

Abstract
The restriction factor TRIM5α binds to the capsid protein of the retroviral core and blocks retroviral replication. The affinity of TRIM5α for the capsid is a major host tropism determinant of HIV and other primate immunodeficiency viruses, but the molecular interface involved in this host–pathogen interaction remains poorly characterized. Here we use NMR spectroscopy to investigate binding of the rhesus TRIM5α SPRY domain to a selection of HIV capsid constructs. The data are consistent with a model in which one SPRY domain interacts with more than one capsid monomer within the assembled retroviral core. The highly mobile SPRY v1 loop appears to span the gap between neighboring capsid hexamers making interhexamer contacts critical for restriction. The interaction interface is extensive, involves mobile loops and multiple epitopes, and lacks interaction hot spots. These properties, which may enhance resistance of TRIM5α to capsid mutations, result in relatively low affinity of the individual SPRY domains for the capsid, and the TRIM5α-mediated restriction depends on the avidity effect arising from the oligomerization of TRIM5α.
Keywords: TRIM5α, HIV, NMR, SPRY
Introduction
Tripartite motif protein TRIM5α of the rhesus monkey potently blocks replication of HIV-1 [1]. TRIM5α acts via direct physical interaction with the mature viral core following the fusion of the virus with the cell membrane [2,3]. There is abundant indirect evidence that the host–pathogen interface involved in TRIM5α-mediated restriction is formed between the SPRY domain of TRIM5α and the N-terminal domain of the capsid (CA-NTD). Even prior to the identification of TRIM5α, experiments with chimeric viruses established that viral determinants of susceptibility to early, postentry blocks are located within the capsid (CA) protein [4]. The shell of the mature viral core is formed by the CA hexagonal assembly, in which the CA-NTD mediates hexamerization, whereas the C-terminal dimerization domain (CA-CTD) links each capsid hexamer to six neighboring hexamers. Incorporation of 12 CA pentamers into the honeycomb-like assembly results in a fully enclosed conical shell of the mature viral core [5–8]. In the assembled structure, the CA-NTD is located on the exterior, thus forming most of the surface exposed to interact with TRIM5α and other cytoplasmic restriction/infectivity factors that affect early events in the viral life cycle.
TRIM5α is a member of the tripartite motif (TRIM) protein family, and it contains the characteristic TRIM domain arrangement in the N-terminal part of the protein consisting of RING, B-box, and coiled-coil domains [9]. The capsid recognition functionality of TRIM5α is contained within the C-terminal SPRY domain, a protein–protein interaction module that is found in approximately half of the TRIM proteins in the human genome [10,11]. The SPRY domain of TRIM5α contains segments that were shaped by very strong positive selection in primate evolution [12]. High variability of the SPRY domain in primates, which most likely is a consequence of the evolutionary antagonism between the virus and the host, is one of the major determinants of host tropism of HIV and related retroviruses [13–15]. Most notably, the poor restriction activity of the human TRIM5α variant against HIV can be dramatically enhanced by mutations of a single amino acid residue within the SPRY domain [15–19].
Early indication that the SPRY domain anchors TRIM5α to the capsid came from the discovery that the owl monkey is protected against HIV by a TRIM5 variant that contains a retrotransposon-mediated substitution of the SPRY domain with cyclophilin A, a cytoplasmic protein that binds HIV capsid in the long loop between helices α4 and α5 of CA-NTD [20,21]. TRIMCyp variants evolved independently in other primate species, providing an example of convergent molecular evolution facilitated by retrotransposon-mediated domain reshuffling [22]. Recently, several artificial TRIM5 variants containing SPRY substitutions with other capsid-interacting protein modules were shown to restrict retroviral replication, providing further support to capsid binding as the principal function of the SPRY domain [23,24].
TRIM5α–capsid binding was demonstrated directly by co-sedimentation of TRIM5α with the mature retroviral cores or with the core-like capsid assemblies formed in vitro [2,3]. These elegant studies revealed that restriction specificity stems from the differences in the binding affinity of TRIM5α to the retroviral cores. For example, the human TRIM5α displays much weaker association with the HIV capsid than the rhesus variant, whereas mutations that restore its restriction activity also restore capsid binding [18,19]. Collectively, these results suggest that structural differences at the TRIM5α–capsid interface that stem from the variations in the amino acid composition of the SPRY and the CA-NTD domains form the molecular basis of the TRIM5α-mediated host tropism.
Capsid recognition by TRIM5α is a challenging system for biochemical and structural studies because TRIM5α binds to the assembled capsid and the high-affinity interaction requires TRIM5α oligomerization mediated by the tripartite motif region [25–30]. A model of TRIM5α–capsid interaction emerged from the pioneering electron microscopy studies of the recombinant chimeric TRIM5α protein revealing that TRIM5α can form two-dimensional hexagonal arrays that match the symmetry of the assembled capsid protein [31]. It was proposed that TRIM5α may act as a pattern recognition molecule through self-assembly and juxtaposition of the capsid-interacting SPRY domains with the regular pattern of epitopes exposed on the surface of the assembled capsid.
We recently solved the structure of the isolated rhesus TRIM5α SPRY domain (rhSPRY) using a combination of X-ray crystallography and NMR spectroscopy [32]. The structure revealed that the putative capsid-interacting surface of SPRY is located on the most structurally variable face of the domain flanked by the highly mobile v1 loop. The interaction of the isolated SPRY with the isolated CA-NTD observed in this study was relatively weak (>400 μM), which is not uncommon for pattern recognition molecules that rely on self-oligomerization and the avidity effect in order to bind to their targets. In this study, we take advantage of the utility of NMR spectroscopy for studies of weak protein–protein interactions to further refine our understanding of SPRY–capsid interactions.
