

Abstract
Microbes or danger signals trigger inflammasome sensors, which induce polymerization of the adapter ASC and assembly of an ASC speck. ASC specks recruit and activate caspase-1, which induces IL-1β cytokine maturation and pyroptotic cell death. Here we show that after pyroptosis ASC specks accumulate in the extracellular space, where they promote further IL-1β maturation. In addition, phagocytosis of ASC specks induces lysosomal damage, nucleation of soluble ASC as well as caspase-1 and IL-1β activation in the recipient cell. ASC specks appear in bodily fluids from inflamed tissues and autoantibodies against ASC specks develop in patients and animals with autoimmune pathologies. Together, these findings reveal extracellular functions of ASC specks and a novel form of cell-to-cell communication.
The activation of most innate immune signaling receptors results in the induction of gene transcription programs leading to production and release of inflammatory mediators via the secretory pathway1. In contrast, the inflammasomes operate on a post-translational level. Inflammasomes trigger the proteolytic maturation of biologically inactive precursor molecules of the IL-1β cytokine family and mediate the release of the active cytokines from the cytosol of cells2. Upon detecting microbial substances or danger signals the inflammasome sensor molecules, such as AIM2 or NLRP3, induce rapid polymerization of the bipartite adapter ASC into large helical filaments by facilitating ASC PYD-PYD self-interactions. The CARD domain of ASC then assembles to nucleate the polymerization and filament formation of pro-caspase-1 leading to its self-activation3-6. Active caspase-1 in turn induces the maturation of IL-1β family cytokines and also mediates their release into the extracellular space4,7. By forming large filamentous signaling platforms within the cytosol of cells, ASC can provide a scaffold for optimal caspase-1 recruitment and activation and thereby trigger a decisive on-off signaling response8. The inflammasome sensor NLRC4, which recognizes flagellin and type III secretion system components, lacks a PYD domain and can activate caspase-1 via direct CARD-CARD interaction. Nevertheless, ASC still forms specks upon NLRC4 activation and enhances caspase-1 and IL-1β activation9,10. Active caspase-1 also regulates the non-canonical release of many other proteins and triggers an inflammatory form of cell death called pyroptosis11,12. Inflammasome activation results in the activation of highly pro-inflammatory cytokines and the death of the activated cell. Hence, inflammasome activation is thought to occur only if a harmful inflammatory response is warranted, such as after tissue damage or persistent cellular invasion by certain bacteria or viruses. Inflammasome activation leads to a rapid recruitment of neutrophils and monocytes to the site of danger, which is important for limiting the spread of infections and for the initiation of repair after tissue damage11. Since inflammasome activation results in cell death and thereby release of cytoplasmic content, it is conceivable that ASC specks could ultimately be found in the extracellular environment13.
We found that ASC specks were released from inflammasome-activated cells and accumulated in the extracellular space, where they retained their ability to mature pro-IL-1β. Extracellular ASC specks were phagocytosed by macrophages resulting in lysosomal damage and IL-1β production in these cells. In mice, injection of ASC specks caused acute inflammatory reactions. Moreover, extracellular ASC specks could be detected after experimental in vivo inflammasome activation or in bronchoalveolar lavage fluids (BALF) from mice and patients with inflammatory airway pathologies. Finally, autoantibodies against ASC specks developed in a fraction of patients or animals with autoimmune pathologies and were able to increase the uptake of ASC specks by macrophages and boost IL-1β activation. Together our results identify ASC specks as an additional endogenous danger signal.
RESULTS
Extracellular ASC specks accumulate after pyroptosis
To visualize inflammasome activation, we generated mouse macrophage reporter cells stably expressing ASC fused with fluorescent proteins (FP)14. ASC-FP was evenly distributed in resting cells, consistent with previous reports of its soluble cytosolic distribution3,4. After inflammasome activation ASC-FP redistributed to form a paranuclear protein speck in activated cells, consistent with the ability of ASC to rapidly oligomerize after inflammasome activation (Fig. 1a and Supplementary movie 1)3-6. Immunofluorescence staining with anti-ASC antibodies (Abs) revealed that endogenous ASC also formed specks after inflammasome activation in human PBMCs and THP-1 cells (Supplementary Fig. 1a). We next activated the NLRC4 inflammasome in vivo by injecting mice with Pseudomonas aeruginosa (P. aeruginosa) into the footpad. Ex vivo staining of permeabilized sections from popliteal lymph nodes (LNs) of these mice revealed the appearance of ASC specks in subcapsular macrophages (Fig. 1b). These data indicate that ASC speck formation is a physiological cellular response to inflammasome activation. Notably, our confocal microscopic analysis of mouse and human ASC-FP reporter cells also revealed the presence of ASC specks in the extracellular space following inflammasome activation (Fig. 1c-d and Supplementary movies 2,3). ASC specks are protein aggregates of considerable size (μ1 μm) and therefore their appearance can be readily quantified by flow cytometry. Using this technique, we observed a time-dependent accumulation of ASC specks in cell supernatants after inflammasome activation (Fig. 1e). To confirm the ASC speck accumulation in cell-free supernatants after inflammasome activation, we performed a biochemical analysis of ASC aggregation. ASC specks can be separated from activated cells and nuclei by a combination of centrifugation and filtration using 5-micron filters that retain nuclei and allow the smaller ASC specks to pass (Supplementary Fig. 1b)4. Using this method, we generated cell-free supernatants of resting or inflammasome-activated cells, performed chemical crosslinking and tested for ASC aggregation by immunoblotting for ASC. This method confirmed the presence of not only monomeric species of ASC (μ22 kDs), but also dimers, trimers and higher order oligomers (Fig. 1f). To investigate whether the activation of other inflammasomes can also trigger ASC speck accumulation in cell-free supernatants we treated cells with poly-dAdT or Anthrax lethal toxin, which are activators of the AIM2 or NLRP1 inflammasome, respectively. ASC specks accumulated in cell-free supernatants of cells activated by NLRP1, NLRP3 and AIM2 inflammasomes (Fig. 1g,h). These data show that activation of all tested inflammasomes leads to ASC specks accumulation in the extracellular space.
