

Abstract
There are many proteomic applications that require large collections of purified protein, but parallel production of large numbers of different proteins remains a very challenging task. To help meet the needs of the scientific community, we have developed a human protein production pipeline. Using high-throughput (HT) methods, we transferred the genes of 31 full-length proteins into three expression vectors, and expressed the collection as N-terminal HaloTag fusion proteins in Escherichia coli and two commercial cell-free (CF) systems, wheat germ extract (WGE) and HeLa cell extract (HCE). Expression was assessed by labeling the fusion proteins specifically and covalently with a fluorescent HaloTag ligand and detecting its fluorescence on a LabChip® GX microfluidic capillary gel electrophoresis instrument. This automated, HT assay provided both qualitative and quantitative assessment of recombinant protein. E. coli was only capable of expressing 20% of the test collection in the supernatant fraction with ≥20 μg yields, whereas CF systems had ≥83% success rates. We purified expressed proteins using an automated HaloTag purification method. We purified 20, 33, and 42% of the test collection from E. coli, WGE, and HCE, respectively, with yields ≥1 μg and ≥90% purity. Based on these observations, we have developed a triage strategy for producing full-length human proteins in these three expression systems.
Keywords: human protein, cell-free protein expression, in vitro transcription translation, Escherichia coli, E. coli expression, wheat germ extract, HeLa cell extract, microfluidic capillary gel electrophoresis, high throughput, protein expression, protein purification, high throughput expression analysis, full-length protein, HaloTag, SIFT
Introduction
The broad goals of proteomics include understanding the composition, structure, and function of all proteins in biological systems, as well as how the various components collectively contribute to phenotype. The technologies used to meet these goals have advanced considerably over the past decade, especially in regard to the throughput capability of sample preparation, assaying, and data analysis. However, high-throughput (HT) protein production continues to be a bottleneck despite the need for purified proteins in many aspects of protein characterization, such as structure determination,1 enzymatic activity analysis,2–4 and protein identification and quantification.5 Purified proteins are also needed for all current methods performing affinity reagents development. Unfortunately, the high cost and limited availability of purified proteins have resulted in a lack of highly specific and high-quality affinity reagents targeting the majority of human proteins.6 Large efforts such as the Protein Capture Reagent Program at the NIH (http://commonfund.nih.gov/proteincapture) aim to address this widespread need. A number of the technologies used by that program require starting with purified antigen in the range of several to tens of micrograms. However, even with such low quantity requirements, the lack of availability and high cost of pure antigens remains a major roadblock toward the goals of producing affinity reagents for the proteome, novel technology development, and scientific knowledge advancement.
Escherichia coli has established itself as the predominant organism for protein production because of the low cost, easy manipulation, and the extensive knowledge accumulated over the past 30 years.7,8 We have previously reported on the in vivo and in vitro high throughput production of human proteins in E. coli.9,10 Although excellent for some proteins, E. coli is poorly suited for producing multidomain full-length eukaryotic proteins that are generally larger than 50 kDa. The problems are often related to poor solubility and ensuing toxicity when overexpressed in bacteria despite extensive effort devoted to circumvent these challenges.8,10–14 As full-length proteins are not a requirement for structural genomics, many protein production centers create many different constructs for each protein of interest (POI). Variants are generated by truncating unstructured regions of protein, parsing domains, using multiple fusion tags and purification strategies, using vectors with varied promoters, and testing different expression and induction conditions, and so forth, to increase the success rate of expression and purification.7,11,12,14–17 Processing multitudes of constructs requires additional time, labor, costs, and bioinformatics support. When multiplied by the scale of the proteome, this approach becomes costly.
The use of insect or mammalian cells for in vivo recombinant protein production has alleviated some key limitations associated with prokaryotic expression systems. Eukaryotic expression systems possess the ability to translate and support correct folding of large multidomain eukaryotic proteins, and to perform eukaryotic posttranslational modifications (PTMs) that are essential for function.10,18–21 However, in vivo eukaryotic expression systems are limited by high cost and labor intensity, difficulty in cell lysis, toxicity, batch variations, and difficulty to automate for HT production.22–24
Significant advances in recent years have been made in the development of commercial eukaryotic CF expression systems. Although, historically the yields of protein were low enough to require isotopic labeling,25 the efficiency of eukaryotic CF expression systems has improved dramatically, substantially lowering the cost per unit of protein produced. Two new systems in particular, the wheat germ extract (WGE)-based WEPRO 7240 series (CellFree Science) and HeLa cell extract (HCE) 1-step human high-yield in vitro translation (IVT) line (Thermo Scientific), have both been reported to express substantially more protein than their predecessors.19,26 A key advantage of CF systems over in vivo eukaryotic expression is that CF expression protocols better facilitate automation, and lack many of the requisite steps of in vivo methods, including the selection of high expression clones, cell culture, and cell lysis, that are associated with in vivo work. Moreover, there is minimal batch-to-batch variation, and protein toxicity is not a concern. These systems have been shown to support some PTMs, including phosphorylation, but most likely have limited capacity to perform other PTMs, such as glycosylation and disulfide bond formation, which makes them less suitable for expressing secreted and membrane proteins.
Very few studies have investigated the suitability of WGE for HT protein production19,27,28 and fewer still have tested HCE systems.5 There is a great disparity between the adoption of CF systems and the use of E. coli for protein expression. In contrast with E. coli, the expression host for 88% of structure determined proteins in the protein data bank, less than 0.5% of protein structures were determined from proteins generated by CF expression.20 Previously reported E. coli protein production pipelines have highly variable success rates, and are largely influenced by each study's target selection criteria and the application-dependent standard of success; collections that are biased toward smaller proteins with predicted similarity to structurally determined proteins or have been domain parsed or truncated, tend to have higher success rates. Reported E. coli expression success rates range from 20 to 80%.9–14,16,28 In some of these works, protein collections that were reported to have low success rates in E. coli had much higher success rates of about 65–95% in WGE.10,16,28 Selection bias for proteins that are likely to express and purify can increase the success rate significantly, but results in many proteins that are untried and uncharacterized. HT methods that succeed in expressing and purifying a high percentage of a large, diverse collection of proteins are highly desired.
Automation of the protein production process, from cloning to assaying purified protein, is essential for HT capacity. Productive pipelines automate the most labor intensive steps, which have traditionally been culturing E. coli, purifying proteins and characterizing expressed and purified recombinant protein. The use of liquid handling robots has become routine at protein production centers to address these needs. Expression and purification are performed in 96-well deepwell block format that conforms to robotic liquid handler operation. HT protein analysis that provides both qualitative and quantitative data has been facilitated with the adoption of microfluidic capillary gel electrophoresis (MCGE). These microfluidic characterization methods, however, analyze total, nonspecific protein. The use of microfluidic systems to evaluate recombinant proteins specifically has not been widely incorporated into protein production center pipelines. Developing protein production pipelines will benefit from the ability to specifically detect recombinant protein with a HT assay.