Results
The capsid-interacting surface of SPRY displays mobility occurring on distinct timescales
NMR spectroscopy is a powerful method for studying protein–protein binding because it can yield site-specific information even for relatively weak intermolecular interactions. We have developed an expression and purification strategy that allows production of sufficient amounts of isotopically enriched SPRY domain of the rhesus TRIM5α protein for NMR studies [32]. Mapping of the interaction surfaces requires assignment of the observed NMR signals to specific atoms in the protein molecule. We used standard triple resonance methods to assign NMR signals of the protein backbone. Approximately 75% of the backbone 15N heteronuclear single quantum coherence (HSQC) signals were assigned, leaving several extended assignment gaps in the protein sequence (Fig. 1). One major assignment gap maps onto the N-terminal backbone segment preceding the v1 variable loop; another one covers the v2 loop with another shorter missing segments in the v3 and v4 loops. These assignment gaps located at or adjacent to the putative capsid-interacting face of the protein limit our ability to map the binding interface because the missing signals may belong to the residues involved in the interaction with the HIV capsid.
Fig. 1.

NMR assignments and protein mobility. Comparison plot of the amide order parameters S2 determined using model-free approach (red circles) and the average B-factors (blue bars) for the backbone heavy atoms (Cα, C′, and N). Protein segments with missing NMR assignments appear as white gaps in the gray background.
Assignment gaps frequently result from the broadening of the NMR signals that weakens them beyond the detection sensitivity of an NMR experiment. Signal broadening can have a variety of mechanisms, but most commonly, it is a consequence of conformational or chemical exchange that occurs with rates comparable to the magnitude of the chemical shift perturbations resulting from this exchange, the situation normally referred to as the intermediate exchange regime in NMR spectroscopy. This is in contrast to the fast protein motions occurring on the picosecond-to-nanosecond timescale, which produce narrow and intense NMR signals and are more amenable to NMR-based approaches. For example, we previously demonstrated that the v1 loop of the rhTRIM5α SPRY domain is very mobile and that its motions are fast as evidenced by the 15N relaxation and the heteronuclear nuclear Overhauser enhancement measurements. The missing signals of the protein backbone segments located in the vicinity of v1 suggest that these regions are also subject to protein motions albeit on a considerably slower timescale. Another indication of protein mobility at the putative capsid-binding face comes from the high-resolution structure of the v1-deleted SPRY variant determined by X-ray crystallography. The high-resolution diffraction data set yields reasonably precise measurements of the atomic B-factors, which reflect molecular motions in the crystal. Figure 1 shows a per-residue plot of the B-factors derived from the crystal structure superposed on the backbone assignment coverage and the order parameters for the backbone amides determined from the NMR experiments. The figure reveals that there is a strong correlation between higher B-factors observed in the crystal and the gaps in the NMR assignment. Furthermore, comparison of the two rhesus SPRY crystal structures, determined independently using distinct protein constructs that crystallize in distinct space groups, reveals that the segments with missing NMR signals display highest deviations between the two structures [32,33]. Collectively, the data indicate that the missing signals are indeed due to the intermediate timescale motions within the backbone segments at or adjacent to the capsid binding interface. Therefore, the highly mobile v1 loop is not the only SPRY segment that can undergo conformational changes upon capsid binding.
Mutations in one variable segment may affect conformation of the adjacent variable loops
The high mobility of the v1 segment is a characteristic feature of the TRIM5α SPRY and has not been observed in other SPRY-containing TRIM proteins. Structural and biophysical characterization of TRIM21–IgG interactions revealed that, in the TRIM21 SPRY, the v1 loop is rather rigid, flanking an extensive preformed binding interface [34,35]. The v1 conformation is essentially identical in the structures of the free and the IgG-bound forms of TRIM21 SPRY and also in the crystal structure of the unliganded TRIM72 SPRY [35,36]. The well-defined conformation adopted by the v1 loop in TRIM21 and TRIM72 is stabilized by several key v1 residues, particularly by the RF motif in the C-terminal portion of v1 (R324 and F325 in TRIM21). In the TRIM21 structure, R324 makes a hydrogen-bonding bridge linking v1 residue N321 to the N-terminal backbone segment of the SPRY domain, whereas F325 is tucked into a hydrophobic pocket formed by the residues of the v2 loop (Fig. 2). F325 is conserved in almost all TRIM proteins including TRIM5 variants (F346 in the rhesus TRIM5α). However, in the TRIM5 structure, the corresponding pocket is occupied by a phenylalanine residue of the v2 loop instead (F383), resulting in the displacement of the v1 phenylalanine F346 and a loss of a defined v1 conformation. Most of the primate TRIM5 variants have an aromatic or a bulky aliphatic side chain in the corresponding v2 position suggesting that v1 mobility is shared by all TRIM5 variants. These observations suggest that v1 mobility may have functional significance and may be associated with the antiretroviral activity of distinct TRIM family members. They also illustrate how mutations in the v2 region can have profound effects on the conformation and mobility of the v1 loop.
Fig. 2.
