

Abstract
The nuclease domain of colicin E7 (NColE7) promotes the nonspecific cleavage of nucleic acids at its C-terminal HNH motif. Interestingly, the deletion of four N-terminal residues (446–449 NColE7 = KRNK) resulted in complete loss of the enzyme activity. R447A mutation was reported to decrease the nuclease activity, but a detailed analysis of the role of the highly positive and flexible N-terminus is still missing. Here, we present the study of four mutants, with a decreased activity in the following order: NColE7 >> KGNK > KGNG ∼ GGNK > GGNG. At the same time, the folding, the metal-ion, and the DNA-binding affinity were unaffected by the mutations as revealed by linear and circular dichroism spectroscopy, isothermal calorimetric titrations, and gel mobility shift experiments. Semiempirical quantum chemical calculations and molecular dynamics simulations revealed that K446, K449, and/or the N-terminal amino group are able to approach the active centre in the absence of the other positively charged residues. The results suggested a complex role of the N-terminus in the catalytic process that could be exploited in the design of a controlled nuclease.
Keywords: DNA cleavage, flow linear dichroism, isothermal calorimetry, positively charged N-terminal residues, Zn2+, binding
Introduction
Colicin E7 (ColE7) is a nuclease toxin produced by Escherichia coli for protection from related bacteria.1,2 It has three functional domains: the receptor binding, the membrane translocation, and the nuclease domain.3,4 The nuclease domain of colicin E7 (NColE7) enters the cytoplasm of the target cell after the cleavage of ColE7 by a specific periplasmic protease recognizing the R447 residue.5 NColE7 kills the attacked cell through the nonspecific digestion of the chromosomal DNA.2 The host cell is protected against nuclease activity by coexpressing an immunity protein (Im7) that inhibits the substrate binding of NColE7.6,7
The mechanism of DNA hydrolysis catalyzed by nuclease colicins was discussed extensively. The proposals largely depend on the quality of the metal ion.6,8–14 The catalytic centre is a ββα-metal binding HNH motif15–17 at the C-terminus of these enzymes (Fig. 1). The metal cofactor in the active site of NColE7 is a Zn2+-ion.9 In contrast, Zn2+ was inhibitory for NColE9—a related nuclease with high sequence identity.11,12 The presence of the metal ion is not necessary for substrate binding, but is essential for the hydrolytic reaction.18 It binds and electrostatically activates the scissile phosphodiester bond. In NColE7, the Zn2+-ion is coordinated by the side-chains of three histidine residues (H544, H569, and H573). A well conserved histidine (H545 in NColE7) is responsible for generating the nucleophilic hydroxide by deprotonating a water molecule (Supporting Information Fig. S1).9,19,20 The general acid residue that protonates the leaving group was not yet identified. In most nucleases it is a water molecule coordinated to the metal ion, which is usually Mg2+.21 However, such a coordinated water molecule was not detected in the distorted tetrahedral arrangement around the Zn2+-ion in NColE7/DNA complex. R538, Q542, and H569 were speculated to provide the proton for the leaving group in NColE722 in analogy to NColE9.23 Recently, a shuttle mechanism was suggested in which the leaving group is protonated by the hydrogen ion originating from the same water molecule that initiated the nucleophilic attack.24
Figure 1.
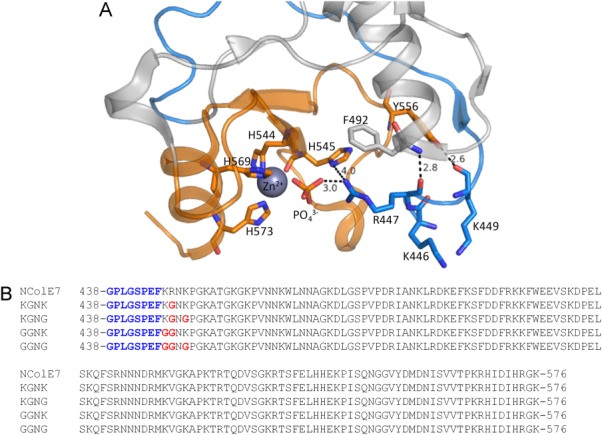
(A) Crystal structure of NColE7 (PDB: 1MZ86) in complex with a Zn2+ and a phosphate ion. The HNH motif is in orange and the N-terminal loop is in blue. Among the N-terminal amino acids, R447 is the closest residue to the phosphate ion that forms a bridge between the Zn2+-ion and R447. Hydrogen bonds of the backbone of the N-terminus are also shown. (B) Sequences of the purified NColE7 variants. The proteins containing the black part of the sequence denoted by NColE7 will be referred to as wild type (WT) NColE7 in the following text. The four mutants are named KGNK, KGNG, GGNK, and GGNG based on the 446–449 segments of the sequence, respectively. The changes compared to the NColE7 sequence are highlighted in red. The remaining sequence after the Human rhinovirus C3 protease cleavage encoded by pGEX-6P-1 plasmid is written in blue.
The N-terminal R447 and the C-terminal HNH motif are close in space in NColE7 [Fig. 1(A)]. This arginine was proposed to increase the DNA binding affinity.5 It is intriguing why this single residue—lying outside the DNA-binding helices—influences the catalytic reaction. In NColE9 the arginine corresponding to R447 in NColE7 was supposed to bind the substrate and stabilize the pentavalent transition state.25,26 Based on the crystal structures of Vvn—an another HNH nuclease—it was also hypothesized that the arginine side-chain binds and stabilizes the cleaved DNA to decelerate the reverse reaction.8,27 R447A mutation in NColE7 significantly reduced its in vitro DNase activity,5 while previously we showed that the cytotoxicity of NColE7 is completely lost upon deletion of the KRNK sequence (residues 446–449) at the N-terminus.28 This suggests that the presence of a positively charged residue at the N-terminus is essential for the catalytic activity and the K446 and/or K449 residues may partly take over the role of the missing arginine. Positively charged residues are often found in a similar orientation close to the active site in the published crystal structures of HNH-nucleases.28 This property could be generally exploited in the design and fine tuning of the catalytic activity of an artificial nuclease with intramolecular allosteric control.29 Therefore, the purpose of this study was to investigate the effect of the mutations of the N-terminal positive residues on the catalytic activity. NColE7 and its four mutants [with KGNK, KGNG, GGNK, and GGNG amino acids within the 446–449 segment of the sequence—see Fig. 1(B)] were expressed, purified and studied by experimental [mass spectrometry (MS), isothermal calorimetric titration, gel mobility shift assays, circular and linear dichroism (LD) spectroscopy], and computational [semiempirical quantum chemical calculations and molecular dynamics (MD)] techniques.