The goal for this study was to establish a pipeline that supports HT full-length human protein production with greater than 90% purity at the scale of tens of micrograms. We compared the performance among two eukaryotic CF expression systems, HCE and WGE, and the conventional E. coli expression strategy. We tested 31 full-length proteins ranging in size from 10 to 120 kDa in expression vectors that utilized Gateway™ technology. We developed a selective binding reagent and an associated method that allows the specific fluorescent evaluation of recombinant protein in a complex protein mixture using MCGE. Using HaloTag as a fusion partner,29,30 POIs were purified in all three expression systems on an automated platform. We successfully purified 45% of these full-length proteins. We demonstrated that both eukaryotic CF systems were far more capable of generating larger proteins than E. coli.
Results
HaloTag expression vector construction
We decided to use a solubility enhancing fusion tag, HaloTag, for our development. Solubility enhancing fusion tags have been reported to improve success rates of E. coli protein production,9,11 but the benefits of these tags for CF expression is less understood. HaloTag binds covalently to a chloroalkane substrate that can be immobilized on beads for purification or linked to various functional groups for downstream assays.29,30 We found that the solubility of several proteins expressed as HaloTag fusion proteins was comparable to other commonly used affinity tags, like glutathione S-transferase (GST) and maltose binding protein (MBP).
Three expression vectors were constructed to support protein expression and purification in the three systems of interest, pCPD_nHalo for E. coli expression, pJSP6_nHalo for WGE, and pJFT7_nHalo for HCE (Supporting Information Fig. S1). These plasmids were adapted from established expression vectors pMCSG32, pEU_HSBC, and pANT7_cGST for E. coli, WGE, and HCE, respectively, to express POIs with an N-terminal HaloTag and tobacco etch virus (TEV) protease cleavage site. The vectors we developed function as Gateway destination vectors, enabling the transfer of full-length human genes in frame through a one-step recombination reaction, and are compatible with tens of thousands of available open reading frame constructs.31,32
Protein selection
We selected 31 full-length proteins (30 human and 1 mouse) listed in Supporting Information Table S1 to evaluate the expression capabilities of the three expression systems. The proteins were chosen if (1) they were involved in cancer pathways,33 (2) their full-length cDNA was available in our plasmid repository of ∼10,000 human genes in a Gateway donor vector,32 and (3) they were not membrane proteins. In addition, several of the selected proteins had previously been produced by a protein structure initiative center (http://www.nesg.org/). The test collection of 31 proteins ranges in size from ∼10 to 120 kDa, and includes seven kinases and four transcription factors. Otherwise, no special selection method was used to create this collection.
Cell free expression systems
The WGE and HCE CF systems differ in several aspects. The WGE system performs transcription and translation in two consecutive steps, whereas HCE is a coupled transcription and translation system. The coupling of transcription and translation enables the use of cDNA as a direct input for protein expression, which is essential for some proteomic applications.26,34 The yield of both systems can be increased by implementing a continuous-exchange CF (CECF) expression format, in which the translation reaction occurs in a semipermeable chamber that is supplied with translation reactants and allows by-products to diffuse away. In addition, WGE can utilize a bilayer format that accomplishes a similar effect without the need of a dialysis membrane, making it more amenable to automation. In this format, the combined volume of the reaction mixture and dialysis buffer is collected for downstream applications such as purification. As illustrated in Supporting Information Figure S2, we used the dialysis format for HCE expression and the bilayer format for WGE expression.
HT HaloTag protein expression analysis
In developing our HT platform, we sought a rapid and simple method to assess production of our recombinant proteins, which is commonly achieved through the use of low-throughput slab gel sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). To observe all proteins, gels are often stained with Coomassie brilliant blue. Visualizing the specific recombinant protein typically requires the complex multistage process of immunoblotting, including transferring the proteins to membranes and probing with specific antibodies. When production shifts to HT, the cumbersome nature of slab gels must give way to automated MCGE systems, such as the LabChip platform. To measure all proteins, these microfluidic systems mix protein samples with an amine-reactive fluorescent dye that labels proteins nonspecifically, and detect proteins with laser-induced fluorescence at the end of the separation channel. This provides an analysis akin to Coomassie-stained SDS-PAGE slab gels. Although LabChip instruments are not typically used to specifically visualize recombinant proteins in complex samples, there are automated immunoblot MCGE systems that perform this function;35 however, these systems are dedicated solely to immuno-MCGE protocols and lack a total protein detection mode, depend on antibody performance, and have lower throughput than LabChip instruments.
To address this shortfall, we developed a HT HaloTag protein detection method without the need for blotting or antibodies. HaloTag fusion proteins in a complex protein mixture can be specifically labeled with a fluorescently conjugated HaloTag ligand that binds covalently to the HaloTag fusion. The protein samples can then be denatured, separated by SDS-PAGE, and the in-gel fluorescent signal measured with a fluorescent imager.29 To develop a method for a microfluidics platform, we combined the HaloTag ligand with Alexa660 and used this to label our recombinant proteins. This was tested on the LabChip GX instrument, which detects the Alexa660 fluorophore, and we observed that HaloTag fusion proteins can be assessed specifically and in parallel with the standard measurements of total protein. We refer to this approach as selective irreversible fluorescence targeting (SIFT). Figure 1 compares total protein and HaloTag-specific signals on a slab gel with the equivalent MCGE assays on a LabChip GX system.
Figure 1.
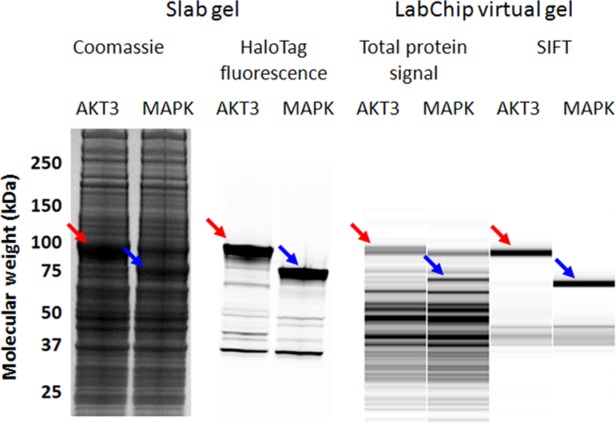
Comparison of slab gel and LabChip expression analysis of AKT3 and MAPK, which were expressed in HCE. Total protein signal can be observed by staining the slab gel with Coomassie, whereas recombinant HaloTag signal can be detected with in-gel fluorescence. For high throughput analysis, the LabChip system can be utilized for measuring total protein content using the Pico Protein Express protocol, whereas HaloTag signal can be distinguished with SIFT.
HT protein expression protocol development
When working with a varied collection of proteins, protein-dependent effects, especially toxicity in E. coli, can cause variation in preinduction growth rate and expression yield. Optimization can be performed individually for each protein, but this is a labor intensive approach and unsuitable for HT protein production. Accordingly, the imperative of HT expression is to express as many proteins as possible using the smallest number of protocols. Many parameters were tested to optimize overall protein expression of the test collection, including evaluating multiple E. coli expression strains, induction strategies, and growth media. Isopropyl β-D-1-thiogalactopyranoside (IPTG)-induced minimal media and lower expression temperatures were found to provide the most consistent induction strategy and resulted with the highest fraction of tested proteins in the soluble fraction compared with either nutrient rich media or auto-inducing media. For CF systems, multiple parameters were assessed, including CF expression formats, transcription template concentration, expression time, and temperature. Manufacturer recommended conditions were found to be optimal for most of these factors. The bilayer format for WGE and dialysis format for HCE both provided the most efficient expression configurations for most proteins, and with these formats, overnight expression gave higher yields than shorter incubations. Using optimal conditions described in Materials and Methods section, the test collection was expressed in the three systems.