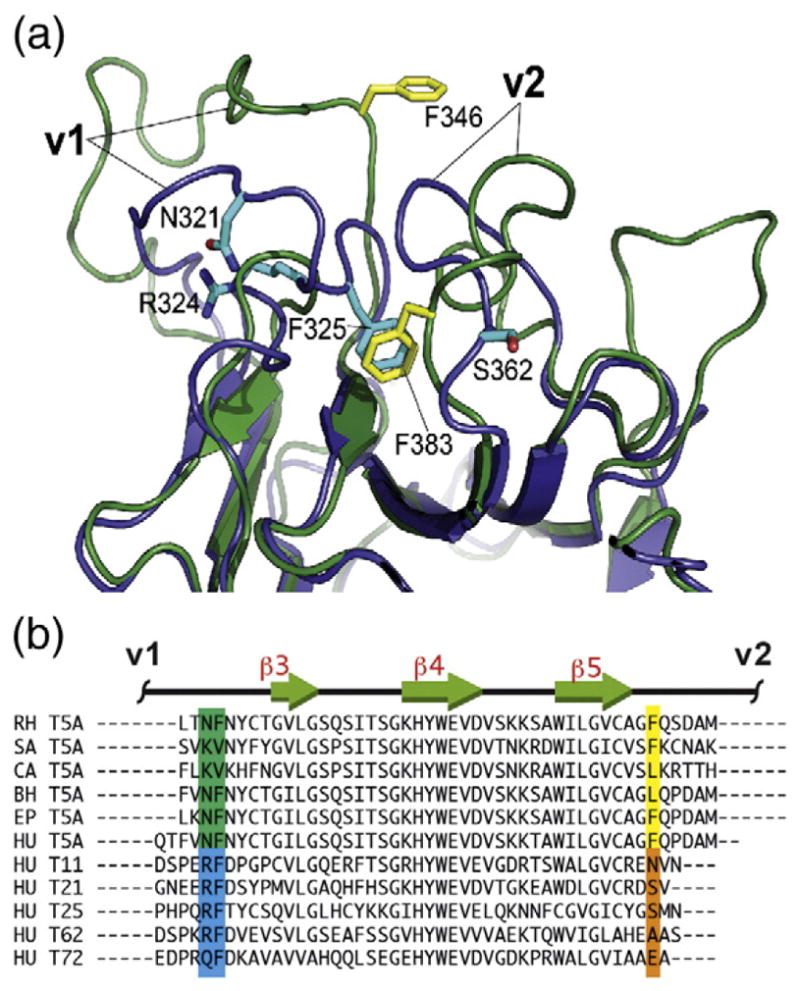
v1 mobility and the associated primary sequence patterns. (a) Superposition of one of the TRIM5 SPRY structures from the NMR ensemble (green, yellow side chains; PDB code: 2LM3) and the TRIM21 SPRY crystal structure (blue, cyan side chains; PDB code: 2IWG). F325 anchors the v1 segment to the protein core in the TRIM21 structure, whereas in the TRIM5 SPRY, the analogous pocket is occupied by the v2 residue F383. (b) Virtually all of the primate TRIM5 variants have an aromatic or large hydrophobic residue in the position corresponding to F383 of the rhesus TRIM5 (highlighted yellow). In contrast, most of the other SPRY-containing TRIM proteins in the human genome contain a small or polar/charged residue in this position (highlighted orange), which correlates with the occurrence of an RF motif in v1 (highlighted blue). Abbreviations: RH, rhesus monkey, Macaca mulatta; SA, white-lipped tamarin, Saguinus labiatus; CA, white-fronted capuchin, Cebus albifrons; BH, hoolock gibbon, Bunopithecus hoolock; EP, patas monkey, Erythrocebus patas; HU, Homo sapiens.
Utility of NMR titrations for mapping interaction surfaces and estimating the affinity of the SPRY–capsid interaction
Our preliminary binding studies with the isolated TRIM5α SPRY domain and the N-terminal domain of the HIV capsid revealed that the interaction between the two proteins is amenable to the NMR-based studies [32]. Standard NMR titration experiments in which the 15N-labeled SPRY domain was titrated with unlabeled CA-NTD demonstrated that the binding is relatively weak and occurs in the intermediate exchange regime. Here we performed NMR titrations to compare the binding of three distinct capsid constructs: the isolated N-terminal domain of the HIV capsid (CA-NTD), the full-length wild-type HIV capsid protein (CA-WT), and the hexameric HIV capsid construct (CA-HEX). Whereas the N-terminal domain of the capsid should contain all of the epitopes exposed on the surface of the mature core, the recognition of the assembled structure may involve binding to multiple epitopes located on the adjacent CA-NTD units. In order to mimic distinct relative arrangements of CA-NTD within the core, we used the disulfide-stabilized hexamer construct CA-HEX that recapitulates the hexameric units formed by the capsid in the assembled structure [6,37] and the full-length capsid that dimerizes in solution via the CA-CTD with Kd ~ 15 μM [38,39].
Figure 3 shows a selection of representative NMR spectra obtained is such titrations. The NMR peaks in the 15N transverse relaxation optimized spectroscopy (TROSY) HSQC spectrum of the SPRY domain are progressively broadened with the addition of the increasing amounts of the capsid. The broadening is not uniform for all the peaks in the spectrum, and the broadening patterns are distinct for the CA-NTD and the CA hexamer. The spectra illustrate that the broadening of the v1 loop region of the SPRY is enhanced compared to other protein signals in the CA-NTD titrations. In contrast, v1 loop is broadened to a significantly lesser extent than the rest of the protein when the hexameric CA construct is added to the SPRY domain. Differences in v1 broadening in different CA constructs are evident in the plots of NMR signal intensity versus CA concentration shown for the N345 residue in the v1 compared to the average broadening of the protein NMR signals (Fig. 4).
Fig. 3.

Signal attenuation patterns observed in NMR titrations of the 15N-labeled SPRY domain with unlabeled capsid constructs. (a) An area of an 15N TROSY spectrum of the free rhesus TRIM5α SPRY domain with some of the signals corresponding to v1 residues labeled. The same spectrum after addition of the 2:1 molar excess of the CA-NTD (b) or CA-HEX (c) capsid constructs. Enhanced attenuation of the v1 signals is observed in the CA-NTD titration, whereas in the CA-HEX titration, the v1 signals are broadened less than the rest of the protein.
Fig. 4.

Sample NMR attenuation curves. Distinct broadening effects of the CA-NTD and CA-HEX constructs on the SPRY spectra are revealed when NMR signal intensities of individual residues are plotted as a function of capsid concentration. In the CA-NTD titration (open circles and triangles), the broadening rate of the residue N345 (triangles) located in the v1 loop is higher than the average protein signal broadening rate (circles). The effect is the opposite in the CA-HEX titration (filled circles and triangles).
When the broadening rates are calculated and plotted individually for each backbone amide, distinctive relaxation patterns can be observed (Fig. 5). At least two distinct mechanisms contribute to signal broadening observed in the NMR titrations. First, the signals are broadened because the molecular weight of the protein complex is at least twice that of the monomeric SPRY domain, resulting in a considerably slower rotational tumbling of the complex in solution. This broadening mechanism affects all protein signals equally, irrespective of the proximity to the binding site. The baseline broadening rate observed for the majority of the protein signals in Fig. 5 arises via this mechanism. Materials and Methods describes how we use the baseline broadening to determine apparent dissociation constants for SPRY–capsid binding. We use the apparent binding constants determined by NMR only as semiquantitative estimates of the binding affinity because of the uncertainties associated with the stoichiometry of the interaction and the expected multiple binding modes described in Discussion.
Fig. 5.