Results
Preparation and DNA cleavage of NColE7 and its mutants
The glutathione-S-transferase (GST) fusion forms of NColE7 and its mutants were coexpressed with the Im7 immunity protein to avoid cytotoxicity. The complexes were purified with GST affinity chromatography and the GST-tag was cleaved on column (Supporting Information Fig. S2.) The nucleases were separated from the immunity protein by adjusting the pH to 3.0.7 At this pH, the His side-chains are protonated and consequently, the purified proteins did not contain metal ion, as confirmed by their mass spectra. Upon addition of one equivalent Zinc(II)-acetate, however, all nucleases were detected in their holo forms (Table I). The GPLGSPEF sequence encoded by pGEX-6P-1 vector remained at the N-terminus of the NColE7 variants after the Human rhinovirus C3 protease cleavage. This sequence was considered not to interfere with the properties investigated below.28
Table I.
Comparison of the Expected and Measured Molecular Masses of the Purified Proteins
| Protein | Expected mass (Da) | Measured mass (Da) |
|---|---|---|
| NColE7 | 15875.2 | 15874.9 |
| KGNK | 15776.1 | 15776.2 |
| KGNG | 15705.0 | 15704.5 |
| GGNK | 15705.0 | 15704.8 |
| GGNG | 15633.9 | 15632.8 |
| Im7 | 10961.1 | 10961.5 / 10412.8a |
The numbers refer to the holo form of the WT NColE7 and its mutants containing one Zn2+-ion. The spectra were recorded after short incubation of the proteins with one equivalent of Zinc(II)-acetate.
The immunity protein was found in two forms corresponding to the full length sequence and with four histidines missing from the His-tag at the C-terminus of the protein.
The cleavage of the pUC19 plasmid by the NColE7 variant proteins in their Zn2+-loaded form was monitored by agarose gel electrophoresis. The supercoiled form of the plasmid already disappeared at the first measurement point (approximately 2 min after mixing the solutions) in the presence of NColE7, while it was still detectable after 140 min in the DNA solutions containing the NColE7 mutants (Fig. 2).
Figure 2.
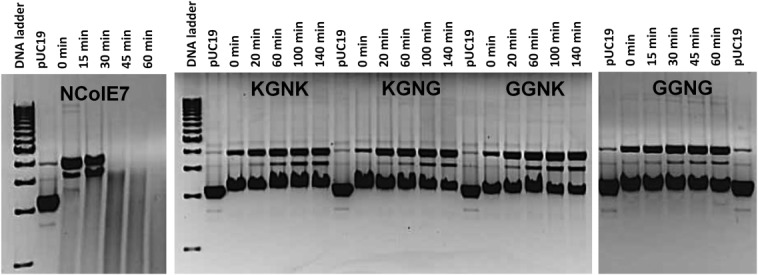
Digestion of 28 nM pUC19 (that is 74 µM calculated for base pairs) by 2.8 µM NColE7 mutants, incubated with one equivalent Zinc(II)-acetate before mixing with DNA. The samples were kept at 37°C and run subsequently on 1% agarose gel. In control experiments (data not shown), cleavage of the plasmid DNA incubated with only Zinc(II)-acetate was not observed. 1 kb Molecular Ruler (BioRad) served as the reference.
Flow linear dichroism (FLD) as a noninvasive technique was simultaneously applied for monitoring the digestion of long linear calf thymus DNA chains. The signal intensity of the FLD spectra depends on the ability of the long filiform DNA molecules to align, that is, on the length, conformation, and rigidity of the double helix.30–32 Due to the cleavage by nucleases, the DNA chains shortened and aquired a lower degree of orientation in the Couette flow cell. Thus, the absolute value of FLD intensity decreased during the measurement (Supporting Information Fig. S3), while the absorbance was also constant during the reaction, indicating that no DNA was lost by precipitation (Supporting Information Fig. S4). The absolute value of FLD intensities at 260 nm plotted as a function of time in Figure 3 allows for the comparison of the nuclease activity of the mutants under the same conditions provided.
Figure 3.
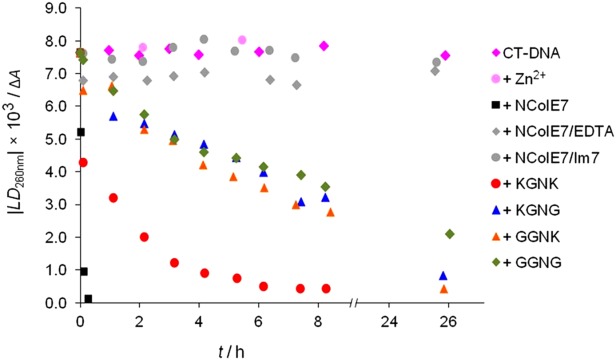
Cleavage of 130 µM (base pairs) CT-DNA by different nucleases (0.5 µM) followed by FLD spectroscopy. The proteins were preincubated for 30 min in the presence of one equivalent ZnCl2. The LD signal intensity at 260 nm is taken from baseline corrected spectra of aliquots of the stock solutions, incubated at 37°C. Control experiments included CT-DNA incubated at 37°C without or in the presence of 0.5 µM ZnCl2, 0.5 µM NColE7, and 60 µM EDTA or 0.5 µM NColE7 and 1 µM Im7.
Folding and Zn2+ binding of the proteins
The synchrotron radiation circular dichroism (SRCD) spectra of the various mutants are shown in Figure 4(A). The shape of the spectra for all studied proteins carrying mutations in the region of KRNK amino acids is similar to the spectrum of NColE7. The interaction with metal ion [Fig. 4(B)] does not yield significant protein refolding. It is likely that the structure of the NColE7 variants is preformed for the metal ion binding, that is, the purified proteins were properly folded and capable of strong Zn2+ binding. The change upon Zn2+ binding was similar for all mutants and to that of NColE9 published earlier.33 Addition of excess Zn2+ did not affect the structure.
Figure 4.