Expression system comparison
Expression of the full-length protein collection in all three systems was assessed using the SIFT assay, and the virtual gels shown in Figure 2 specifically show the recombinant protein in all supernatant fractions. A comparison of slab gel and LabChip virtual gel for the HCE collection is shown in Supporting Information Figure S3. LabChip-SIFT protein quantification was found to be comparable to slab-gel based quantification (Supporting Information Fig. S4). Nearly, all proteins migrate in the LabChip assay as they do in a slab gel. Several proteins were expressed at high levels in all three expression systems; however, these proteins tend to be the smaller members of the collection, due primarily to the low expression success rate of E. coli for larger proteins. Many proteins did express in E. coli, but approximately half of these were insoluble: the total fractions of these samples show fluorescently labeled HaloTag proteins, but the signal is absent in the supernatant fraction. This indicates that these insoluble fusion proteins contain a functional HaloTag. With the exception of MYOT, which had low solubility in all three systems, every other protein that expressed in a CF system was soluble. There is a clear trend that E. coli does not efficiently produce larger proteins (>80 kDa), whereas WGE and HCE systems express these better. As can be seen in the heat map of recombinant fusion protein quantification, expression yield was more variable in E. coli and HCE, whereas WGE expressed the collection at more homogenous levels. Average expression yields are shown in Table I.
Figure 2.
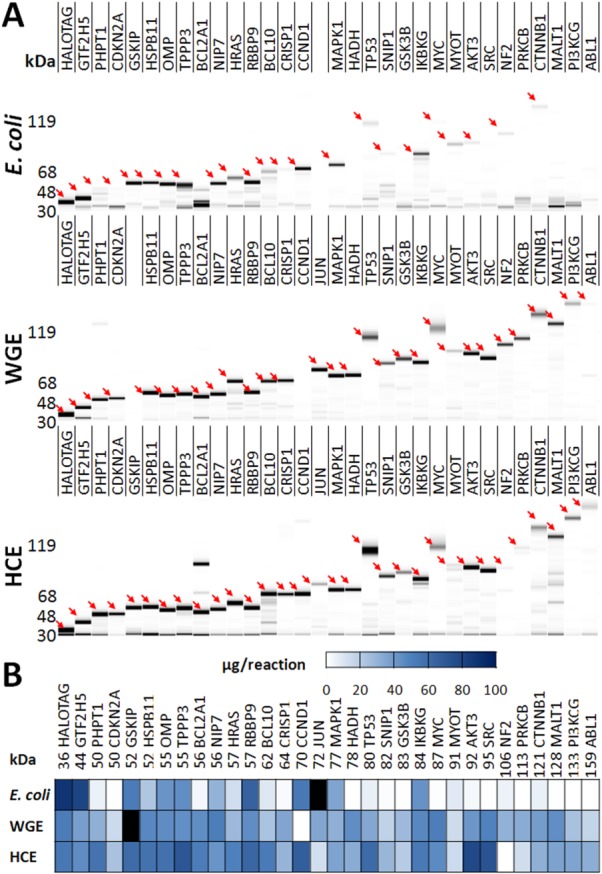
SIFT expression analysis of HaloTag fusion proteins. (A) Virtual gel images of the full-length human protein collection expressed as N-terminal HaloTag fusion proteins in E. coli, WGE, and HCE systems. Following expression, supernatant fractions of reactions were diluted 1:20, 1:5, and 1:10 for E. coli, WGE, and HCE, respectively, incubated with the SIFT Ligand, and analyzed using a LabChip GX instrument. Arrows point to the POI. (B) The heat map displays yield per reaction. Proteins that were not determined are shaded black.
Table I.
Summary of High Throughput Protein Expression and HaloTag Purification Using Three Expression Systems
| Expression system | E. coli | WGE | HCE |
|---|---|---|---|
| Reaction volumea | 2 mL | 226 μL | 100 μL |
| Expression success rateb | 22/30 (73%) | 26/30 (87%) | 27/31 (87%) |
| Soluble expression success rateb | 10/30 (33%) | 25/30 (83%) | 26/31 (84%) |
| Mean soluble expression yield (μg) | 47.5 | 35.1 | 45.5 |
| HaloTag purification success ratec | 6/30 (20%) | 10/30 (33%) | 13/31 (42%) |
| Mean HaloTag purification yield (μg) | 5.8 | 4.1 | 3.5 |
| Mean HaloTag purification recoveryd | 42% | 24% | 15% |
WGE was performed in a bilayer CECF format; HCE was performed in a dialysis CECF format.
≥20 μg. The HaloTag control is not included in this count.
≥1 µg, ≥90% purity.
Recovery is calculated for each protein as (purification yield/expression yield) × (fusion protein mass/target mass).
Degradation of full-length protein into a stable HaloTag end product was observed in both E. coli and HCE expression systems. This proteolysis has been observed in similar studies with different fusion tags.9 This degradation, along with overall expression full-length protein yield, coincided with longer incubation times. Notably, this degradation was minimal for bilayer WGE produced protein.
Full-length human protein purification with HaloTag
Expression reactions were bound to magnetic HaloTag ligand resin. Using a liquid handling robot, the resin was washed to remove unbound proteins, and POIs were released by cleavage with a HaloTag fusion TEV protease that itself also bound to available ligand on the resin. To optimize protein purification, numerous parameters were tested, including the amount of protease, resin-to-sample ratio, binding time, number of washes, cleavage time, resin equilibration buffers, and type of detergent. In general, the HaloTag purification strategy was found to be robust, as yield and purity changed little with respect to choice of buffer [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), phosphate buffered saline (PBS), and tris buffered saline (TBS)], reducing reagent [dithiothreitol (DTT) and tris(2-carboxyethyl)phosphine (TCEP)], or detergent (IGEPAL CA 630 and Tween 20). Resin and protease quantity significantly impacted the yield of purified protein, and notably, excess resin decreased yield. When the resin was washed less than five times after the binding step, overall protein purity was lower. The collection of proteins was purified using the optimal conditions described in Materials and Methods section.
SDS-PAGE analysis of the purified test collection is shown in Figure 3. The TEV-cleaved HaloTag (∼34 kDa) from HaloTag fusion proteins and HaloTEV protease (∼80 kDa) were sometimes observed as weak contaminants. It is not clear why both of these proteins were not removed by the Halo resin. We confirmed that the eluted HaloTag contaminants were functional because they reacted with the SIFT ligand, but they were not selectively depleted from the elution fraction after rebinding eluate to fresh Halo resin. The HaloTag band was more prominently observed with multiple proteins that are known to oligomerize when properly folded, such as WGE- and HCE-produced IKBKG, HADHSC, and p53. Conceivably, the protein may have been captured through the HaloTag of one subunit of the oligomer, and the other members were captured by virtue of their oligomerization, leaving the HaloTag fusion of one or more of the subunits unbound to the resin and releasable by the TEV protease. If this is the case, the abundance of cleaved HaloTag in the CF purifications and its absence in elution fractions of E. coli implies that proteins are better folded in the CF systems than in E. coli. However, this band was also observed with some proteins not expected to form homooligomers, such as HCE-produced BCL2A1, SNIP1, and GSK3B. It is possible that some of these proteins may either stick to the resin nonspecifically, or that aggregates may form around the POI and elute off when the POI is cleaved from the resin. The presence of the HaloTag band and the absence of POI, seen predominantly with HCE-produced CCND1, SNIP1, and CDKN2A, suggest successful binding and cleavage of fusion protein, but failed release of the POI from the resin.