NMR titrations of the 15N-labeled SPRY. The wild-type rhesus SPRY construct (a and b) and the v1C-5A mutant SPRY (c and d) were titrated with three different capsid constructs: CA-NTD, CA-WT, and CA-HEX. Capsid concentration is given as the concentration of the capsid monomer for all titrations. The intensity attenuation upon addition of increasing amounts of capsid was converted to fraction bound as described in Materials and Methods. (a and c) The binding curves and the derived apparent dissociation constructs determined in the titrations of the WT SPRY (a) and v1C-5A SPRY (c) with the three capsid constructs. (b and d) The broadening rates of the backbone amide NMR signals plotted for all assigned SPRY residues. The enhanced broadening of the signals in the loops v1 and v2 is apparent when the WT SPRY is titrated with the CA-NTD and CA-WT construct, whereas it is not observed in the CA-HEX titration. The enhanced v1/v2 broadening is also not observed in the titrations performed with the v1C-5A SPRY mutant.
Second, interaction with the binding partner can change the chemical environment of the spin due to direct proximity to the ligand, allosteric effects, and/or altered molecular mobility. The effect of the chemical shift perturbation on the NMR signals depends on the rate of exchange between the two states. If the exchange is much faster than the chemical shift difference between the free and bound form, then the signals shift gradually from the free to the bound positions as the titration is performed. If the exchange is much slower than the chemical shift difference, then the signals of the free protein gradually disappear while a new set of signals corresponding to the bound form emerges. When the two values are comparable—the situation referred to as the intermediate exchange regime—the broadening of the signals is observed, which eventually weakens the signals beyond the detection sensitivity. This is the situation we encounter in the case of the SPRY–capsid interaction: we only observe the broadening/weakening of the signals with no significant peak shifts as the titration is performed (Supplemental Fig. S2). This additional broadening mechanism (exchange broadening) arising from the chemical shift perturbations results in enhanced broadening rates for a subset of NMR signals. Patterns of residues with enhanced relaxation rates can be used to identify interacting surfaces in very much the same way it is done with chemical shift changes.
In summary, we use NMR titrations to obtain two distinct properties of the SPRY–capsid binding: the apparent dissociation constants (1) and site-specific signatures of the interaction (2).
Variable loops v1 and v2 are involved in binding to CA-NTD and CA-WT, but not CA-HEX
Segments of the protein most affected by the interaction can be identified when the broadening rates are plotted individually for each residue (Fig. 5). For example, when the SPRY domain binds to the CA-NTD, a subset of peaks displays intensity decay rates substantially higher than the baseline broadening arising from the slower rotational diffusion of the complex. Residues in the v1 loop (particularly the backbone amides of L337, T339, S342, L343, and T344) are most affected by the interaction with CA-NTD, indicative of a significant conformational change in the v1 loop upon binding. Figure 3b illustrates that many v1 signals are broadened beyond detection limit when CA-NTD is added to SPRY in a 2:1 molar excess, whereas signals arising from other parts of the protein are still clearly visible. In addition, we observe enhanced broadening of v2 residues (G382, D386, N394, and E395) and residues in the N-terminal segment preceding v1 (A306 and A308). The v2 and pre-v1 residues affected by binding are located proximal to v1; thus, their relaxation enhancements can be due to direct contact with the capsid or, alternatively, can be caused by the v1 conformational change. When titrations are performed with the full-length HIV capsid CA-WT, a similar broadening of the v1 segment is observed. In contrast, the capsid hexamer CA-HEX displays a strikingly different broadening profile (Fig. 5b, bottom panel). In these titration series, no significant enhancement of the broadening rates is observed for the v1 and v2 loops. On the contrary, the v1 segment is broadened less than the rest of the protein (Fig. 3c), suggesting that, in the SPRY–CA-HEX complex, the v1 region remained mobile. Enhanced v1 broadening is partially restored when the titration is performed with the reduced, monomeric form of the CA-HEX protein construct confirming that the effect is linked to protein hexamerization (Supplemental Fig. S3). Thus, these findings indicate that the binding modes are similar for the CA-NTD and full-length CA constructs, whereas the interaction with the hexamer is distinct and does not appear to involve the v1 loop.
Titrations of a v1 mutant SPRY confirm that binding to the CA-HEX does not require v1-mediated interactions
Previously, we described a SPRY mutant with five-alanine substitution of the NFNYC segment located in the C-terminal portion of the v1 loop (v1C-5A). This mutant was chosen because of its very strong defect in HIV restriction and its impaired capsid binding observed in the sedimentation assays, as compared to point mutations of surface residues, most of which display only modest reductions in restriction efficiency [32]. The v1C-5A mutation has minimal effect on the integrity of the SPRY structure: with the exception of the mutated residues and the residues in the immediate vicinity, the NMR signals are identical in the spectra of the mutant and wild-type protein. In order to evaluate the effect of the v1C-5A substitution in our NMR-based binding assays, we performed titrations of the v1C-5A mutant with the three capsid constructs described above (Fig. 5). First, the broadening profiles indicate that the mutant does not display enhanced broadening in the v1–v2 regions observed in the binding of the wild-type SPRY to the CA-NTD and CA-WT constructs. This is consistent with a defect in the v1 loop that blocks v1 involvement in the binding. Second, the apparent dissociation constants observed in NMR titrations of the v1C-5A SPRY with the three capsid constructs reveal that the effect of the v1C-5A mutation on the apparent dissociation constants is the smallest in the CA-HEX construct. This supports a model in which the v1 region is not required for interaction of the isolated SPRY domain with an isolated capsid hexamer.
CA-WT residues most perturbed by SPRY binding map to the outer edge of the CA hexamer
We then investigated whether NMR titrations of the 15N-labeled capsid constructs with the unlabeled rhesus SPRY could help identify capsid surfaces involved in the interaction. Titration of the 15N-labeled CA-NTD produced significant fluctuations in the broadening rates and did not reveal any residues with the broadening rates significantly higher than the background variation. This may be due to the less significant chemical exchange contribution to the broadening of the capsid NMR signals and the low affinity of the CA-NTD interaction with SPRY. We repeated the experiment with the 15N-labeled CA-WT, which binds significantly tighter than CA-NTD, and were able to identify some residues with enhanced broadening (Supplemental Fig. S4). To further increase the signal-to-noise ratio of this experiment, we repeated the titration using a fully deuterated, 15N-labeled CA-WT sample, in which the relaxation contribution arising from slower molecular tumbling is reduced. The plot of the broadening rates versus the residue number reveals a subset of residues with clearly enhanced broadening rates compared to the rest of the protein (Fig. 6a). When the most perturbed residues are highlighted on the crystal structure of the capsid hexamer, they map on the outer edge of the hexamer, consistent with the model that places SPRY domain over the interhexamer gaps in the capsid lattice (Fig. 6b).