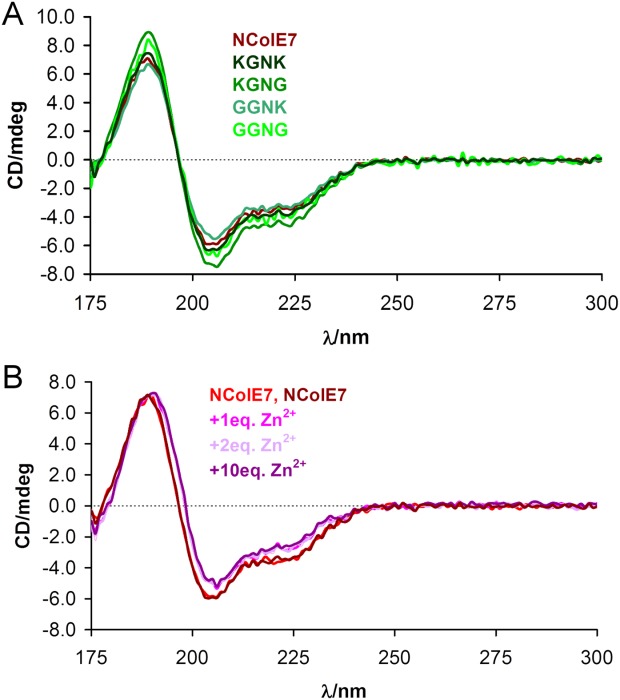
(A) SRCD spectra of the NColE7 variant proteins. (B) SRCD spectra of WT NColE7 in the absence (two independent measurements) and upon the addition of Zn2+-ions. All spectra were normalized to the same 31.6 µM protein concentration.
Quantitative data on the Zn2+ binding of mutants were obtained by isothermal microcalorimetric (ITC) titrations. An exotherm heat effect was observed at the beginning of the titrations. This effect could not be avoided and no suitable model for analysis was found that could well describe the whole curve. Therefore, in each case the first part of the curve was ignored during the analysis (Fig. 5). Similar curves with even more intense changes at the beginning of the titration of the zinc transporter YiiP protein with ZnCl2 34 were observed, presumably due to the formation of oligomeric complexes. Although dimerization of the NColE7 variants may also occur,7 further investigations are needed to understand this phenomenon.
Figure 5.
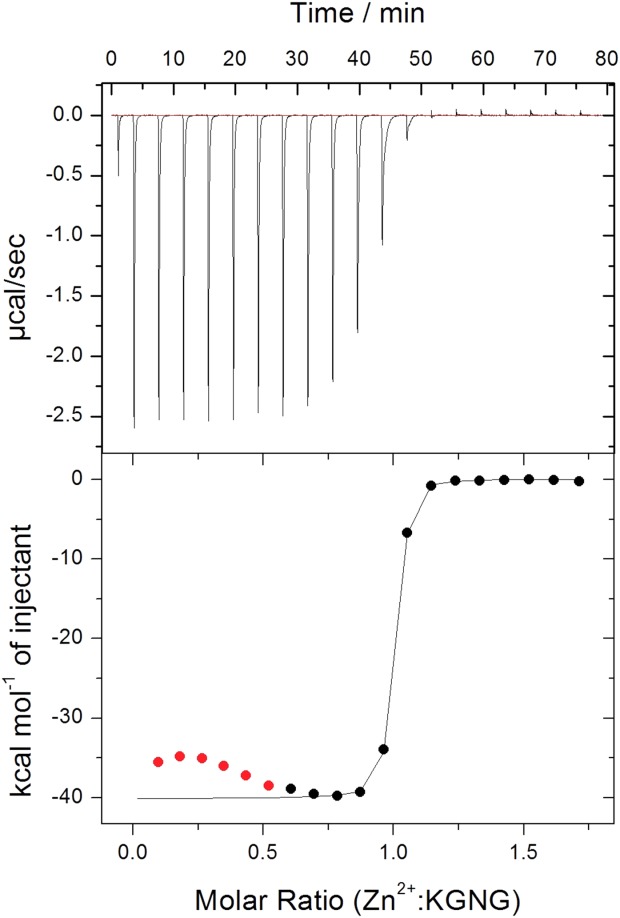
Microcalorimetric titration of 50 µM KGNG in 20 mM cacodylate buffer, pH = 7.0. The red points at the beginning were ignored during the curve evaluation.
DNA binding
Based on the earlier results, we expected that the N-terminal mutations will significantly influence the DNA binding of the proteins. The presence of the metal ion in the active site of NColE7 is necessary for the catalytic activity, but it is not required for the DNA binding.18 Accordingly, EDTA was applied to prevent the cleavage of the DNA in the experiments below. CT-DNA solution was titrated with the proteins (Fig. 6). Interaction with proteins leads to a decreased FLD signal of DNA due to the change in the overall shape and thus, to the decrease of the orientation ability of the molecules. The cleavage of DNA was excluded based on the comparison with gel electrophoresis experiments. The UV absorption spectra confirmed that precipitation of the DNA-protein complexes did not significantly affect the data (Supporting Information Fig. S5). The order of the DNA binding based on the protein concentration required to decrease the FLD signal of DNA to its half intensity is as follows: NColE7 (1.0 µM) > KGNG (1.8 µM) ∼ KGNK (2.0 µM) ∼ GGNK (2.4 µM) > GGNG (3.7 µM). The FLD data, however, reflect the combined effect of the formation of the protein/DNA complexes and the subsequent conformation change of the DNA. Therefore, the apparent Kd values related to DNA binding were calculated from the gel electrophoresis experiments carried out with a 13bp DNA fragment (Supporting Information Fig. S6). Kd values between 0.15 and 0.32 µM were obtained for the DNA binding affinities of NColE7 and its mutants against a 13 bp fragment.
Figure 6.
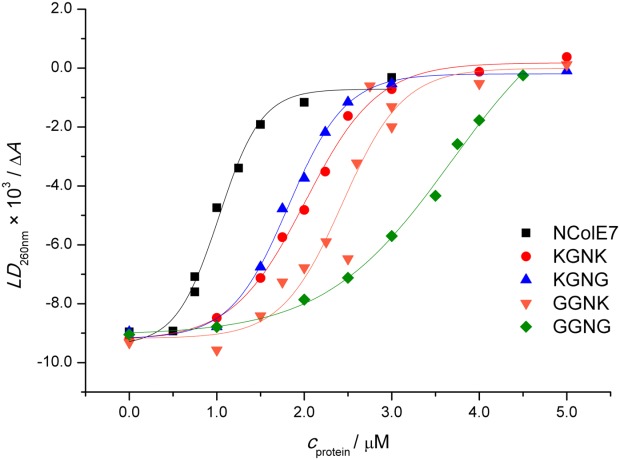
The effect of the CT-DNA binding of NColE7 and the four mutants on the FLD signal intensity at 260 nm. 130 µM CT-DNA (calculated for base pairs) was incubated with 0–5 µM protein and 60 µM EDTA in each case.