Figure 3.
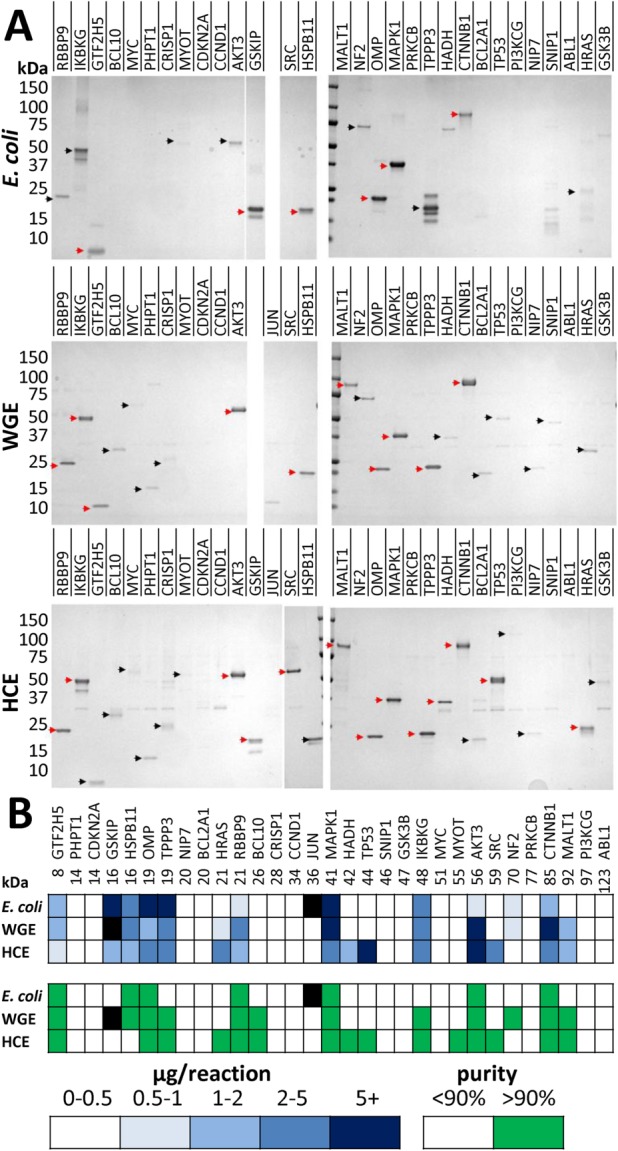
Purification analysis of 31 full-length proteins in E. coli, WGE, and HCE systems. (A) Coomassie stain of 5/100 μL elution fraction. Red arrows indicate sufficient yield (>1 μg) and high (>90%) purity, whereas black arrows indicate low yield and/or purity. Proteins that were not determined are shaded black. (B) Heat map showing quantification of yield and purity of elution fractions. Note: the GSKIP construct that was used contains a second TEV site immediately upstream of gene start codon, and the smaller band seen in these elution fractions corresponds to the twice TEV cleaved product.
Using the HaloTag purification strategy, 14 of the 31 (45%) proteins in the collection had reasonable protein purification yield (≥1 μg) and high purity (>90%). Of these 14, HCE was the most successful at making purified human protein with 13/31 (42%) successes. Notably, the enrichment of protein purified from HCE is the greatest of the three systems, as HCE is by far the densest milieu of lysate proteins. WGE and E. coli systems were less successful than HCE, with 10/30 (33%) and 6/30 (20%) proteins achieving these standards, respectively.
Several POIs failed to purify, despite evidence of expression in at least two expression systems, such as CDKN2A, NIP7, CCND1, and MYC. In some cases, the cause for failure was low purity. To improve purity, several proteins were purified while testing more stringent wash conditions. As wash stringency was increased to denaturing conditions, several proteins were found to stick more to the resin, suggesting that unfolded protein has higher nonspecific affinity for the Halo resin. Increasing wash stringency tended to lower purification yield, and therefore was not regarded as a suitable means to improve purity.
To assess whether purified proteins were properly folded, several proteins within the test collection with known function were selected for functional analysis. ABL1 is a nonreceptor tyrosine kinases that autophosphorylates when it is functionally active.36 A kinase activity assay was performed with purified ABL1. Figure 4 panel A shows that HaloTag-ABL1 was phosphorylated during expression in HCE, and subsequently dephosphorylated by phosphatase treatment while bound to Halo resin. Following TEV cleavage and release, purified ABL1 auto-phosphorylated in the presence of adenosine triphosphate (ATP). Two other proteins in our test set, MAPK1 and p53, have been shown formerly to interact.37 To demonstrate that the purified proteins behave accordingly, a coimmunoprecipitation-like assay was performed. MAPK1 was expressed in HCE as a C-terminal GST fusion protein, and following incubation of crude MAPK1-cGST with p53 purified from all three systems, the mixture was bound to glutathione resin. Panel B shows that p53 purified from E. coli, WGE, and HCE all bound to MAPK1.
Figure 4.
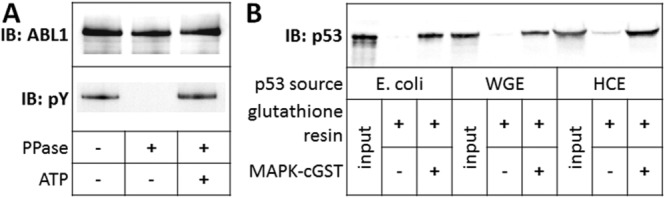
Functional assays of selected purified proteins. (A) Auto-phosphorylation of purified, dephosphorylated ABL1. ABL1 kinase was prepared by expression in HCE as an N-terminal HaloTag fusion protein, binding to resin, dephosphorylation and cleavage from the resin with TEV protease. Purified protein was then incubated with and without ATP. Phosphotyrosine signal was detected by immunoblotting with antiphosphotyrosine. (B) Interaction of purified p53 with MAPK1. The MAPK1-p53 interaction was queried by incubating HCE-produced MAPK1-cGST with p53 purified from E. coli, WGE, and HCE. The MAPK1-cGST and p53 mixtures were bound onto glutathione resin, washed, and boiled in SDS-PAGE sample buffer. Glutathione resin was incubated with p53 alone as a negative control. Immunoblotting was performed to detect p53 signal.