Fig. 6.

NMR titrations of the perdeuterated, 15N-labeled CA-WT. Titrations were performed and analyzed in exactly the same way as the titrations of the 15N-labeled SPRY. (a) The broadening rate plot reveals that a subset of CA-WT residues displays enhanced signal attenuation. (b) In the crystal structure of the capsid hexamer, residues with elevated broadening rates (red and orange) map onto the outer edge of the hexamer.
Discussion
The evolutionary law proposed by Leigh Van Valen in 1973 and now commonly referred to as the Red Queen Hypothesis postulated that coupled evolution of antagonistic species underlies the extinction probabilities observed in the fossil record [40]. TRIM5α-mediated recognition of the retroviral capsid is a particularly well documented case of molecular Red Queen evolution manifested by the positive selection in the protein segments that map onto the C-terminal SPRY domain [12]. The structure of the rhesus monkey TRIM5α SPRY domain revealed that the structurally divergent, capsid-binding surface of the SPRY is formed by the hypervariable loops v1 through v4 [32,33]. Our analysis of the X-ray, NMR, and the protein sequence data indicates that the high mobility of the v1 loop is very likely shared by most primate TRIM5α variants. The primary amino acid signature of the v1 mobility described here is not very common in other SPRY-containing TRIM proteins in the human genome, which suggests that only a small subset of the human TRIM proteins may be involved in the direct recognition of retroviruses or other pathogens. The structural plasticity of the protein segments involved in capsid recognition may increase resistance of TRIM5α to capsid mutations; therefore, protein mobility at host–pathogen protein–protein interfaces may be a common outcome of antagonistic co-evolution at the molecular level.
Another characteristic feature of the TRIM5α–capsid binding apparent from our NMR studies is the relatively low affinity of the interaction between the isolated SPRY domain of rhTRIM5α and the various HIV capsid constructs. Even the lowest apparent dissociation constants that we determined are in the high micromolar range. These findings agree with previous studies demonstrating that co-sedimentation of TRIM5α with the capsid requires TRIM5 self-association mediated by the tripartite motif [25,29]. Capsid recognition by TRIM5α is, therefore, a high-avidity interaction that requires multiple copies of the capsid-binding module tethered together in order to achieve physiologically relevant affinity.
Although the relationship between TRIM5 oligomerization and SPRY–capsid binding remains to be elucidated, it is most likely that individual SPRY domains recognize capsid epitopes independently, unlike, for example, antibodies, in which two distinct domains, VH and VL, are held together and interact with a single antigen epitope. TRIM5α dimerizes via the coiled-coil region [27], but it is not known whether the coiled-coil dimer is parallel or antiparallel and whether the two SPRY domains within the dimer are positioned in close proximity. Independent binding by individual SPRY modules is supported by what we know about target recognition by other SPRY domains [34,41,42] and about the restriction properties of TRIMCyp [20,21] and other artificial TRIM5-derived chimeric proteins [23,24]. Experiments with isolated SPRY units should, therefore, be informative of the interfaces formed between the full-length TRIM5 and the assembled capsid, whereas the relative distances between individual epitopes on the surface of the assembled capsid and between individual SPRY modules in the oligomeric TRIM5α determine the coupling between binding and oligomerization.
The inherently weak affinity of the individual SPRY domains for their epitopes on the capsid surface makes it a challenging system for mechanistic and structural investigation. The goal of this study was to evaluate whether NMR-based approaches could be used to identify and characterize capsid epitopes recognized by the SPRY domain. We demonstrate here that, despite the complications associated with the intermediate exchange regime, NMR-based titrations can be used to extract two distinct types of information about the interaction: chemical exchange broadening can be used to identify residues that experience significant chemical shift changes upon binding (Fig. 5a), whereas the broadening that results from slower molecular tumbling can be used to extract apparent dissociation constants (Fig. 5b) (see Results and Materials and Methods for a more detailed explanation).
The apparent Kd values should be interpreted with caution because they do not necessarily represent binding affinities for specific epitopes. There is inherent uncertainty in the apparent Kd measurements associated with the unknown stoichiometry of the binding and the possibility of multiple overlapping epitopes. All binding curves shown in Results plot signal intensities versus the monomeric capsid concentration. In the case of the capsid hexamer, for example, the apparent Kd will be accurate only in the case when each hexamer contains six independent epitopes that can bind six SPRY molecules simultaneously, which is not likely given the relative sizes of the two molecules. Therefore, the apparent Kd values for the capsid hexamer and the wild-type capsid dimer are the high limit values of the actual dissociation constants. Even these high estimates of the Kd are lower than the apparent Kd values measured for the isolated CA-NTD. These findings confirm that an isolated SPRY domain interacts with multiple CA-NTD epitopes displayed on the surface of the assembled capsid.
The presence of multiple epitopes on the CA-NTD surface and the possibility of distinct relative arrangements of the two proteins in the complex further complicate interpretation of the NMR titration data. When distinct binding modes have similar affinities, they make comparable contributions to the measured apparent Kd values (Fig. 5a) and to the chemical shift perturbation patterns (Fig. 5b). In the situation when a particular binding mode has significantly higher affinity over others, it will dominate both types of data. These ambiguities can be partially or fully resolved in experiments with distinct capsid constructs or with specific mutations on the capsid–SPRY interface. For examples, titrations with the CA-NTD and CA-WT constructs reveal that one of the dominant binding modes engages the v1 segment of SPRY. In contrast, the highest-affinity binding mode between the SPRY and the cross-linked capsid hexamer (CA-HEX) does not involve v1. This was further confirmed by the titrations using the v1C-5A SPRY mutant. Collectively, the broadening profiles and the apparent Kd measurements reported here can be best explained by the interaction model shown in Fig. 7. In this model, the SPRY domain is positioned on the surface of the assembled capsid in a way that allows it to interact with more than one monomer within one hexamer and with the v1 reaching across the interhexamer gap and making contacts with an adjacent hexamer. The figure illustrates the multivalent nature of the interaction and how overlapping binding epitopes on the CA-NTD should be taken into account in the interpretation of the NMR titration data.