Molecular dynamics
MD simulations were performed with the NColE7, KGNK, KGNG, GGNK, and GGNG protein sequences starting from the residue 446, ignoring the eight amino acids part from the GST cleavage in the experiments [Fig. 1(B)]. The models contained Zn2+-bound proteins, with the metal ion in the HNH motif also coordinated by a phosphate ion. The root mean square deviation (RMSD) from the reference structure at 0 ps is higher for the mutants than for NColE7 (Supporting Information Fig. S7). The lysine containing mutants are similar to each other, but the increased RMSD for the GGNG mutant indicated that this structure changed to the largest extent. It was shown that in the KGNK and KGNG mutants the side-chain of K446, while in GGNK and GGNG the N-terminal amino group approached the phosphate ion (Figs. 7(A) and Supporting Information Fig. S7). This supports the experimental observation that these positively charged groups might be able to partially take over the role of the arginine and can promote the catalytic activity. However, for GGNG only a few structures with such conformation were found, and due to the high flexibility of this chain, the N-terminus often turned out from the active site.
Figure 7.
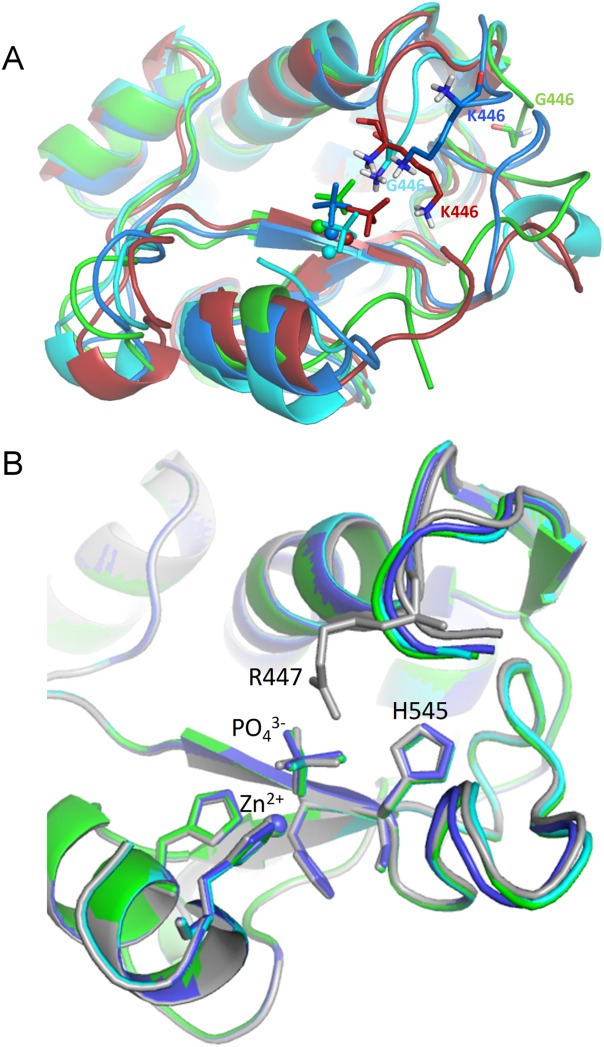
(A) Snapshots at 20 ns of the simulations of the mutant proteins: KGNK in red, KGNG in blue, GGNK in cyan, and GGNG in green. The Zn2+-ions are shown by spheres and the phosphate ion by sticks. (B) Optimized structure of the mutants and the WT NColE7. NColE7 is in grey, KGNG in blue, GGNK in cyan, and GGNG in green. Molecules were aligned by PyMOL.
Semiempirical calculations
To get more information about the fine structure of the active site, we optimized the geometry of the proteins by semiempirical quantum chemical calculations, handling the whole molecule on the PM6 level in MOPAC2009 (MOZYME method) with implicit water surroundings. The alignment to the optimized structure of NColE7 yielded an RMSD of 0.086 nm for KGNG, 0.087 nm for GGNK, and 0.113 nm for GGNG. Small deviations from NColE7 occurred mainly at the mutated N-termini, but could also be detected in the loop between the two β-strands of the HNH motif. The phosphate ion mimicking the scissile phosphodiester bond was also displaced [Fig. 7(B)]. This effect can be related to the decreased activity and supports the experimental data.
Discussion
The nuclease domain of ColE7 (NColE7) and its four N-terminal mutants [Fig. 1(B)] were expressed and purified. Gel electrophoresis and FLD spectroscopic experiments demonstrated that the catalytic activity of the mutants significantly decreased compared to NColE7. The results revealed a substantial decrease of the enzymatic activity (Figs. 2 and 3) due to the R447G mutation, in agreement with the previous observations with R447E and R447A mutants.5 According to LD results, the observed order of nuclease activity was NColE7 >> KGNK > GGNK ∼ KGNG > GGNG (Fig. 3). This order implies that K446 and K449 can promote the reaction in the absence of R447 to a low extent. The catalytic activity was not completely abolished in any mutant. The activity of the GGNG mutant in contrast to the ΔN4-NColE7 protein28 can be explained either by the positive N-terminal amino group at position G438 and/or the interactions of the backbone. The N-terminal part interacts directly with the DNA-binding helix, as well as with the loop of HNH motif [Fig. 1(A)]. The structure of this loop is essential for cleavage.35–37
The reason for the dramatic change in enzymatic activity was systematically studied. As shown by CD-spectroscopy (Fig. 4), the mutations did not significantly affect the distribution of the secondary elements in the structure of the protein, suggesting that all four mutants maintained the native NColE7 folding. Similar result was obtained by the comparison of CD spectra of the WT ColE7 with those of the K446E, R447E, and K446E/R447E mutants in the presence of Im7.5
According to the ITC results (Fig. 5), the Kd values are in nM range, similarly, as it was estimated for the Zn2+ binding of NColE9.38 The data collected in Table II demonstrate that all the mutants bound Zn2+-ion as strongly as NColE7 with 1:1 stoichiometry. The binding was enthalpy-favored resulting in large ΔH values ranging from −40 to −30 kcal/mol. The mutations had no obvious effect on Zn2+-binding: Kd ∼ 13 nM was obtained for all of the proteins within the experimental error suggesting that the difference in metal binding can not be the reason for the decreased activity of the mutants compared to NColE7.