Table I summarizes the purification performance of the three expression systems. Among the successfully purified proteins, the mean recovery for the three systems was 42, 22, and 15%, for E. coli, WGE, and HCE, respectively. Although HaloTag fusion protein specifically binding to Halo ligand on the magnetic resin was estimated to be highly efficient and consistent among the collection, the amount of POI that eluted was highly variable and protein dependent.
Discussion
A paramount obstacle for proteomic studies, such as the generation of affinity reagents to the human proteome, is the difficulty of high throughput recombinant protein production.6 This challenge is rooted in the need to accommodate many proteins with staggering biochemical diversity using a minimal set of protocols. Functional proteomics, and in particular microarray based functional studies, can utilize purified protein at microgram scales. A key goal of these applications is to have a very high protein production success rate, which contributes to lowering the cost of HT purification.
We have attempted to establish a cost effective triage protocol to produce full-length proteins at high purity and a high success rate using in vivo E. coli expression, and two eukaryotic CF systems: WGE and HCE. To this end, we compared the expression capabilities of three systems with 31 full-length proteins. CF expression systems had a much higher success rate than E. coli, making them highly suitable for producing full-length eukaryotic proteins. For the collection of proteins we assessed, 25/30 (83%) could be expressed in excess of 20 μg in WGE and 26/31 (84%) in HCE. In comparison, only 10/30 (33%) E. coli expressed proteins met these criteria, a subset that is biased toward smaller proteins in the collection. Among other rationales, slower translation rate in the presence of eukaryotic chaperones to support proper folding of complex eukaryotic proteins may contribute to high CF success rate.18,38 An advantage of the high expression rate of CF eukaryotic expression is a reduced need for a factorial approach involving many fusion tags, expression conditions, promoters, expression strains, and so forth, that are a part of many E. coli based production pipelines. Adding these many constructs and protocols to a protein production pipeline greatly increases workload of protein production centers.
The SIFT protocol we developed has several advantages over alternative protocols such as methods that couple immuno-precipitation with MCGE or automated CGE immunoblot methods for recombinant fusion protein analysis in complex biological samples. Without the need for direct detection by antibodies or enrichment of nonspecifically labeled protein, SIFT omits long incubation and washing steps. The miniaturization of the microfluidic capillary channel in the chip reduces separation medium volume and separation time. Each sample is processed in about 40 s, allowing the complete processing of a 96-well plate in about 90 min. In comparison, automated antibody-based CGE methods typically take upwards of 16 h to process a 96-well plate. SIFT can provide qualitative and quantitative measurement of specific protein concentration that is on-par with in-gel fluorescence imaging. A significant benefit to the SIFT approach is that it can be performed on existing LabChip systems with only minimal alteration of the sample preparation protocol. SIFT can be easily extended to other affinity tags that, like HaloTag, bind specifically and irreversibly to a fluorescently conjugated substrate, including SNAP and CLIP tags.
There are alternative methods to label recombinant protein specifically, such as those that add fluorescent amino acid-charged tRNA, like FluoroTect GreenLys, into CF expression reactions.39 Although this method is a convenient approach to label all synthesized proteins, a relatively small fraction of proteins will be labeled and variable labeling can occur because there is a mixture of both natural aminoacyl-tRNA and the fluorescent variant in the expression reaction. Furthermore, the number of fluorophores that different proteins incorporate will vary depending on the lysine composition of each protein. An advantage of SIFT is that recombinant proteins are labeled through a highly efficient reaction with the fusion tag, and binding occurs with a stoichiometry of exactly one fluorophore per protein, allowing fluorescent signal to be correlated precisely with protein concentration.
At the purification stage, 6/30 proteins were purified from E. coli, 10/30 purified from WGE, and 13/31 purified from HCE with yields ≥1 μg and ≥90% purity. The average purification yield of E. coli was 5.8 μg, WGE 4.1 μg, and HCE 3.5 μg per reaction. A number of proteins resulted in inefficient overall recovery. One cause may be that the POI became insoluble after cleavage of the HaloTag fusion with TEV protease and, in turn, may cause some of these to bind nonspecifically to the Halo resin. Cleaved POI was observed on Halo resin by boiling the resin in SDS sample buffer after TEV cleavage and washing. For these recalcitrant proteins, none of the conditions tested during optimization amended this problem.
The HaloTag purification strategy achieved a combined 45% purification success rate with this diverse and challenging test collection. Although we tested numerous parameters to optimize the HaloTag purification protocol, the purification efficiency from these expression systems could be further improved, possibly with new Halo resin surface chemistry, or using alternative affinity tags. Functional assays revealed that purified proteins were active and therefore properly folded.
One major limitation of CF systems is cost, which can be tens of times more expensive than in vivo E. coli protein expression. Producing protein in cultured mammalian or insect cells remains both expensive and cumbersome, requiring long periods of cell growth, preparation of many cells, and laborious cell lysis methods that are not conducive to HT production. The low success rate of prokaryotic systems, and the low throughput and high cost of in vivo eukaryotic protein expression, make eukaryotic CF systems attractive choices for HT protein production for proteomic studies. Ongoing technical developments in structural biology are reducing the amount of material needed for protein structure determination,40,41 and may lead to increased CF system usage within structural biology.19,27
CF systems are capable of producing the amount of protein required by proteomic applications, such as emergent affinity reagent development pipelines. For applications that require quantities of protein near the 100 μg protein scale, a range that is generally considered necessary for current affinity reagent selection and characterization techniques, CF expression volumes can be increased as needed. When doing this, the overall cost of producing protein increases with CF expression volume. However, some proteins cannot be produced using E. coli, and there may be no low cost and HT alternatives for these targets. As with any production pipeline, throughput decreases substantially when working with volumes that exceed the capacity of a deepwell block format, although in some cases it may be reasonable to use multiple deepwell blocks in parallel and combine the relevant wells. Otherwise, highly specialized equipment is required to maintain throughput of larger scale work.
Based on these results, our proposed full-length human protein production triage strategy is shown in Figure 5. The order is dependent on the application and required scale. For larger quantities of protein, E. coli is well worth screening first for expression and purification, to find proteins that can be produced cheaply before moving onto the more costly CF systems. However, for applications that can utilize smaller amounts of protein, CF systems' high success rate makes them a good first choice. Our automated pipeline, including SIFT, enables efficient assessment of this triage strategy for a large number of proteins. Proteome-level protein production remains an unsolved challenge. We have reported that the incorporation of CF expression into a high throughput pipeline can expand the scope of proteins that can be produced and isolated for proteomic research. Our ultimate goal is to further improve our pipeline to eventually support proteome-level full-length protein production for various proteomics applications.
Figure 5.
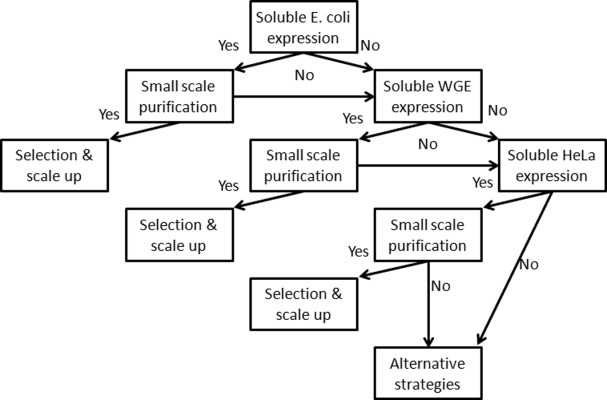
Decision tree of full-length human protein purification in E. coli, WGE, and HeLa lysate systems for larger scale applications.