Fig. 7.

Low-resolution model of the SPRY–capsid interaction. (a) HIV capsid contains two independently folded domains CA-NTD and CA-CTD connected by a flexible linker. The CA-CTD-mediated dimerization and CA-NTD-mediated hexamerization are two major interfaces formed in the hexagonal-like assembly of the capsid in the mature viral core. (b) All the epitopes recognized by the SPRY domain are located within the CA-NTD. Our data suggest that there are several distinct and possibly overlapping CA-NTD epitopes that the SPRY can possibly bind to. (c) Dimerization of CA-WT can mimic relative orientation of two CA-NTDs located in the neighboring hexamers within the assembled core. Binding of the SPRY domain to CA-WT involves the v1 loop and has higher affinity than binding to the isolated CA-NTD. (d) Isolated capsid hexamers (CA-HEX) produced by disulfide cross-linking of the CA-NTD (yellow bars) and by disruption of the CA-CTD dimerization interface (red stars) binds to the SPRY with higher affinity than CA-NTD, but the binding does not involve the v1 loop. (e) Our data are best explained by a binding model that positions the SPRY domain over the interhexamer gap with the v1 loop spanning the gap. (f) Alternative model to account for the lack of v1 involvement in binding to CA-HEX. v1 may interact with capsid surfaces that are no longer accessible in the cross-linked hexamer.
One can envision an alternative model to explain the apparent lack of v1 involvement in the binding of the SPRY to the capsid hexamer in our NMR titration experiments (Fig. 7e). The v1 loop may interact with the capsid surfaces involved in capsid hexamerization and, therefore, not exposed in the cross-linked capsid hexamer. Although we cannot rule out this possibility based on our data, it is not likely because TRIM5α is capable of binding to the tubular in vitro assemblies of the capsid and the v1 segment is essential for the interaction with that intact assembled structure that does not display any free hexamerization surfaces [3]. Furthermore, TRIM5α was also shown to bind to and destabilize tubular CA structures, which contained intrahexamer cross-linking preventing the possibility of even transient opening of the hexamer interface [43].
The interaction model we propose, which positions SPRY domains over the interhexamer gaps in the capsid lattice making contacts with epitopes on neighboring hexamers, is further supported by the titrations of the labeled capsid with the unlabeled SPRY domain, which reveal that residues most perturbed by the titration are located on the outer edges of the hexamer. The relative sizes of the SPRY monomer and the capsid lattice make it very unlikely for a SPRY domain to span the interhexamer gap and, at the same time, make contacts with the residues located close to the 6-fold symmetry axis of the hexamer. Mutations in the N-terminal β-hairpin of the capsid were shown to affect MLV and SIVmac restriction by the rhesus TRIM5α [44,45], but we see no chemical shift perturbation of the N-terminal β-hairpin region of the HIV CA-WT construct upon capsid binding. It is feasible that β-hairpin mutations have an indirect, allosteric effect on SPRY binding resulting from small changes in the relative angle between capsid monomers within the hexamer that may produce significant relative displacement of the SPRY-binding epitopes located at the outer edge of the hexamer.
The inherent limitation of the chemical shift perturbation measurements is that they only allow approximate mapping of the binding surfaces but are insufficient for a high-resolution reconstruction of the complex. We, therefore, do not attempt to generate higher-resolution models based on the data presented here. Further refinement of the relative orientation of the SPRY and the capsid in the complex requires pair-wise distance restraints between defined sites on the capsid and the SPRY domain. Obtaining such distance restraints for weak protein–protein interactions requires specialized NMR approaches, and this work is currently in progress.
Our results illustrate the utility of NMR spectroscopy for the study of TRIM5α–capsid binding despite the complications arising from the intrinsically weak interaction of the individual SPRY domains with the assembled retroviral capsid. The emerging picture is that of an interaction mediated by mobile protein segments that involves a pattern of multiple epitopes presented by several neighboring capsid units within the assembled mature core of the HIV retrovirus. The observation that the distinct epitope arrangements presented by the dimeric and hexameric capsid constructs did not produce a dramatic increase in apparent affinity suggests that the interaction interface is devoid of interaction hot spots and that the binding energy is distributed relatively evenly between individual epitopes. Studies with other retroviral capsids and distinct restriction-competent TRIM5 variants will reveal whether these features are general properties of TRIM5α-mediated restriction and whether protein mobility at the host–pathogen interface is a common outcome of the antagonistic co-evolution of retroviruses and their hosts.
Materials and Methods
Protein expression and purification
15N-labeled rhSPRY was expressed and purified as described previously [32]. The v1C-5A mutant was created using the standard PCR-based mutagenesis method and confirmed by DNA sequencing. For the expression and purification of the v1C-5A mutant, the same protocol was followed as in the rhSPRY. CA-NTD, CA-WT, and CA-HEX were all expressed in BL21 (DE3) cells (New England Biolabs) in LB media at 37 °C until the cell density reached A600 ~ 0.6. Then, the temperature was lowered to 22 °C, and protein expression was induced with 1 mM IPTG. The cells were harvested 12–14 h post induction. All capsid constructs were incubated with 30% saturated ammonium sulfate at 4 °C for 2 h. In the case of CA-NTD and CA-WT, the precipitate was dissolved in 50 mM Tris (pH 7.5), 100 mM NaCl, and 10 mM DTT while the CA-HEX precipitate was dissolved in 50 mM 4-morpholineethanesulfonic acid (pH 6.5), 50 mM NaCl, and 25 mM DTT buffer; all proteins were purified by size-exclusion chromatography on the Superdex 75 column (GE Life Sciences) yielding >95% pure protein. In order to assemble the CA-HEX in its hexameric form, the protein was concentrated to 15 mg/ml using Amicon Ultra concentrators (Millipore, Billerica, MA, USA) and a series of dialyses was performed. First, it was dialyzed against 50 mM Tris (pH 8.0), 1 M NaCl, and 200 mM β-mercaptoethanol buffer followed by a second dialysis against the same buffer with 25 mM β-mercaptoethanol and a final dialysis against 50 mM Tris (pH 8.0). Each dialysis step was conducted at 4 °C for 24 h. Finally, the hexameric capsid construct was purified by size-exclusion chromatography on the Superdex 200 column (GE Life Sciences).