Table II.
Zn2+ and DNA Binding Affinities of NColE7 Variants
| Zn2+ binding | DNA binding | ||
|---|---|---|---|
| Protein | Stoichiometry | Kd (nM) | Kd (µM) |
| NColE7 | 1.2 | 16.6 ± 3.5 | 0.16 ± 0.05 |
| KGNK | 0.9 | 13.2 ± 1.5 | 0.15 ± 0.05 |
| KGNG | 1.0 | 13.4 ± 1.4 | 0.32 ± 0.07 |
| GGNK | 1.2 | 11.9 ± 2.6 | 0.30 ± 0.06 |
| GGNG | 0.9 | 12.8 ± 1.9 | 0.30 ± 0.06 |
Micro ITC titrations of 50 µM proteins with ZnCl2 stock solution were performed in 20 mM cacodylate buffer at pH = 7.0. DNA binding of the 13 bp DNA was monitored by gel electrophoresis. For the analysis a model with one Zn2+ and one DNA binding site was applied. Kd is the apparent dissociation constant under the given condition. The fitting errors are also provided.
In an earlier publication, the significantly increased KM value of the R447A mutant compared to NColE7 was related to its decreased DNA binding affinity.5 The Kd values range between ∼ 0.15 and 0.32 µM for NColE7 and its variants (Table II), revealing that the changes at the N-terminus did not significantly affect the DNA binding affinity. This is in accordance with the fact that the strong DNA binding part of NColE7 is the central helical section.9,35,36,39 Only small differences were detected by FLD spectroscopy and gel mobility shift assays in the DNA binding of the mutants and NColE7 (Fig. 6). These results imply the functional role of the N-terminal positively charged groups either by taking part in the molecular mechanism of the reaction, or influencing the conformation and electronic properties of the bound DNA molecule.
The computational results showed that there is cooperation between the N- and C-termini of the protein. MD simulations pointed out that the N-terminal amino group and the lysine side-chains are capable to bind the scissile phosphate in the absence of R447 [Figs. 7(A) and Supporting Information Fig. S7]. According to semiempirical quantum chemical calculations, the active site of the mutants is slightly distorted, and thus the scissile phosphate is displaced [Fig. 7(B)].
Both experimental and computational data revealed that R447 is the most efficient, but the K446 and K449 side-chains and/or the N-terminal amino group can also promote the catalytic reaction. The phenomenon resembles to an intramolecular allosteric process in that the N-terminal flexible chain has to approach the C-terminal active site during reaction. This knowledge may be applied to modulate the catalytic activity of an NColE7 based artificial nuclease.
Materials and Methods
Cloning, protein expression and purification
The pQE70 plasmid containing the NColE7 and Im7 immunity protein genes was a generous gift from Prof. K.-F. Chak, Institute of Biochemistry and Molecular Biology, National Yang Ming University, Taipei, Taiwan.1,2 From this template, the primers applied in PCR (collected in Supporting Information Table S1.) amplified DNA segments including the genes of the native or mutated NColE7 as well as the Im7 protein. The obtained fragments were cloned into a pGEX-6P-1 vector (GE Healthcare BioSci.) providing an N-terminal GST affinity fusion tag. The plasmids were transformed into E. coli DH10B cells for DNA cloning and into E. coli BL21 (DE3) cells for protein production in 3 × 650 mL LB/Amp culture. At OD600 ∼ 0.6–0.7 the protein expression was induced with isopropyl β-D-1-thiogalactoside (IPTG, final concentration 0.1 mM), and the incubation was continued for further 2 h at 25°C. Cells were then sedimented by centrifugation and the pellets were resuspended in 50 mL PBS buffer (0.14 M NaCl, 2.7 mM KCl, 10.0 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3) by sonication. The soluble fractions were loaded on a GST affinity chromatography column (GSTPrepFF16/10, GE Healthcare BioSci.). The fusion proteins were cleaved on column with Human rhinovirus C3 protease40—sold as PreScission protease by GE Healthcare—to remove the GST tag. Twenty milliliter of 10 µM protease in PBS was loaded on the column and the reaction was continued overnight at 4°C or 2 h at room temperature followed by the elution with PBS. The fractions containing the nuclease mutants in complex with the immunity protein as well as the protease were collected. To disrupt the interaction between the NColE7 mutants and the Im7 protein, the pH was adjusted to 3.0 after a 3× dilution with a 20 mM Gly/HCl buffer. The components were then separated on a Sepharose SP FF 16/10 cation exchange column with a binding buffer 20 mM Gly/HCl pH = 3.0 and a gradient of 0–2 M NaCl in 30×column volume. The immunity protein was eluted at pH = 8.0 with PBS conatining 2M NaCl. The fractions of the nucleases or Im7 were concentrated by Amicon ultrafilter with 5 kDa cut-off, and the buffer was exchanged to 20 mM HEPES, pH = 7.7. The sequences of the purified mutant proteins in comparison with NColE7 are shown in Figure 1(B).
Nano-electrospray ionization mass spectrometry (nano-ESI-MS)
Mass spectra were obtained on a LCT Premier (Waters) instrument equipped with a Nanoflow Electrospray Ionization source and a time-of-flight analyzer. The instrument was operated in positive ion mode and it was calibrated using 100 mg/mL CsI in 50% 2-propanol in the m/z range from 600 to 12,000. Samples were sprayed from middle size Au/Pd-coated borosilicate glass capillary needles (Proxeon) loaded with 3 µL protein solution. The protein concentration was between 10 and 20 µM in 500 mM ammonium acetate (Sigma) buffer. For the study of metal binding, one equivalent Zn2+-acetate was added to the protein samples before measurement. The desalting of the protein solution and buffer exchange to the volatile buffer was done using Micro BioSpin chromatography column (BioRad). The needle voltage was typically around 1200 V and 50 V cone voltage was applied, with a cone gas maintained at 20 L/h and the source temperature was maintained at 50°C. The recorded m/z data were deconvoluted using the MassLynx™ v4.1 (Waters) software equipped with the MaxEnt1 algorithm. The high charge states of the multiply charged spectrum, ranging from +10 to +17, were used to calculate the apparent mass.