Materials and Methods
Cloning
Three Gateway compatible destination vectors were cloned as illustrated in Supporting Information Figure S1. The HaloTag7 and TEV protease cleavage site linker encoding sequence was amplified from pFN22A (Promega) with a 5′ NdeI site and 3′ BsrGI site, and ligated into pCPD_nMBP (DNASU.org) linearized by NdeI and BsrGI, to generate pCPD_nHalo_empty (Supporting Information Fig. S1). The Gateway death cassette was amplified from a modified pANT7_cGST destination vector with 5′ and 3′ BsrGI sites, and ligated into pCPD_nHalo_empty linearized by BsrGI, to generate pCPD_nHalo_DC, the Gateway compatible destination vector for E. coli expression. To create pJSP6_nHalo_empty, the HaloTag sequence was amplified with 5′ NcoI and 3′ PmeI sites, and ligated into NcoI and PmeI linearized pEU_HSBC. WGE destination vector pJSP6_nHalo_DC was generated by ligating the death cassette, amplified with 5′ and 3′ SgfI and PmeI restriction sites, into SgfI and PmeI linearized pJSP6_nHalo_empty. To generate pJFT7_nHalo_empty, the HaloTag sequence was amplified from pCPD_nHalo with a 5′ NcoI site, and a 3′ SpeI site, and ligated into a modified version of pANT7_cGST with an NcoI site introduced immediately after the EMCV IRES, which was linearized with NcoI and SpeI. The death cassette with 5′ and 3′ BsrGI sites was ligated into BsrGI linearized pJFT7_nHalo_empty to generate pJFT7_nHalo_DC for HCE expression. Maps and sequences of these vectors can be found at http://dnasu.org/. The cDNA for the test collection was acquired from DNASU and transferred into the three destination vectors via LR cloning (Supporting Information Fig. S1). All constructs were sequence verified. CF expression plasmids were prepared using NucleoBond Xtra midi or maxi-prep kits (Macherey Nagel).
E. coli in vivo expression
Expression plasmids were transformed into E. coli strain Rosetta2 (DE3) (EMD Millipore) and cultured in a 96-well format as previously described.9 The expression protocol was adapted from a previously described protocol.16 Briefly, isolated colonies were grown in 1 mL LB media + 100 μg/mL Ampicilin, 34 μg/mL chloramphenicol, 0.4% glucose, in a 96 well, 2-mL deepwell block (R.K. Manufacturing), overnight at 37°C. The cultures were diluted 1:20 into 2-mL supplemented MJ9 media and grown in 24-well, 10-mL deepwell blocks (Seahorse Bioscience), for ∼2.5 h at 37°C until OD600 = 0.5–0.6. Cultures were cooled to 18°C for 15 min, and expression was induced with 1-mM IPTG. After 18 h, expression cultures were pelleted at 6000g for 10 min, and resuspended in 250-μL lysis buffer (50 mM HEPES, 150 mM NaCl, pH 7.8, 0.01% IGEPAL, 1 mM DTT, 2 mg/mL lysozyme, 25 μg/mL DNase, 5 mM Mg2+, 100 μM PMSF). Lysates were kept frozen at −20°C and rethawed for purification.
CF expression
WGE expression was performed with WEPRO 7240 (CellFree Sciences) in a 226-μL bilayer format using flat-bottom 96-well plates (Greiner) following the manufacturer's protocol, for 20 h at 15°C. For HCE expression, reaction mix was prepared according to the manufacturer's instructions (Thermo-Pierce). The reaction mixture was centrifuged at 10,000g for 2 min, and 105.6 μL of supernatant was aliquoted per reaction into a 96-well polymerase chain reaction (PCR) plate. For each reaction, 4.4 μL of 1 μg/μL expression plasmid was mixed with the clarified IVT mixture, and 100 μL of the IVT reaction was transferred into a dialysis chamber of a 96-well microdialysis plate (Thermo-Pierce). The dialysis chambers were equilibrated with 20 min soaking in 1.8 mL, 30°C, 1X dialysis buffer (Thermo-Pierce) prior to adding the IVT reaction. Following 20 h, 30°C expression, lysates were transferred to a 96-well PCR plate, and centrifuged for 6000g for 10 min.
HaloTag Alexa660 ligand synthesis
One milligram NHS-activated Alexa660 (Life Technologies) was incubated with 0.6 mg HaloTag amine ligand (Promega) in anhydrous DMF containing 50 mM DIPEA (diisopropylethyl amine) at 37°C overnight, purified by reverse phase HPLC on a C18 column with a 0–96% MeOH in water, 2% per min gradient, and characterized by monitoring the absorbance at 660 nm.
SDS-PAGE expression analysis and HaloTag-specific detection
E. coli, WGE, and HCE expression reactions were diluted in 1X HEPES (50 mM HEPES, 150 mM NaCl, pH 7.8) 1:10, 2:5, and 1:5, respectively, to normalize the concentration of HaloTag protein. Samples were split into supernatant and total fraction plates. Supernatant plates were centrifuged 5000g for 20 min, and the supernatants were transferred into new plates. These samples were analyzed with SDS-PAGE by incubating fractions of lysate with 4 μM Alexa660-conjugateded Halo ligand (Promega) for 20 min at room temperature, and denaturating in Laemmli sample buffer with boiling at 85°C for 3 min. The ratio of diluted sample to 4 μM Alexa660-HL to 4X Laemmli sample buffer (Bio-Rad) was 2:1:1, making the final E. coli, WGE, and HCE dilutions 1:20, 1:5, and 1:10, respectively. Gels were loaded with 10 μL of sample and run until separation was satisfactory. Gels were imaged using a Typhoon fluorescent imager (GE Life Sciences). Gels were subsequently stained with Simply Blue Coomassie stain (Life Technologies). Gels images were analyzed using ImageQuantTL software (GE Life Sciences). A HaloTag protein standard was used to create a molar calibration curve. Molarity was converted to protein mass using the theoretical mass of each fusion protein.
LabChip HaloTag-specific expression analysis
Lysed E. coli, WGE, and HCE samples, arrayed in 96-well PCR plates, were diluted 1:10, 2:5, and 1:5 in 50 mM HEPES, 150 mM NaCl, pH 7.8. The plates were centrifuged 5000g for 20 min, and the supernatants were transferred to new plates. These samples were diluted 2:3 with 4 μM Alexa660-HaloTag ligand, and incubated for 20 min at room temperature. Sample plates were frozen at −80°C for storage and transportation. Samples were thawed, and 2 μL of sample was added to 17 μL of denaturating Pico Protein Sample buffer containing DTT, and prepared according to the manufacturer's instructions. Samples were centrifuged at 2000g for 2 min, denatured at 95°C for 5 min, and 35 μL of MilliQ water was added to each sample. Samples were centrifuged at 2000g for 2 min, and the sample plates were run sequentially on a LabChip GXII instrument using the Pico Protein 200 assay. The Pico Protein ladder was prepared in Pico Labeling Buffer, and a HT Protein chip was prepared, both according to the manufacturer's instructions. Electropherograms and virtual gels were analyzed using LabChip GX software.