Figure 1.

ASC specks accumulate in the extracellular space after inflammasome activation. (a) Confocal imaging of untreated or ATP-activated (5 mM, 40 min) ASC-mCerulean expressing iMøs, Scale bar: 22 μm. (b) Ex-vivo immunostaining of the subcapsular sinus from permeabilized sections of popliteal LNs from mice 4 h after injection with PBS, or P. aeruginosa. Scale bar: 15 μm and 19 μm. (c) Confocal imaging of ASC-mCerulean iMøs treated as in (a). Arrows indicate extracellular ASC specks. Scale bars: 4.4 μm and 3 μm. (d) Fluorescence imaging of LPS-primed (1 μg/ml) and nigericin-activated (10 μM) ASC-mCerulean expressing THP-1 monocytes. Arrows indicate extracellular ASC specks. Scale bar: 10 μm. (e) Flow cytometry and (i) quantification of extracellular specks in cell-free supernatants of ASC-mCerulean THP-1s treated as in d. (f) Immunoblot of DSS cross-linked endogenous ASC in cell pellets, or 5 μm filtrates of cell-free supernatants from WT THP-1 monocytes treated as in d. (g) Quantification of extracellular specks and (h) immunoblot of ASC in LPS-primed (250 ng/ml, 3h) iMøs treated as indicated. (i) Assessment of LDH release in cell-free supernatants of ASC-mCerulean THP-1s treated as in d, calculated from cells treated with 1% Triton X100. (j) Confocal time lapse of ASC-mCerulean THP-1 monocytes treated as in d in the presence of propidium iodide (1 μg/ml). Scale bars 8 μm, time (min). (k) Confocal time lapse of ATP-activated ASC-mCerulean iMøs in presence of directly conjugated anti-GFP Alexa 555 mAb (1 μg). Scale bars 33.0 μm, time (min). Data are representative of three (a,c-e,i) or two independent experiments (b,f-h,j,k); (e)Technical triplicates (colored lines) from one representative of three independent experiments; (i) combine data from two independent experiments (mean + SD of the number of specks per μl of acquired sample); g, mean + SD of triplicates from a representative of two independent experiments.
We next assessed whether extracellular ASC speck accumulation is a result of inflammasome-mediated cell death. Indeed, the appearance of extracellular ASC specks in cell-free supernatants correlated with the release of LDH from activated cells (Fig. 1i). Consistent with the induction of cell death after inflammasome activation and plasma membrane destabilization being a key feature of pyroptotic cell death11, inflammasome reporter macrophages failed to exclude propidium iodide (PI) shortly after ASC speck formation triggered by inflammasome activation (Fig. 1j and Supplementary Movie 4). Thus, ASC specks are likely not actively released but rather remain left over in the extracellular space after cellular demise. However, since pyroptosis also induces membrane disintegration it appeared conceivable that extracellular fluid could also have access to ASC specks shortly after pyroptotic cell death. To test this hypothesis, we added directly conjugated fluorescent anti-GFP Abs to the supernatants and activated NLRP3 in ASC-FP reporter cells. Live cell time-lapse imaging showed that fluorescent anti-GFP Abs were excluded from live cells but positively stained ASC-FP specks shortly after their appearance in activated cells (Fig. 1k and Supplementary movie 5). These data confirm that ASC speck formation precedes cell death and that extracellular fluid gains access to ASC specks within pyroptotic cell remnants shortly after formation of the ASC speck.