Isothermal calorimetry
Isothermal calorimetric titrations were performed on a MicroCal Auto ITC-200 (GE) instrument. The protein samples (∼50 µM) were prepared by 12 h dialysis in 7000 MWCO Thermo Scientific Slide-A-Lyzer casettes, against 20 mM cacodylate buffer, pH = 7.0. ZnCl2 was dissolved in the same buffer. The ionization enthalpy of cacodylate—applied as buffer—is close to zero (−0.47 kcal/mol41), so the contribution of protonation/deprotonation processes, if there were any present, was negligible in the observed ΔH. The dilution heat of ZnCl2 with the buffer was determined for each experiment and the integrated data of dilution heats were subtracted from the corresponding data of protein titrations. A control titration of the KGNK mutant with plain buffer was done showing no significant effects (data not shown). The enthalpy change during the titrations of 200 µL protein solutions with 2 µL aliquots of 400 µM ZnCl2 up to 40 µL (spacing of 240 s) can be consequently attributed to the metal binding or competition processes. Instead of degassing the samples before titration, the plates were shortly centrifuged.
Gel mobility shift assays
In the protein-DNA binding studies, the concentration of the 13 base pair oligonucleotide was 0.2 µM, and the protein final concentration ranged between 0 and 7 µM. The 31P-radiolabelled DNA was hybridized from one single oligonucleotide with complementary sequence at each end forming a loop. The samples were run on 6% native polyacrylamide gel at 4°C. The reaction mixture contained 4 mM NaCl, 4 mM HEPES buffer (pH = 7.9), and 100 µM EDTA to inhibit DNA digestion. For DNA cleavage studies, 450 ng/well 2686 base pairs pUC19 plasmid DNA was applied, and the protein concentration was 2.8 µM. Zn2+-ions were added to proteins prior the reaction in 1:1 molar ratio. Ten microliter of the reaction mixture was loaded onto an ethidium bromide containing 1% agarose gel. The electrophoresis was performed in a Bio-Rad Wide Mini Sub Cell® GT system at 6.7 V/cm in TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA, pH = 8.0). For comparison 6 µL of the Bio-Rad 1 kbp marker DNA was also loaded to the gel.
Circular dichroism spectroscopy
The SRCD spectra were recorded at the SRCD facility at the CD1 beamline at the Institute for Storage Ring Facilities (ISA), University of Aarhus, Denmark.42,43 All spectra were recorded with 1 nm steps and a dwell time of 2 s per step, in 100.4 µm quartz cells (SUPRASIL, Hellma GmbH, Germany). The concentration of the protein solutions was 3.2 × 10−5 M in 10 mM HEPES, pH = 7.7.
Linear dichroism spectroscopy
FLD spectra were measured on a Jasco-815 CD spectrophotometer equipped for LD spectroscopy, using a microvolume Couette flow device as described in.44 An additional quartz lens was mounted to allow for focusing onto the sample in the Couette cell, which was positioned as close to the photomultiplier as possible in the J-815 sample compartment. In the Couette cell, an outer quartz cylinder rotates, and an inner quartz rod is static, the annular gap is 0.25 mm giving a combined light path of 0.5 mm. Water circulation through the metal block of the flow device thermostated the cell to 298.0 K. The sample volume was 70 µL and 3000 rpm rotation was applied. The optical bandwidth was 1 nm and the spectra were recorded in continuous mode between 190 and 400 nm with 50 nm/min scanning speed, 1 s integration time, 0.5 nm data pitch, and with 15 L/min nitrogen flow. LD of double stranded DNA samples yields a characteristic negative signal at the absorbance maximum of the DNA bases (ca. 260 nm) since the transition moments of the base π- π* transitions are located in the plane of the bases, and as these are oriented orthogonally with respect to the axis of DNA helix.45
Protein-DNA binding was studied with a 130 µM (final concentration calculated for base pairs) calf thymus CT-DNA sample. The mixture contained 60 µM EDTA, 17 mM KH2PO4 and 2.4 mM HEPES (the pH of both buffers was adjusted to 7.7), and 0–5 µM protein. The solutions were incubated for 10 min before recording the spectra. The incubation time did not influence the results.
The cleavage of 130 µM (bp) CT-DNA by different mutants was followed for 8 h (1 spectrum/h). The proteins (40 µM) were incubated with one equivalent of Zn(Ac)2 (40 µM) for 30 min at room temperature before the reaction started. The final concentration of both the protein and Zn2+-ions was 0.5 µM. 720 µL of reaction mixture was incubated at 37°C and in each hour 70 µL aliquot was taken for LD test. All solutions were in mixed buffers of 17 mM KH2PO4 and 2.4 mM HEPES, the pH of both buffers was adjusted to 7.7. The spectra were smoothed with the means-movement method, convolution width of 11.
Computational methods
Initial conformation of NColE7 and the shortened mutants was obtained from a crystal structure 1M08.7 The original N-terminus of NColE7 was restored by an M446K mutation. In all models, the proteins had uncapped termini (i.e., NH3+ and COO− groups).
MD calculations were performed with GROMACS 4.05,46 with the force field Gromos 53a6.47 The ionizable residues were charged according to the default pKa values at pH = 7.2 detected by PropKa 3.0.48 Each protein was placed in a cubic box with edge size of about 8 nm, and solvated by explicit SPC/E water model containing about 16,000 equilibrated water molecules. The system was neutralized with Cl− ions replacing water molecules according to the electrostatic potential of the system points. Energy minimization was carried out with the steepest descent method. Two hundred picoseconds position restrained dynamics was performed in NVT ensemble to equilibrate the system (solvate and generate initial velocities with Maxwell distribution) including explicit water molecules. Twenty-five nanoseconds MD simulations were performed in the NPT ensemble with periodic box conditions. The integration step was 2 fs. The temperature was set to 300 K and isotropic Berendsen p-coupling and T-coupling was applied. For Coulomb interactions, Particle Mesh Ewald Method (PME) was applied with 0.9 nm cut-off for electrostatic and 1.6 nm for van der Waals interactions. The dielectric constant was set to 1.0. The Linear Constraint Solver (LINCS) constraint algorithm was used. Trajectories were analyzed starting at 500 ps.
