, Winger AM, Gualberto JM, Corbier C, Decottignies P, Jacquot JP, Lemaire SD, Didierjean C, Rouhier N. Structure-function relationship of the chloroplastic glutaredoxin S12 with an atypical WCSYS active site. J Biol Chem. 2009;284:9299–9310. doi: 10.1074/jbc.M807998200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda K, Hägglund P, Finnie C, Svensson B, Henriksen A. Structural basis for target protein recognition by the protein disulfide reductase thioredoxin. Structure. 2006;14:1701–1710. doi: 10.1016/j.str.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Kodali VK, Thorpe C. Quiescin sulfhydryl oxidase from Trypanosoma brucei: catalytic activity and mechanism of a QSOX family member with a single thioredoxin domain. Biochemistry. 2010;49:2075–2085. doi: 10.1021/bi902222s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israel BA, Kodali VK, Thorpe C. Going through the barrier: coupled disulfide exchange reactions promote efficient catalysis in quiescin sulfhydryl oxidase. J Biol Chem. 2014;289:5274–5284. doi: 10.1074/jbc.M113.536219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba K, Takahashi Y, Ito K, Hayashi S. Critical role of a thiolate-quinone charge transfer complex and its adduct form in de novo disulfide bond generation by DsbB. Proc Natl Acad Sci USA. 2006;103:287–292. doi: 10.1073/pnas.0507570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Cheng W, Grider RF, Shen G, Li W. Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nature Commun. 2014;5:3110. doi: 10.1038/ncomms4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeMaster DM, Springer PA, Unkefer CJ. The role of the buried aspartate of Escherichia coli thioredoxin in the activation of the mixed disulfide intermediate. J Biol Chem. 1997;272:29998–30001. doi: 10.1074/jbc.272.48.29998. [DOI] [PubMed] [Google Scholar]
- 26.Chivers PT, Raines RT. General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry. 1997;36:15810–15816. doi: 10.1021/bi971504l. [DOI] [PubMed] [Google Scholar]
- 27.Starks CM, Francois JA, Macarthur KM, Heard BZ, Kappock TJ. Atomic-resolution crystal structure of thioredoxin from the acidophilic bacterium Acetobacter aceti. Protein Sci. 2007;16:92–98. doi: 10.1110/ps.062519707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haebel PW, Goldstone D, Katzen F, Beckwith J, Metcalf P. The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: crystal structure of the DsbC-DsbDα complex. EMBO J. 2002;21:4774–4484. doi: 10.1093/emboj/cdf489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Åslund F, Berndt KD, Holmgren A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem. 1997;272:30780–30786. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 30.Araki K, Iemura S, Kamiya Y, Ron D, Kato K, Natsume T, Nagata K. Ero1-α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J Cell Biol. 2013;202:861–874. doi: 10.1083/jcb.201303027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt.4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Afonine PV, Grosse-Kunstleve RW, Adams PD. The Phenix refinement framework. CCP4 Newsletter. 2005;42 contribution 8. [Google Scholar]
- 36.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, Hung L-W. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr. 2008;D64:515–524. doi: 10.1107/S0907444908004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. 1976;A32:922–923. [Google Scholar]
- 38.Spassov VZ, Flook PK, Yan L. LOOPER: a molecular mechanics-based algorithm for protein loop prediction. Protein Eng Des Sel. 2008;21:91–100. doi: 10.1093/protein/gzm083. [DOI] [PubMed] [Google Scholar]
- 39.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 40.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcook HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen MP, Tildesley DJ. Computer simulation of liquids. Oxford: Oxford University Press; 1987. pp. 156–162. [Google Scholar]
- 42.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 43.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim Th, Suhai S, Seifert G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys Rev. 1998;B58:7260–7268. [Google Scholar]
- 44.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem. 1998;B102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 45.MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 46.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 48.Cui Q, Elstner M, Kaxiras E, Frauenheim T, Karplus M. A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J Phys Chem. 2000;B105:569–585. doi: 10.1002/jcc.10201. [DOI] [PubMed] [Google Scholar]
- 49.Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comput Phys. 1977;23:187–199. [Google Scholar]
- 50.Doron D, Kohen A, Major DT. Collective reaction coordinate for hybrid quantum and molecular mechanics simulations: a case study of the hydride transfer in dihydrofolate reductase. J Chem Theory Comput. 2012;8:2484–2496. doi: 10.1021/ct300235k. [DOI] [PubMed] [Google Scholar]
- 51.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. J Comput Chem. 1992;13:1011–1021. [Google Scholar]