ASC specks remain active in the extracellular space
Since the accumulation of ASC specks in cell-free supernatants of inflammasome-activated cells correlated with cell death, we next investigated whether caspase-1 is required for this process. Using flow cytometry, we found that ASC speck accumulation after inflammasome activation in cell-free supernatants was greatly reduced in Casp1−/− cells when compared to WT cells (Fig. 2a). Similarly, we failed to detect ASC speck accumulation in supernatants from inflammasome-activated WT cells, in which caspase-1 activity was inhibited with z-YVAD-FMK (Fig. 2b). Chemical crosslinking of filtered cell-free supernatants and cell lysates from resting or inflammasome-activated cells followed by immunoblotting for ASC confirmed that caspase-1 activity was dispensable for ASC speck formation in cells, yet caspase-1 activity was required for ASC speck release (Fig. 2c). To assess the formation ASC specks from endogenous sources and to follow extracellular ASC speck accumulation after inflammasome activation, we stimulated WT, Asc−/−, Nlrp3−/−, or Casp1−/− macrophages with lipolysaccharide (LPS) and ATP. We then isolated endogenous ASC specks from either cell-free supernatants or cell pellets of the activated cells and probed for ASC and NLRP3 by immunoblotting. We found that endogenous ASC specks assembled in WT and in Casp1−/− cells but not in Nlrp3−/− cells, which is consistent with NLRP3 signaling upstream of ASC (Supplementary Fig. 2a, upper panel). However, we could detect ASC speck accumulation in cell-free supernatants of inflammasome-activated in WT but not in Casp1−/− cells (Supplementary Fig. 2a, lower panel). Together these findings confirm that caspase-1 mediated cell death is required for the accumulation of extracellular ASC specks formed from endogenous ASC.
Figure 2.

Extracellular specks remain active in the extracellular space. Flow cytometry (a) and quantification of extracellular ASC specks (b) in cell-free supernatants of LPS-primed ATP activated ASC-mCerulean expressing iMøs from either Casp1−/− or WT mice, treated or not with 10 μM of YVAD for 30 min. (c) Immunoblot of DSS cross-linked-ASC in 5 μm filtered cell-free supernatants and whole cell lysates of WT or Casp1−/− iMøs treated as in a. (d) Immunoblot of IL-1β and caspase-1 in cytosols prepared from untreated or LPS-primed Pycard−/− iMøs and left untreated (None) or incubated with in vitro-assembled ASC specks. Data are representative of four (a-c) or two (d) independent experiments.
We next hypothesized that one function of extracellular ASC specks could be to promote caspase-1 and IL-1β processing in the extracellular space. Indeed, it is known that pro-caspase-1 and pro-IL-1β can accumulate in supernatants of inflammasome-activated macrophages15,16, and thus it appeared plausible that ASC specks could further promote caspase-1 and IL-1β maturation. Fluorescent ASC specks spontaneously assembled upon heating of cytosolic extracts (S100 lysates) to 37°C allowing us to prepare recombinant ASC specks from macrophages (Supplementary Fig. 2b)4. Immunoblot and flow cytometric analysis revealed that these recombinant ASC specks contained NLRP3, ASC and caspase-1 (Supplementary Fig. 2c,d). Furthermore, confocal imaging of antibody stained endogenous proteins in activated THP-1 cells confirmed that both extracellular and intracellular ASC specks contained both NLRP3 and ASC (Supplementary Fig 2e). NLRP3 was found to be in the core of the endogenously formed ASC specks, similar to previous findings with in vitro reconstituted inflammasomes using purified proteins5. Of note, imaging of inflammasome-activated cells revealed that only a portion of NLRP3 was found associated to ASC specks, although the entire cellular ASC was recruited to the speck (Supplementary Fig. 2f). To characterize whether ASC specks can activate pro-IL-1β or pro-caspase-1 in cell-free systems, we next isolated cytosols from LPS-primed Asc−/− macrophages and incubated them at 37°C in absence or presence of recombinant ASC specks purified from ASC-FP cells. This analysis showed that processing of pro-caspase-1 and pro-IL-1β into their mature forms only occurred when Asc−/− cytosols were incubated with ASC specks (Fig. 2d). Of note, no IL-1β or caspase-1 processing was detectable in cytosol from LPS-primed, ATP-activated Asc−/− macrophages (Fig. 2d).
To assess whether ASC specks could also act to mature IL-1β from its pro-form when it is released from cells, we next generated cell-free supernatants from resting or inflammasome-activated macrophages. To additionally assess the role of caspase-1 present on the speck in this process, we isolated ASC specks from WT or Casp1−/− macrophages and incubated them with cell-free supernatants from inflammasome activated macrophages at 37°C. We observed that both WT and Casp1−/− ASC specks promoted further cleavage of pro-IL-1β that had been released from inflammasome-activated cells (Supplementary Fig. 3a). These data show that ASC specks have the capacity to induce processing of pro-IL-1β released from cells. Additionally, the fact that pro-IL-1β processing was also observed when Casp1−/− ASC specks were added to supernatants suggested that these specks could recruit and activate pro-caspase-1 from cellular supernatants, which in turn was sufficient to mediate pro-IL-1β processing. Indeed, Casp1−/− ASC specks carried pro-IL-1β, which was not observed on WT ASC specks. This implies that WT ASC specks induce pro-IL-1β cleavage in a process that required caspase-1 on the ASC specks. How pro-IL-1β is bound to the Casp1−/− ASC- specks remains to be investigated. To assess whether Casp1−/− ASC specks can indeed recruit and activate pro-caspase-1 from cellular supernatants, we added Casp1−/− ASC specks to cell-free supernatants of inflammasome-activated macrophages and incubated the mixture as indicated. The Casp1−/− ASC specks were subsequently pelleted by centrifugation, washed and assessed for caspase-1. Anti-GFP immunoblots served as a loading control since the Casp1−/− ASC specks were isolated from ASC-FP expressing Casp1−/− cells. This analysis revealed that Casp1−/− ASC specks are able to recruit and mature procaspase-1 released from inflammasome-activated cells (Supplementary Fig. 3b). Together, these data show that extracellular ASC specks have the ability to recruit and activate both pro-caspase-1 and pro-IL-1β in cell cytosols or cell-free supernatants.