Semiempirical quantum chemical computations were performed on the proteins including a Zn2+ and a phosphate ion with the PM6 method implemented in MOPAC2009.49–52 Localized molecular orbitals were applied by the MOZYME52 model. The solvation was considered by COSMO method53 with the dielectric constant of 78.4. The geometry optimization was carried out by the L-Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm method after the initial minimization of the hydrogen positions. The gradient norm was set to 1.0 kcal/mol/Å. The thermodynamic parameters were computed with the PM6 method, and then recalculated in one self-consistent field method (SCF) cycle with the PM6-DH2 correction.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Chak K, Kuo W, Lu F, James R. Cloning and characterization of the Cole7 plasmid. J Gen Microbiol. 1991;137:91–100. doi: 10.1099/00221287-137-1-91. [DOI] [PubMed] [Google Scholar]
- 2.Lin Y, Liao C, Liang P, Yuan H, Chak K. Involvement of colicin in the limited protection of the colicin producing cells against bacteriophage. Biochem Biophys Res Commun. 2004;318:81–87. doi: 10.1016/j.bbrc.2004.03.184. [DOI] [PubMed] [Google Scholar]
- 3.Liao C, Hsiao K, Liu Y, Leng P, Yuen HS, Chak K. Processing of DNase domain during translocation of colicin E7 across the membrane of Escherichia coli. Biochem Biophys Res Commun. 2001;284:556–562. doi: 10.1006/bbrc.2001.5016. [DOI] [PubMed] [Google Scholar]
- 4.Cheng YS, Shi Z, Doudeva LG, Yang WZ, Chak KF, Yuan HS. High-resolution crystal structure of a truncated ColE7 translocation domain: implications for colicin transport across membranes. J Mol Biol. 2006;356:22–31. doi: 10.1016/j.jmb.2005.11.056. [DOI] [PubMed] [Google Scholar]
- 5.Shi Z, Chak K, Yuan H. Identification of an essential cleavage site in ColE7 required for import and killing of cells. J Biol Chem. 2005;280:24663–24668. doi: 10.1074/jbc.M501216200. [DOI] [PubMed] [Google Scholar]
- 6.Sui M, Tsai L, Hsia K, Doudeva L, Ku W, Han G, Yuan H. Metal ions and phosphate binding in the H-N-H motif: crystal structures of the nuclease domain of ColE7/Im7 in complex with a phosphate ion and different divalent metal ions. Protein Sci. 2002;11:2947–2957. doi: 10.1110/ps.0220602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y, Hsia K, Doudeva LG, Chak K, Yuan HS. The crystal structure of the nuclease domain of Colicin E7 suggests a mechanism for binding to double-stranded DNA by the H–N–H endonucleases. J Mol Biol. 2002;324:227–236. doi: 10.1016/s0022-2836(02)01092-6. [DOI] [PubMed] [Google Scholar]
- 8.Hsia K, Li C, Yuan H. Structural and functional insight into sugar-nonspecific nucleases in host defense. Curr Opin Struct Biol. 2005;15:126–134. doi: 10.1016/j.sbi.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 9.Doudeva L, Huang D, Hsia K, Shi Z, Li C, Shen Y, Cheng Y, Yuan H. Crystal structural analysis and metal-dependent stability and activity studies of the ColE7 endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+ Protein Sci. 2006;15:269–280. doi: 10.1110/ps.051903406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pommer AJ, Cal S, Keeble AH, Walker D, Evans SJ, Kühlmann UC, Cooper A, Connolly BA, Hemmings AM, Moore GR, James R, Kleanthous C. Mechanism and cleavage specificity of the H-N-H endonuclease colicin E9. J Mol Biol. 2001;314:735–749. doi: 10.1006/jmbi.2001.5189. [DOI] [PubMed] [Google Scholar]
- 11.Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, Cavard D. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papadakos G, Wojdyla JA, Kleanthous C. Nuclease colicins and their immunity proteins. Q Rev Biophys. 2012;45:57–103. doi: 10.1017/S0033583511000114. [DOI] [PubMed] [Google Scholar]
- 13.Galburt EA, Stoddard BL. Catalytic mechanisms of restriction and homing endonucleases. Biochemistry. 2002;41:13851–13860. doi: 10.1021/bi020467h. [DOI] [PubMed] [Google Scholar]
- 14.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 15.Mehta P, Katta K, Krishnaswamy S. HNH family subclassification leads to identification of commonality in the His-Me endonuclease superfamily. Protein Sci. 2004;13:295–300. doi: 10.1110/ps.03115604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:3552–3569. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veluchamy A, Mary S, Acharya V, Mehta P, Deva T, Krishnaswamy S. HNHDb: a database on pattern based classification of HNH domains reveals functional relevance of sequence patterns and domain associations. Bioinformation. 2009;4:80–83. doi: 10.6026/97320630004080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku W, Liu Y, Hsu Y, Liao C, Liang P, Yuan H, Chak K. The zinc ion in the HNH motif of the endonuclease domain of colicin E7 is not required for DNA binding but is essential for DNA hydrolysis. Nucleic Acids Res. 2002;30:1670–1678. doi: 10.1093/nar/30.7.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eastberg JH, Eklund J, Monnat R., Jr Stoddard BL. Mutability of an HNH nuclease imidazole general base and exchange of a deprotonation mechanism. Biochemistry. 2007;46:7215–7225. doi: 10.1021/bi700418d. [DOI] [PubMed] [Google Scholar]
- 20.Midon M, Gimadutdinow O, Meiss G, Friedhoff P, Pingoud A. Chemical rescue of active site mutants of S. pneumoniae surface endonuclease EndA and other nucleases of the HNH family by imidazole. Chembiochem. 2012;13:713–721. doi: 10.1002/cbic.201100775. [DOI] [PubMed] [Google Scholar]
- 21.Dupureur CM. Roles of metal ions in nucleases. Curr Opin Chem Biol. 2008;12:250–255. doi: 10.1016/j.cbpa.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Ko T, Liao C, Ku W, Chak K, Yuan HS. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure. 1999;7:91–102. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- 23.Garinot-Schneider C, Pommer AJ, Moore GR, Kleanthous C, James R. Identification of putative active-site residues in the DNase domain of Colicin E9 by random mutagenesis. J Mol Biol. 1996;260:731–742. doi: 10.1006/jmbi.1996.0433. [DOI] [PubMed] [Google Scholar]
- 24.Bueren-Calabuig JA, Coderch C, Rico E, Jimenez-Ruiz A, Gago F. Mechanistic insight into the catalytic activity of betabetaalpha-metallonucleases from computer simulations: vibrio vulnificus periplasmic nuclease as a test case. Chembiochem. 2011;12:2615–2622. doi: 10.1002/cbic.201100485. [DOI] [PubMed] [Google Scholar]