HaloTag purification
Magne HaloTag resin (Promega) was equilibrated with HaloTag purification buffer (HPB): 50 mM HEPES, 150 mM NaCl, pH 7.8, 1 mM DTT, 0.01% IGEPAL, supplemented with 2 mM ATP and 10 mM MgSO4. Lysed E. coli, WGE, and HCE reactions were clarified at 5000g for 20 min prior to binding to equilibrated resin. For each sample, 382 μL equilibrated-resin slurry, which contained 10 μL of settled resin, was aliquoted into a 2-mL deepwell block. Protein was bound for 2 h with vigorous shaking at 1000 rpm at room temperature. Resin was washed five times with 500 μL of HPB, with 5 min 1000 rpm shaking intervals, using a Biomek FX liquid handling robot (Beckman Coulter). Protease cleavage was performed by adding 100 μL of 30 ng/μL HaloTEV protease (Promega), diluted in HPB, to each sample, and shaking for 2 h at room temperature. The 100 μL of elution fraction was collected and analyzed by SDS-PAGE.
SDS-PAGE purification analysis
Elution fractions were collected and diluted in 2X Laemmli sample buffer (Bio-Rad), boiled for 3 min at 85°C, and separated on Criterion 4–20% TGX gels (Bio-Rad). Coomassie stained gels were image quantified using bovine serum albumin (BSA) protein standard (Thermo).
ABL1 kinase assay
Following expression in HCE, 2 μL of ABL1-nHalo in HCE lysate was bound on 2.5 μL of HaloTag resin equilibrated and suspended in 100 μL HPB, at room temperature for 1 h with shaking. Resin was washed 5X with 100 μL HPB with 5 min shaking intervals between washes. The resin was suspended in 100 μL of HPB and was split into three 30 μL aliquots. One of the aliquots was left in HPB, and the other two were incubated in 30 μL PPase reaction buffer [10% λ-PPase, 50 mM HEPES, 10 mM NaCl, 2 mM DTT, 0.01% Brij 35, pH 7.5, 1 mM MnCl2 (NEB)] added. The PPase reaction was performed at 30°C with shaking for 1 h. Resin was washed 5X with 100 μL HPB with 5 min shaking intervals between washes. All fractions were cleaved with 30 μL 30 ng/μL TEV protease in HPB at room temperature for 1 h with shaking. One of the PPase treated fractions was incubated with 30 μL kinase buffer [25 mM Tris, pH 7.5, 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2, 0.5 mM ATP (cell signaling)], at 30°C with shaking for 1 h. All aliquots of resin were boiled for 5 min at 85°C in Laemmli sample buffer and 10 μL was loaded and run on an SDS-PAGE gel, transferred to a PVDF membrane, and blotted with anti-Abl1 or anti-pY (cell signaling).
MAPK1 and p53 protein–protein interaction assay
MAPK1 was expressed in HCE using the pANT7_cGST as the expression vector. A 20-μL aliquot of MAPK1-cGST expressed in HCE was mixed with 0.5 μg of purified p53 from E. coli, WGE, and HCE, with the volume finalized to 85 μL with protein-protein interaction (PPI) buffer (PBS, 0.05% TritonX-100, 5 mM MgCl2, 10 mg/mL BSA, 0.5 mM DTT). For no-bait controls, 20 μL of PPI buffer was substituted for MAPK1-cGST. The mixtures were incubated for 2 h at 4°C and bound to 25 μL PPI equilibrated 50% glutathione resin slurry, overnight at 4°C with rotating. The resin was washed 7X with 500 μL PPI buffer. The resin was suspended in a final volume of 75 μL Laemmli sample buffer and boiled for 10 min at 85°C. Immunoblotting was performed on 10 μL loaded sample with anti-p53 D01 monoclonal antibody (Santa Cruz).
Acknowledgments
The authors gratefully acknowledge the discussions and advice offered by Penny Jensen and Krishna Vattem at Thermo Fisher Scientific; and Robin Hurst and Rachel Ohana at Promega Inc.
Glossary
- HT
high throughput
- WGE
wheat germ extract
- HCE
HeLa cell extract
- CF
cell free
- CECF
continuous exchange cell free
- POI
protein of interest
- MCGE
microfluidic capillary gel electrophoresis
- TEV
tobacco etch virus
- SIFT
selective irreversible fluorescence targeting
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Terwilliger TC, Stuart D, Yokoyama S. Lessons from structural genomics. Annu Rev Biophys. 2009;38:371–383. doi: 10.1146/annurev.biophys.050708.133740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fasolo J, Sboner A, Sun MG, Yu H, Chen R, Sharon D, Kim PM, Gerstein M, Snyder M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011;25:767–778. doi: 10.1101/gad.1998811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 5.Stergachis AB, MacLean B, Lee K, Stamatoyannopoulos JA, MacCoss MJ. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat Methods. 2011;8:1041–1043. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marx V. Calling the next generation of affinity reagents. Nat Methods. 2013;10:829–833. doi: 10.1038/nmeth.2607. [DOI] [PubMed] [Google Scholar]
- 7.Vincentelli R, Romier C. Expression in Escherichia coli: becoming faster and more complex. Curr Opin Struct Biol. 2013;23:326–334. doi: 10.1016/j.sbi.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–222. doi: 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- 9.Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J. Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci USA. 2002;99:2654–2659. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langlais C, Guilleaume B, Wermke N, Scheuermann T, Ebert L, LaBaer J, Korn B. A systematic approach for testing expression of human full-length proteins in cell-free expression systems. BMC Biotechnol. 2007;7:64. doi: 10.1186/1472-6750-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bird LE. High throughput construction and small scale expression screening of multi-tag vectors in Escherichia coli. Methods. 2011;55:29–37. doi: 10.1016/j.ymeth.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Bussow K, Scheich C, Sievert V, Harttig U, Schultz J, Simon B, Bork P, Lehrach H, Heinemann U. Structural genomics of human proteins—target selection and generation of a public catalogue of expression clones. Microb Cell Fact. 2005;4:21. doi: 10.1186/1475-2859-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, Hammarstrom M, Johansson I, Kotenyova T, Nilsson M, Nordlund P, Weigelt J. The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif. 2008;58:210–221. doi: 10.1016/j.pep.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Pacheco B, Crombet L, Loppnau P, Cossar D. A screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr Purif. 2012;81:33–41. doi: 10.1016/j.pep.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Dessailly BH, Nair R, Jaroszewski L, Fajardo JE, Kouranov A, Lee D, Fiser A, Godzik A, Rost B, Orengo C. PSI-2: structural genomics to cover protein domain family space. Structure. 2009;17:869–881. doi: 10.1016/j.str.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acton TB, Xiao R, Anderson S, Aramini J, Buchwald WA, Ciccosanti C, Conover K, Everett J, Hamilton K, Huang YJ, Janjua H, Kornhaber G, Lau J, Lee DY, Liu G, Maglaqui M, Ma L, Mao L, Patel D, Rossi P, Sahdev S, Shastry R, Swapna GV, Tang Y, Tong S, Wang D, Wang H, Zhao L, Montelione GT. Preparation of protein samples for NMR structure, function, and small-molecule screening studies. Methods Enzymol. 2011;493:21–60. doi: 10.1016/B978-0-12-381274-2.00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao R, Anderson S, Aramini J, Belote R, Buchwald WA, Ciccosanti C, Conover K, Everett JK, Hamilton K, Huang YJ, Janjua H, Jiang M, Kornhaber GJ, Lee DY, Locke JY, Ma LC, Maglaqui M, Mao L, Mitra S, Patel D, Rossi P, Sahdev S, Sharma S, Shastry R, Swapna GV, Tong SN, Wang D, Wang H, Zhao L, Montelione GT, Acton TB. The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J Struct Biol. 2010;172:21–33. doi: 10.1016/j.jsb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takai K, Sawasaki T, Endo Y. Practical cell-free protein synthesis system using purified wheat embryos. Nat Protoc. 2010;5:227–238. doi: 10.1038/nprot.2009.207. [DOI] [PubMed] [Google Scholar]