Extracellular specks constitute a danger signal
Phagocytosis of extracellular protein aggregates, such as amyloid-β and islet amyloid polypeptide (IAPP), can activate the NLRP3 inflammasome by inducing lysosomal damage17-19. Since ASC specks are large protein aggregates, this raised the possibility that they could also be sensed as a danger signal by phagocytic immune cells. Indeed, we observed that macrophages ingested ASC specks, which remained stable in phagolysosomal compartments of cells over several hours before being degraded (Fig. 3a,b). To investigate whether the ingestion of ASC specks results in lysosomal damage, similar to what is observed with crystals or peptide aggregates17,20-25, we next assessed phagolysosomal stability. We loaded intracellular compartments with fluorescent dextran25 and subsequently added fluorescent ASC specks or controls. Incubation of macrophages with ASC specks or the lysosomal damaging peptide LeuLeu-OMe caused lysosomal swelling and leakage of fluorescent dextran into the cytosol (Fig. 3c-d), suggesting that ASC specks can trigger lysosomal damage. To directly test whether the uptake of exogenous ASC specks induces inflammasome activation in recipient cells, we incubated ASC-mCherry expressing macrophages with ASC-mCerulean specks so that we could simultaneously track both the added aggregated ASC specks and the soluble ASC inside macrophages. We observed that uptake of ASC-mCerulean specks by ASC-mCherry cells resulted in the formation of mCherry positive specks within the cells taking up ASC-mCerulean specks (Fig. 3e). Consistent with this observation, ASC specks - but not a mock preparation of cytosolic lysates of ASC deficienc (Pycard−/−) macrophages - induced IL-1β release from recipient macrophages in a dose- and time-dependent manner (Fig. 3f,g). Collectively, these results indicate that inflammasome-activated macrophages release ASC specks which are perceived as an endogenous danger signal with the potential to propagate inflammatory signals to the surrounding immune cells.
Figure 3.

Extracellular specks represent a cell-derived danger signal. (a) Confocal imaging of iMøs incubated with in vitro-assembled ASC-mCerulean specks for 2 hours. Scale bar 9 μm. (b) Immunoblot of ASC-mCerulean (anti-GFP) in lysates of BMDMs incubated with ASC-mCerulean specks. Non-phagocyted specks were washed away. (c) Confocal imaging of iMøs incubated with Dextran-A488 (20 μg/ml) alone or together with the lysosomal damaging LeuLeu-O-Me (0.5 μM), or ASC-mCerulean specks (100 μg/ml) for 4 hours. Scale bars: 7.0 μm top left, 4.9 μm top right, 6.0 μm bottom left, and 2.9 μm bottom right. (d) Average size of lysosomes in cells treated as indicated. (e) Percentage of recipient ASC-mCherry cells containing ASC-mCherry specks after incubation with silica crystals (100 μg/ml), nigericin (10 μM) or ASC-mCerulean specks (100 μg/ml). (f-g) ELISA of IL-1β in supernatants of LPS-primed (200 ng/ml, 2 h) BMDMs that were either left untreated or stimulated with cholesterol crystals (250 μg/ml), ASC specks, or a mock preparation from Pycard−/− iMøs (200 μg/ml). Data are representative from three independent experiments (a-e); d, mean + SD of combined data from two independent experiments, in which at least 5 fields/condition were analyzed (Mann-Whitney test, **P < 0.0057); e, combined data from multiple fields (n = 8) from two independent experiments (mean ± SD, Mann-Whitney test, *P < 0.05, **P < 0.006); f, Mean + s.e.m. of combined data from four independent experiments; g, mean + SD of combined data from two independent experiments.