- 25.Walker DC, Georgiou T, Pommer AJ, Walker D, Moore GR, Kleanthous C, James R. Mutagenic scan of the H-N-H motif of colicin E9: implications for the mechanistic enzymology of colicins, homing enzymes and apoptotic endonucleases. Nucleic Acids Res. 2002;30:3225–3234. doi: 10.1093/nar/gkf420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maté MJ, Kleanthous C. Structure-based analysis of the metal-dependent mechanism of H-N-H endonucleases. J Biol Chem. 2004;279:34763–34769. doi: 10.1074/jbc.M403719200. [DOI] [PubMed] [Google Scholar]
- 27.Li CL, Hor LI, Chang ZF, Tsai LC, Yang WZ, Yuan HS. DNA binding and cleavage by the periplasmic nuclease Vvn: a novel structure with a known active site. EMBO J. 2003;22:4014–4025. doi: 10.1093/emboj/cdg377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czene A, Németh E, Zóka IG, Jakab-Simon NI, Körtvélyesi T, Nagata K, Christensen HEM, Gyurcsik B. The role of the N-terminal loop in the function of the colicin E7 nuclease domain. J Biol Inorg Chem. 2013;18:309–321. doi: 10.1007/s00775-013-0975-7. [DOI] [PubMed] [Google Scholar]
- 29.Gyurcsik B, Czene A. Towards artificial metallonucleases for gene therapy: recent advances and new perspectives. Future Med Chem. 2011;3:1935–1966. doi: 10.4155/fmc.11.139. [DOI] [PubMed] [Google Scholar]
- 30.Matsuoka Y, Nielsen PE, Norden BJ. On the structure of active chromatin. A flow linear dichroism study of chromatin fractionated by nuclease digestion. FEBS Lett. 1984;169:309–312. doi: 10.1016/0014-5793(84)80340-3. [DOI] [PubMed] [Google Scholar]
- 31.Gololobov GV, Chernova EA, Schourov DV, Smirnov IV, Kudelina IA, Gabibov AG. Cleavage of supercoiled plasmid DNA by autoantibody Fab fragment: application of the flow linear dichroism technique. Proc Natl Acad Sci USA. 1995;92:254–257. doi: 10.1073/pnas.92.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hicks MR, Rodger A, Thomas CM, Batt SM, Dafforn TR. Restriction enzyme kinetics monitored by UV linear dichroism. Biochemistry. 2006;45:8912–8917. doi: 10.1021/bi0601712. [DOI] [PubMed] [Google Scholar]
- 33.van den Bremer ET, Jiskoot W, James R, Moore GR, Kleanthous C, Heck AJ, Maier CS. Probing metal ion binding and conformational properties of the colicin E9 endonuclease by electrospray ionization time-of-flight mass spectrometry. Protein Sci. 2002;11:1738–1752. doi: 10.1110/ps.0200502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao Y, Fu D. Thermodynamic studies of the mechanism of metal binding to the Escherichia coli zinc transporter YiiP. J Biol Chem. 2004;279:17173–17180. doi: 10.1074/jbc.M400208200. [DOI] [PubMed] [Google Scholar]
- 35.Hsia K, Chak K, Liang P, Cheng Y, Ku W, Yuan HS. DNA binding and degradation by the HNH protein ColE7. Structure. 2004;12:205–214. doi: 10.1016/j.str.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Yang W, Li C, Doudeva LG, Yuan HS. Structural basis for sequence-dependent DNA cleavage by nonspecific endonucleases. Nucleic Acids Res. 2007;35:584–594. doi: 10.1093/nar/gkl621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang H, Yuan HS. The conserved asparagine in the HNH motif serves an important structural role in metal finger endonucleases. J Mol Biol. 2007;368:812–821. doi: 10.1016/j.jmb.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 38.Pommer AJ, Kuhlmann UC, Cooper A, Hemmings AM, Moore GR, James R, Kleanthous C. Homing in on the role of transition metals in the HNH motif of colicin endonucleases. J Biol Chem. 1999;274:27153–27160. doi: 10.1074/jbc.274.38.27153. [DOI] [PubMed] [Google Scholar]
- 39.Levin KB, Dym O, Albeck S, Magdassi S, Keeble AH, Kleanthous C, Tawfik DS. Following evolutionary paths to protein-protein interactions with high affinity and selectivity. Nat Struct Mol Biol. 2009;16:1049–1055. doi: 10.1038/nsmb.1670. [DOI] [PubMed] [Google Scholar]
- 40.Walker P, Leong L, NG P, Tan S, Waller S, Murphy D, Porter A. Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Biotechnology. 1994;12:601–605. doi: 10.1038/nbt0694-601. [DOI] [PubMed] [Google Scholar]
- 41.Fukada H, Takahashi K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins. 1998;33:159–166. [PubMed] [Google Scholar]
- 42.Miles AJ, Hoffmann SV, Tao Y, Janes RW, Wallace BA. Synchrotron radiation circular dichroism (SRCD) spectroscopy: new beamlines and new applications in biology. J Spectroscopy. 2007;21:245–255. [Google Scholar]
- 43.Miles AJ, Janes RW, Brown A, Clarke DT, Sutherland JC, Tao Y, Wallace BA, Hoffmann SV. Light flux density threshold at which protein denaturation is induced by synchrotron radiation circular dichroism beamlines. J Synchrotron Radiat. 2008;15:420–422. doi: 10.1107/S0909049508009606. [DOI] [PubMed] [Google Scholar]
- 44.Marrington R, Dafforn T, Halsall D, Rodger A. Micro-volume Couette flow sample orientation for absorbance and fluorescence linear dichroism. Biophys J. 2004;87:2002–2012. doi: 10.1529/biophysj.103.035022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norden B, Kubista M, Kurucsev T. Linear dichroism spectroscopy of nucleic-acids. Q Rev Biophys. 1992;25:51–170. doi: 10.1017/s0033583500004728. [DOI] [PubMed] [Google Scholar]
- 46.Hess B, Kutzner C, van dS, Lindahl E. GROMACS 4:   algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 47.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 48.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 49.Stewart JJP. Application of the PM6 method to modeling proteins. J Mol Model. 2009;15:765–805. doi: 10.1007/s00894-008-0420-y. [DOI] [PubMed] [Google Scholar]
- 50.Stewart JJP. 2008. MOPAC2009, Stewart computational chemistry. Colorado Springs. Available at: http://openmopac.net.
- 51.Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model. 2007;13:1173–1213. doi: 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stewart JJP. Application of localized molecular orbitals to the solution of semiempirical self-consistent field equations. Int J Quantum Chem. 1996;58:133–146. [Google Scholar]
- 53.Klamt A, Schüürmann G. Cosmo—a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans. 1993;2:799–805. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information