NMR titrations
NMR titrations of 15N-labeled rhSPRY and v1C-5A mutant (0.18–0.25 mM) with unlabeled capsid constructs (CA-NTD, CA-WT, CA-HEX) were conducted at 298 K on a Bruker Avance 700-MHz spectrometer by recording a series of 1H–15N TROSY HSQC spectra containing molar ratios from 1:0 up to 1:2.5. All samples were prepared in 50 mM sodium phosphate (pH 7.0), 50 mM NaCl, and 7% (v/v) D2O. The NMR data were processed using NMRPipe [46]. For each cross-peak, the intensity was evaluated using NMRView [27] and plotted as a function of the monomeric capsid concentration. When signal attenuation is evaluated quantitatively in NMR titrations, it is important to accurately determine the protein dilution factor for each titration point. This was done in two independent ways. First, 1 mM 15N-labeled urea was added to the initial labeled protein sample as internal reference. As the binding partner was titrated into the NMR sample, the intensity of the urea NMR signal was used to determine the dilution factor. Second, intensities of the NMR signals from the disordered C-terminal segment of the SPRY domain were used as an alternative way to determine dilution factors. NMR signals of this highly mobile protein segment are not broadened or attenuated by capsid binding; thus, their intensity is directly proportional to the concentration of the labeled protein. The two methods yielded very similar dilution factor values.
Residue-specific broadening rates
Residue-specific broadening rates were used to identify protein segments most affected by protein binding. Broadening rates R for each peak were determined by fitting signal intensity plots of rhSPRY or v1C-5A mutant with simple exponential decays I(c) = Ioe−Rc, where c is the capsid construct concentration (CA-NTD, CA-WT, or CA-HEX). This is an oversimplification because the intensity decay is not a simple exponential function of the titrant concentration. However, it provides a robust and consistent way of comparing the broadening of NMR signals arising from distinct residues within the same protein.
We do not include error bars in the plots of broadening rates because errors arising from the noise in the individual titration experiments (reported by the fitting program) are generally smaller than the irreproducibility among separate titration experiments. Figure S1 in Supplemental Data illustrates variation observed in separate titration experiments. Here we only discuss enhancements that clearly exceed background variation in the broadening rates.
Apparent dissociation constants
NMR titrations are commonly used to determine dissociation constants of weak protein–protein interactions. We used one of the standard approaches of fitting NMR titration data [47]. In our system, the apparent dissociation constant values should be used with caution because of unknown stoichiometry of the binding and the possibility of multiple overlapping epitopes (see Discussion). We use monomeric capsid concentration for fitting NMR binding curves for all capsid constructs (CA-NTD, CA-WT, and CA-HEX). The apparent dissociation constants for the WT rhSPRY and v1C-5A mutant binding with the different capsid species were calculated by using nonlinear fitting of the equation:
where I is the average intensity of the backbone NMR signals of the protein (excluding v1 and v2 loops and other residues with abnormally high broadening rates due to chemical exchange; see the previous section) measured as a function of the total capsid concentration CAt. Total concentration of the SPRY domain SPt was determined from UV absorbance at 280 nm. The boundary NMR intensity values for the free and fully bound SPRY (Ifree and Ibound) were allowed to vary in the fit. This formula is only an approximation for the NMR broadening arising from slower molecular tumbling of the protein complex, but the errors involved are smaller than the uncertainties arising from the unknown stoichiometry and overlapping binding sites. Thus, these systematic errors in the determined values do not affect the interpretation of the binding data presented in Discussion.
The plots of the bound fraction versus capsid concentration shown in Fig. 5 were obtained from signal intensities using the following formula:
Model-free analysis
ModelFree4 program was used to derive and optimize the internal dynamics and global tumbling parameters using the model-free approach [48]. Residues with 1H–15N nuclear Overhauser enhancement value less than 0.65 or with R2/R1 value exceeding one standard deviation from the mean, which indicate large-amplitude motion on the nanosecond-to-picosecond timescale or exchange, were excluded from the fitting. Overall correlation time and rotational diffusion tensors for isotropic, axially symmetric, and fully anisotropic models were estimated from the R2/R1 ratios of the remaining backbone amides and the structure of rhSPRY protein (PDB code: 2LM3) using the program TENSOR2 [45]. Backbone relaxation data were fit to the five standard Lipari–Szabo model-free formalism models: S2 (model 1); S2 and Te (model 2); S2 and Rex (model 3); S2, Te, and Rex (model 4); and S2, S2f, and Te (model 5).
Supplementary Material
Acknowledgments
Support for the UTHSCSA NMR Core Facility is provided by The University of Texas Health Science Center Executive Research Committee and the Cancer Therapy Research Center (P30 Cancer Center Support Grant from the National Cancer Institute CA054174). The Scholar Award from the Cancer Prevention and Research Institute of Texas (CPRIT) to D.I. and an NIH R01 AI087390 to F.D.-G. are gratefully acknowledged.