ASC specks have prionoid activities
While analyzing the uptake of ASC-mCerulean specks into ASC-mCherry reporter macrophages by confocal microscopy, we noticed that ASC-mCherry was recruited to a fraction of the phagocytosed ASC-mCerulean specks (Fig. 4a, empty arrows vs. white arrow). We noted that after longer incubation periods ASC-mCerulean specks had recruited the entire soluble cytosolic ASC of the recipient ASC-mCherry reporter macrophages (Fig. 4b). This data suggested that in addition to inducing IL-1β release from macrophages, extracellular ASC specks may also aggregate cytosolic soluble ASC from recipient cells after their escape from phagosomes. A similar ‘seeding’ behavior is commonly observed with prion or prionoid proteins which are able to aggregate their cytosolic soluble counterparts once they escape endocytic vesicles and gain access to the host cell cytosol26-28.. We next investigated whether ASC specks in cells or purified from activated cells have prion-like features, similar to what has recently been shown using purified proteins5,6. Imaging of fluorescent ASC specks using confocal scanning microscopy and super resolution stimulated emission depletion (STED) microscopy revealed the presence of long fibrillar structures (Fig. 4c,d). We next generated recombinant ASC specks from WT cells or, as a control, subjected Pycard−/− cells to the same protocol. Electron microscopy of ASC speck material isolated from WT cells revealed the presence of fibrillar structures (Fig. 4e,f). Furthermore, ASC speck preparations obtained from ASC-FP inflammasome reporter cells and stained with gold-labeled anti-GFP antibodies identified the imaged structures as ASC specks (Fig. 4g). Cryo-electron microscopy further confirmed the presence of fibrillar structures in ASC specks preparations isolated from macrophages (Supplementary Fig. 4a).
Figure 4.

ASC has prionoid features. Confocal imaging of ASC-mCherry expressing iMøs incubated overnight (a), or 36 hours (b) with in vitro-assembled ASC-mCerulean specks. Open triangles indicate ASC-mCerulean specks that recruited ASC-mCherry and closed triangle indicate ASC-mCerulan specks that have not recruited ASC-mCherry. Scale bars: 2.6 μm. (c) Confocal imaging of untreated or LPS-primed, poly dAdT-activated ASC-mCerulean expressing iMøs. Scale bar: 4.9 μm. (d) Stimulated emission depletion (STED) microscopy of ASC specks purified from ASC-mCerulean expessing iMøs stimulated with ATP and stained with anti-GFP and Atto 647N-conjugated secondary Abs. Scale bar: 0.5 μm. (e) Electron microscopy (EM) of ASC specks prepared from WT or mock preparations from Pycard−/− iMøs. Scale bars: 100 nm. (f) EM of in vitro-assembled ASC-mCerulean specks. Scale bars: 0.5 μm. (g) EM of ASC-mCerulean specks isolated from LPS-primed, nigericin activated iMøs stained with anti-GFP Abs directly conjugated to 10 nm gold nanoparticles. Data (a-g) are representative of at least two independent experiments.
Most proteins that form prions have the ability to promote their own polymerization in a process called seeding, where preformed fibers provide templates for fiber elongation. This process potentially drives prion infectivity and the development of associated diseases26,29,30. To assess the propensity of purified ASC specks isolated from cells to act as a seed for further aggregation of soluble ASC in more detail, we added ASC-mCerulean specks to soluble ASC-mCherry cytosolic extracts and examined the recruitment of the soluble ASC-mCherry to ASC-mCerulean specks over time. This analysis revealed that ASC specks recruited soluble ASC within seconds (Supplementary Fig. 4b, and Supplementary movie 6) leading to the formation of an outer layer of ASC-mCherry around a core of ASC-mCerulean (Supplementary Fig. 4c). Moreover, the rapid recruitment of soluble ASC-mCherry to ASC-mCerulean specks could also be followed by flow cytometry (Supplementary Fig. 4d). Of note, ASC was specifically required for nucleation of its soluble counterpart, since soluble Fc-mCherry protein could not be aggregated by incubation with ASC-mCerulean specks (Supplementary Fig. 4e). Hence, these data suggest that ASC specks isolated from cells act as a seed to further aggregate soluble ASC in a cell free system or within cytosols after ASC specks escape the phagosome. These features are shared with other prionoid proteins26 and these data suggest that ASC specks are able to ‘infect’ bystander cells and propagate nucleation of soluble ASC.