- 19.Guild K, Zhang Y, Stacy R, Mundt E, Benbow S, Green A, Myler PJ. Wheat germ cell-free expression system as a pathway to improve protein yield and solubility for the SSGCID pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:1027–1031. doi: 10.1107/S1744309111032143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez FJ, Vega MC. Technologies to keep an eye on: alternative hosts for protein production in structural biology. Curr Opin Struct Biol. 2013;23:365–373. doi: 10.1016/j.sbi.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Wellensiek BP, Larsen AC, Flores J, Jacobs BL, Chaput JC. A leader sequence capable of enhancing RNA expression and protein synthesis in mammalian cells. Protein Sci. 2013;22:1392–1398. doi: 10.1002/pro.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barford D, Takagi Y, Schultz P, Berger I. Baculovirus expression: tackling the complexity challenge. Curr Opin Struct Biol. 2013;23:357–364. doi: 10.1016/j.sbi.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunt I. From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Protein Expr Purif. 2005;4:1–22. doi: 10.1016/j.pep.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Possee RD, Hitchman RB, Richards KS, Mann SG, Siaterli E, Nixon CP, Irving H, Assenberg R, Alderton D, Owens RJ, King LA. Generation of baculovirus vectors for the high-throughput production of proteins in insect cells. Biotechnol Bioeng. 2008;101:1115–1122. doi: 10.1002/bit.22002. [DOI] [PubMed] [Google Scholar]
- 25.Seal AJ, Collingridge GL, Henley JM. An investigation of the membrane topology of the ionotropic glutamate receptor subunit GluR1 in a cell-free system. Biochem J. 1995;312:451–456. doi: 10.1042/bj3120451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Festa F, Rollins SM, Vattem K, Hathaway M, Lorenz P, Mendoza EA, Yu X, Qiu J, Kilmer G, Jensen P, Webb B, Ryan ET, Labaer J. Robust microarray production of freshly expressed proteins in a human milieu. Proteomics Clin Appl. 2013;7:372–377. doi: 10.1002/prca.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beebe ET, Makino S, Nozawa A, Matsubara Y, Frederick RO, Primm JG, Goren MA, Fox BG. Robotic large-scale application of wheat cell-free translation to structural studies including membrane proteins. Nat Biotechnol. 2011;28:239–249. doi: 10.1016/j.nbt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tyler RC, Aceti DJ, Bingman CA, Cornilescu CC, Fox BG, Frederick RO, Jeon WB, Lee MS, Newman CS, Peterson FC, Phillips GN, Jr., Shahan MN, Singh S, Song J, Sreenath HK, Tyler EM, Ulrich EL, Vinarov DA, Vojtik FC, Volkman BF, Wrobel RL, Zhao Q, Markley JL. Comparison of cell-based and cell-free protocols for producing target proteins from the Arabidopsis thaliana genome for structural studies. Proteins. 2005;59:633–643. doi: 10.1002/prot.20436. [DOI] [PubMed] [Google Scholar]
- 29.Ohana RF, Encell LP, Zhao K, Simpson D, Slater MR, Urh M, Wood KV. HaloTag7: a genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr Purif. 2009;68:110–120. doi: 10.1016/j.pep.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Ohana RF, Hurst R, Vidugiriene J, Slater MR, Wood KV, Urh M. HaloTag-based purification of functional human kinases from mammalian cells. Protein Expr Purif. 2011;76:154–164. doi: 10.1016/j.pep.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G, Hu Z, Venkatesan K, Bethel G, Martin P, Rogers J, Lawlor S, McLaren S, Dricot A, Borick H, Cusick ME, Vandenhaute J, Dunham I, Hill DE, Vidal M. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J, Throop A, Fiacco M, Labaer J. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42:D1253–1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Barker K, Steel J, Park J, Saul J, Festa F, Wallstrom G, Yu X, Bian X, Anderson KS, Figueroa JD, LaBaer J, Qiu J. A versatile protein microarray platform enabling antibody profiling against denatured proteins. Proteomics Clin Appl. 2013;7:378–383. doi: 10.1002/prca.201200062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rustandi RR, Loughney JW, Hamm M, Hamm C, Lancaster C, Mach A, Ha S. Qualitative and quantitative evaluation of Simon, a new CE-based automated Western blot system as applied to vaccine development. Electrophoresis. 2012;33:2790–2797. doi: 10.1002/elps.201200095. [DOI] [PubMed] [Google Scholar]
- 36.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275:35631–35637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 37.Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem. 2000;275:35778–35785. doi: 10.1074/jbc.M004267200. [DOI] [PubMed] [Google Scholar]
- 38.Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: applications come of age. Biotechnol Adv. 2012;30:1185–1194. doi: 10.1016/j.biotechadv.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L, Zhao KQ, Hurst R, Slater MR, Acton TB, Swapna GV, Shastry R, Kornhaber GJ, Montelione GT. Engineering of a wheat germ expression system to provide compatibility with a high throughput pET-based cloning platform. J Struct Funct Genomics. 2010;11:201–209. doi: 10.1007/s10969-010-9093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aramini JM, Rossi P, Anklin C, Xiao R, Montelione GT. Microgram-scale protein structure determination by NMR. Nat Methods. 2007;4:491–493. doi: 10.1038/nmeth1051. [DOI] [PubMed] [Google Scholar]
- 41.Boutet S, Lomb L, Williams GJ, Barends TR, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, Messerschmidt M, Barty A, White TA, Kassemeyer S, Kirian RA, Seibert MM, Montanez PA, Kenney C, Herbst R, Hart P, Pines J, Haller G, Gruner SM, Philipp HT, Tate MW, Hromalik M, Koerner LJ, van Bakel N, Morse J, Ghonsalves W, Arnlund D, Bogan MJ, Caleman C, Fromme R, Hampton CY, Hunter MS, Johansson LC, Katona G, Kupitz C, Liang M, Martin AV, Nass K, Redecke L, Stellato F, Timneanu N, Wang D, Zatsepin NA, Schafer D, Defever J, Neutze R, Fromme P, Spence JC, Chapman HN, Schlichting I. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337:362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information