Abbreviations used
- HSQC
heteronuclear single quantum coherence
- TROSY
transverse relaxation optimized spectroscopy
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2013.07.025
References
- 1.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–53. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 2.Sebastian S, Luban J. TRIM5α selectively binds a restriction-sensitive retroviral capsid. Retrovirology. 2005;2:40. doi: 10.1186/1742-4690-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, et al. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc Natl Acad Sci USA. 2006;103:5514–9. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owens CM, Yang PC, Gottlinger H, Sodroski J. Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J Virol. 2003;77:726–31. doi: 10.1128/JVI.77.1.726-731.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131:70–9. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, et al. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–92. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pornillos O, Ganser-Pornillos BK, Yeager M. Atomic-level modelling of the HIV capsid. Nature. 2011;469:424–7. doi: 10.1038/nature09640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–6. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–51. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNab FW, Rajsbaum R, Stoye JP, O’Garra A. Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol. 2011;23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 11.Han K, Lou DI, Sawyer SL. Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 2011;7:e1002388. doi: 10.1371/journal.pgen.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 2005;102:2832–7. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohkura S, Yap MW, Sheldon T, Stoye JP. All three variable regions of the TRIM5α B30.2 domain can contribute to the specificity of retrovirus restriction. J Virol. 2006;80:8554–65. doi: 10.1128/JVI.00688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song BW, Gold B, O’hUigin C, Javanbakht H, Li X, Stremlau M, et al. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5α exhibits line age-specific length and sequence variation in primates. J Virol. 2005;79:6111–21. doi: 10.1128/JVI.79.10.6111-6121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stremlau M, Perron M, Welikala S, Sodroski J. Species-specific variation in the B30.2(SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J Virol. 2005;79:3139–45. doi: 10.1128/JVI.79.5.3139-3145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pham QT, Bouchard A, Grutter MG, Berthoux L. Generation of human TRIM5α mutants with high HIV-1 restriction activity. Gene Ther. 2010;17:859–71. doi: 10.1038/gt.2010.40. [DOI] [PubMed] [Google Scholar]
- 17.Yap MW, Nisole S, Stoye JP. A single amino acid change in the SPRY domain of human Trim5α leads to HIV-1 restriction. Curr Biol. 2005;15:73–8. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 18.Pham QT, Veillette M, Brandariz-Nunez A, Pawlica P, Thibert-Lefebvre C, Chandonnet N, et al. A novel aminoacid determinant of HIV-1 restriction in the TRIM5α variable 1 region isolated in a random mutagenic screen. Virus Res. 2013;173:306–14. doi: 10.1016/j.virusres.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Li X, Stremlau M, Lee M, Sodroski J. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J Virol. 2006;80:6738–44. doi: 10.1128/JVI.00270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–73. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 21.Nisole S, Lynch C, Stoye JP, Yap MW. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci USA. 2004;101:13324–8. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoye JP, Yap MW. Chance favors a prepared genome. Proc Natl Acad Sci USA. 2008;105:3177–8. doi: 10.1073/pnas.0800667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee K, Mulky A, Yuen W, Martin TD, Meyerson NR, Choi L, et al. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J Virol. 2012;86:3851–60. doi: 10.1128/JVI.06607-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Javanbakht H, Yuan W, Yeung DF, Song B, Diaz-Griffero F, Li Y, et al. Characterization of TRIM5α trimerization and its contribution to human immunodeficiency virus capsid binding. Virology. 2006;353:234–46. doi: 10.1016/j.virol.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5α protein. J Virol. 2008;82:11669–81. doi: 10.1128/JVI.01559-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, et al. Biochemical characterization of a recombinant TRIM5α protein that restricts human immunodeficiency virus type 1 replication. J Virol. 2008;82:11682–94. doi: 10.1128/JVI.01562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yap MW, Mortuza GB, Taylor IA, Stoye JP. The design of artificial retroviral restriction factors. Virology. 2007;365:302–14. doi: 10.1016/j.virol.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, et al. A B-box 2 surface patch important for TRIM5α self-association, capsid binding avidity, and retrovirus restriction. J Virol. 2009;83:10737–51. doi: 10.1128/JVI.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Sodroski J. The TRIM5α B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol. 2008;82:11495–502. doi: 10.1128/JVI.01548-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. Hexagonal assembly of a restricting TRIM5α protein. Proc Natl Acad Sci USA. 2011;108:534–9. doi: 10.1073/pnas.1013426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, et al. Structure of the rhesus monkey TRIM5α PRYSPRY domain, the HIV capsid recognition module. Proc Natl Acad Sci USA. 2012;109:13278–83. doi: 10.1073/pnas.1203536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, et al. Structural insight into HIV-1 capsid recognition by rhesus TRIM5α. Proc Natl Acad Sci USA. 2012;109:18372–7. doi: 10.1073/pnas.1210903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc Natl Acad Sci USA. 2007;104:6200–5. doi: 10.1073/pnas.0609174104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeble AH, Khan Z, Forster A, James LC. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci USA. 2008;105:6045–50. doi: 10.1073/pnas.0800159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park EY, Kwon OB, Jeong BC, Yi JS, Lee CS, Ko YG, et al. Crystal structure of PRY-SPRY domain of human TRIM72. Proteins. 2010;78:790–5. doi: 10.1002/prot.22647. [DOI] [PubMed] [Google Scholar]
- 37.Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol. 2010;401:985–95. doi: 10.1016/j.jmb.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, et al. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–53. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 39.Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, Sundquist WI. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J Mol Biol. 1997;269:780–95. doi: 10.1006/jmbi.1997.1051. [DOI] [PubMed] [Google Scholar]
- 40.Van Valen L. A new evolutionary law. Evol Theory. 1973;1:1–30. [Google Scholar]
- 41.Filippakopoulos P, Low A, Sharpe TD, Uppenberg J, Yao S, Kuang Z, et al. Structural basis for Par-4 recognition by the SPRY domain- and SOCS box-containing proteins SPSB1, SPSB2, and SPSB4. J Mol Biol. 2010;401:389–402. doi: 10.1016/j.jmb.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo JS, Suh HY, Park SY, Oh BH. Structural basis for protein recognition by B30.2/SPRY domains. Mol Cell. 2006;24:967–76. doi: 10.1016/j.molcel.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, et al. Rhesus TRIM5α disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011;7:e1002009. doi: 10.1371/journal.ppat.1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 2011;7:e1002011. doi: 10.1371/journal.ppat.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarthy KR, Schmidt AG, Kirmaier A, Wyand AL, Newman RM, Johnson WE. Gain-of-sensitivity mutations in a Trim5-resistant primary isolate of pathogenic SIV identify two independent conserved determinants of Trim5α specificity. PLoS Pathog. 2013;9:e1003352. doi: 10.1371/journal.ppat.1003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Fielding L. NMR methods for the determination of protein–ligand dissociation constants. Prog Nucl Magn Reson Spectrosc. 2007;51:219–42. [Google Scholar]
- 48.Lipari G, Szabo A. Model-Free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–70. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.