ASC specks induce inflammatory responses in vivo
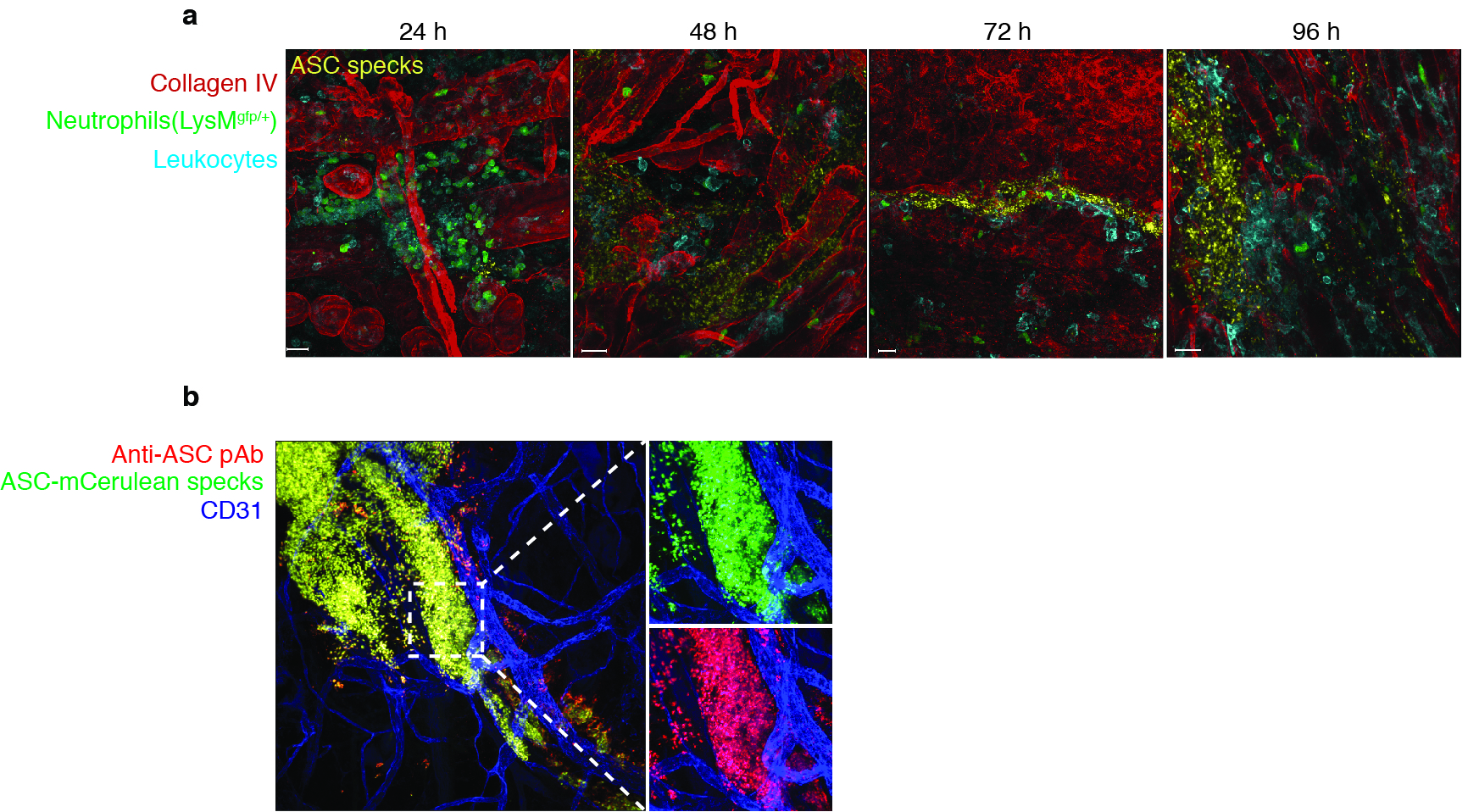
Activation of the NLRP3 inflammasome in vivo results in neutrophil and inflammatory monocyte recruitment in a largely IL-1R-dependent fashion20,21. To assess the ASC speck-induced inflammatory response in vivo we injected fluorescent ASC specks or, as a control, fluorescent beads into the ear of mice and visualized subdermal neutrophil recruitment using two different mouse models. Compared to injection of fluorescent beads, injection of ASC specks led to a strong recruitment of GFP-positive neutrophils in LysMgfp/gfp transgenic reporter mice (Fig. 5a). We next examined neutrophil recruitment following ASC speck injection in WT mice. ASC speck injection into the subdermis resulted in increased neutrophil diapedesis and attachment of neutrophils to venules in areas close to the deposited ASC specks (Fig. 5b). Of note, imaging of the subdermal injection site for up to 96 hours revealed that injected ASC specks remained visible which indicates that they are highly resistant to extracellular proteases (Supplementary Fig 5a). This protease insensitivity can typically be observed with other prionoid proteins27,28,31.
Figure 5.

ASC specks propagate inflammation in vivo. (a) Confocal imaging of whole mount ear skin of LysMgfp/gfp mice 4 h after subdermal injection of ASC-mCerulean specks (5 μg in 10 μl of PBS) or 10 μl of PE calibrate beads. (b) Confocal microscopy imaging of whole mount ear skin of WT mice 4 h after subdermal injection of ASC-mCerulean specks. Neutrophils (anti-Ly6G), endothelial/stomal cells (ECs) (anti-CD31). (c) Gating strategy and quantification of peritoneal lavage cells CD11b+Gr-1+F4/80− neutrophils and CD11b+Ly6C+Ly6G−F4/80− inflammatory monocytes in the peritoneum of WT mice 16 h after intraperitoneal injection of silica crystals (250 μg) or ASC specks (200 μg). (d) Quantification as in (e) of peritoneal lavage cells in WT mice or Nlrp3−/− and Il1r−/− mice injected with ASC specks (20 μg). (e) ELISA of IL-1β in supernatants of LPS-primed WT, Nlrp3−/−, Asc−/− or Casp1−/− iMøs that were left untreated (None) or stimulated with in vitro-assembled ASC specks (100 μg/ml). Representative of two (a-b) independent experiments; c, mean ± s.e.m. of pooled data from three independent experiments. Each symbol represents one mouse (n = 8 per group). Neutrophils: **P = 0.0006, Mann Whitney test; **P = 0.0018, unpaired two-tailed Student's t test. Inflammatory monocytes: **P = 0.0021, Mann Whitney test; **P = 0.0023, Mann Whitney test). d, mean ± SD from combined data from two independent experiments. PBS (n = 4), WT (n = 15), Nlrp3−/− (n = 8), Il1r−/− (n = 4). Neutrophils: **P = 0.0081, Inflammatory Monocytes: **P = 0.0040 , Mann Whitney test; (e) Mean ± s.e.m. of combined data from three independent experiments, Mann Whitney test, p < 0.05.
Consistent with the inflammatory properties of ASC specks injected into the subdermis, intraperitoneal injection of ASC specks in WT mice promoted the development of a sterile peritonitis, characterized by the recruitment of neutrophils and inflammatory monocytes (Fig. 5c). The recruitment of these inflammatory cells after injection of ASC specks was partially impaired in Nlrp3−/− mice and entirely blocked in Il1r−/− mice, implying that ASC specks elicit IL-1β maturation in vivo (Fig. 5d). Furthermore, these data suggest that ASC specks can induce caspase-1 activation in an NLRP3-independent manner, likely due to its propensity to activate ASC and caspase-1 in recipient cells. Consistent with this, ASC specks elicited only reduced IL-1β release in Nlrp3−/− macrophages, but the response was dependent on both ASC and caspase-1 (Fig. 5e). Thus, ASC specks cause inflammatory responses in vivo and in vitro and the activation of IL-1β is not fully dependent on NLRP3 inflammasome activation.
Extracellular specks accumulate in inflammatory diseases
We next investigated whether inflammasome activation in vivo can lead to the appearance of extracellular ASC specks. Immunostaining of fixed and permeabilized LN sections revealed that ASC specks were present in the majority of subcapsular macrophages after infection with P. aeruginosa (Fig. 1b). To investigate whether this infection also induces the appearance of extracellular ASC specks in vivo, we modified the detection protocol and first infected mice via injection of P. aeruginosa in the footpad followed by injection of fluorescently conjugated anti-ASC Abs, 4 hours later, via the same route. We performed this procedure to be able to stain only ASC specks appearing in the extracellular space (in the absence of detergents). The draining popliteal LNs were then harvested and visualized by confocal microscopy. Indeed, using this technique fluorescently we detected extracellular ASC specks in the subcapsular macrophage area of infected LNs. In contrast, no staining was found in the same area of LNs obtained from the mock infected (i.e. PBS injected) mice (Fig. 6a). As P. aeruginosa is a known activator of the NLRC4 inflammasome32, these data suggest that in vivo activation of the NLRC4 inflammasome can also result in release of ASC specks into the extracellular space. Furthermore, these data show that ASC specks are accessible to Abs in vivo after inflammasome activation. We confirmed that the anti-ASC antibody specifically stained ASC specks deposited in tissues (Supplementary Fig. 5b).
Figure 6.

Extracellular ASC specks are formed in vivo and accumulate during human chronic inflammatory disease. (a) Confocal imaging of subcapsular sinus macrophages of popliteal draining LNs from P. aeruginosa infected mice injected 4 h later via the same route with 4 μg of PE-labeled anti-ASC Abs. Scale bars: 53 μm left panels 37 μm right panels, 9 μm inserts. (b) Flow cytometry of ASC-mCerulean specks pre-incubated with two monoclonal anti-ASC Abs (top histograms) or IgG1 isotype controls (bottom histograms) directly labeled with A488 and A647 fluorophores. Amount of specks per μl of sample was determined by subtracting A488+A647+ events in anti-ASC stained sample from those in IgG1 control stained samples. (c) Adjusted quantification of A488+A647+ ASC specks in cell-free BALF from WT BALB/c mice exposed to cigarette smoke or normal air for 8 weeks. (d) Flow cytometry of BALF samples from patients with COPD (5 ml/patient) before and after removal of dead cells by centrifugation (400 x g, 5 min) and size filtration (5 μm). (e) Immunoblotting for ASC (right) of cell-free filtered BALF samples from patients with COPD (n = 4), Pneumonia (n = 4), Pulmunary hypertension (n =2) or healthy donors (n =2) after chemical crosslinking with 1 mM of DSS. Cell-free sups from untreated (–) or LPS-primed, nigericin-activated THP-1s were used as controls. Data show representative images (a) flow cytometry (b, d), and immunoblotting (e) analysis from one out of two independent experiments; (c) cumulative from one experiment; each symbol represents an individual mouse; horizontal and vertical lines indicate mean + SD, *P = 0.0317, Mann-Whitney test.
Our data showing a role of extracellular ASC specks in driving inflammatory responses opened the possibility that ASC specks can accumulate in inflamed tissues. Chronic obstructive pulmonary disease (COPD) is an inflammatory airway disease, in which inflammasome activation is thought to play a role33,34. COPD develops due to chronic inflammatory responses to noxious particles or gases in lung tissues. To test the hypothesis that ASC specks accumulate in chronic lung inflammation, we made use of a mouse model of smoke-induced COPD, in which mice develop hallmark features of the human disease35. Mice were subjected to air or cigarette smoke five days per week for a total of eight weeks, after which we bronchoalveolar lavage fluid (BALF) was collected. We found that ASC specks can be quantified by flow cytometry after staining with two anti-ASC mAbs labeled each with a different fluorescent dye (Fig. 6b). Using this method, we observed that mice that inhaled cigarette smoke for 8 weeks had significantly higher counts of extracellular ASC specks in the collected BALF compared to mice exposed to air for the same period (Fig. 6c). These data indicate that ASC specks may be part of a chronic inflammatory response to smoke-induced cell and tissue damage. We next tested whether ASC specks could also be identified in BALF from healthy human volunteers or patients with COPD, pneumonia or pulmonary hypertension. BALF samples from patients contained a large numbers of cells (Fig. 6d, top). Therefore, we removed cells by centrifugation at 400 x g and further filtered the supernatants through 5 micron filters similar to the protocol established to separate ASC specks from cellular debris in cell culture supernatants (Supplementary Fig 1b). This procedure yielded cell-free BALF, yet the smaller material was still available for further analysis in the filtrate (Fig. 6d, middle and bottom). To investigate whether extracellular ASC specks found in BALF obtained from humans was in an active state (i.e., containing ASC monomers, dimers and oligomers), the filtrate was pelleted by centrifugation and subjected to chemical cross-linking (Fig. 6e). This analysis revealed that BALF from patients with COPD and pneumonia contained extracellular pre-assembled ASC specks, while BALF from patients with pulmonary hypertension or from healthy donors did not (Fig. 6e). Cell-free supernatants from inflammasome-activated THP-1 cells were used as a positive control. Together, these data show that ASC specks are present in the extracellular space in lungs from patients with inflammatory pulmonary diseases and suggest that they could be part of the chronic inflammatory response observed in these patients.
Anti-ASC Abs opsonize ASC specks and increase inflammation
We next sought to investigate whether extracellular ASC specks modify the inflammatory response after inflammasome activation in vivo. We therefore intravenously injected anti-ASC or IgG isotype control Abs into mice and subsequently induced peritonitis by intraperitoneal injection of silica crystals. The systemically available anti-ASC Abs should bind to the extracellular ASC speck appearing after in vivo inflammasome activation and thereby alter the biological response. Indeed, mice treated with anti-ASC Abs prior to activation of inflammasomes by intraperitoneal silica crystal injection had considerably increased neutrophil and inflammatory monocyte recruitment to the peritoneum compared to control IgG Abs treated mice (Fig. 7a). These data confirm that ASC specks are released after inflammasome activation in vivo and become accessible to anti-ASC Abs. The observed increased inflammatory response in vivo to silica crystals in the presence of anti-ASC Abs pointed to the possibility that these Abs could opsonize ASC specks and thereby induce increased rather than decreased inflammation. To test this, we pre-incubated fluorescent ASC specks with anti-ASC or anti-GFP Abs and compared phagocytosis of these ASC specks by macrophages. We found that antibody-opsonized ASC specks were taken up more efficiently by macrophages compared to ASC specks that had not been opsonized by Abs (Fig. 7b-d). Consistent with this notion, antibody-opsonization of ASC specks increased the release of IL-1β from macrophages (Fig. 7e). Together these data suggest that anti-ASC Abs can contribute to inflammatory responses by opsonizing ASC specks.
Figure 7.

(a) Quantification of peritoneal neutrophils and inflammatory monocytes in peritoneal lavage from C57BL/6 mice intravenously injected with anti-ASC pAb (100 μg) or purified rabbit IgG (100 μg) and intraperitoneally injected 2 h later with silica crystals (250 μg). (b) Fluorescence microscopy of BMDMs incubated with untreated or antibody coated (1 μg) ASC-mCerulean specks. Scale bars: 90 μm. (c) Percentages of macrophages with phagocytosed ASC-mCerulean specks. (d) Quantification of mCerulean positive lysosomes. (e) ELISA of IL-1β in supernatants of LPS-primed BMDMs left untreated, or stimulated with in vitro-assembled ASC specks pre-coated with 1 μg of anti-ASC Abs. (f) Flow cytometry screening method and median fluorescence intensity (graph) for the reactivity of sera from healthy donors (n = 7) or ANA+ autoimmune patients (n = 80) to ASC-mCerulean specks. (g) ELISA of IL-1β in supernatants of LPS-primed BMDMs left untreated, or stimulated with in vitro-assembled ASC specks coated with anti-ASC pAb or pooled (n = 4) sera from pristane-induced SLE or control mice. Data are representative of three (b-d) or two independent experiments (a,f,g). a culmulative from one experiment (mean + SD) PBS + IgG (n = 3), PBS + anti-ASC (n = 3), silica + IgG (n = 4), silica + anti-ASC (n = 7). Neutrophils: **P = 0.0061, Mann Whitney test; Inflammatory monocytes: **P = 0.0028, unpaired two-tailed Student's t test; Culmulative from one experiment in which 3 fields/condition (c) and 20 cells/condition (d) were analyzed. Each symbol represents one cell (mean ± SD); e, Combined data from two independent experiments performed in triplicates (Mean + s.em); g, mean + SD of one representative out of two independent experiments.
Several autoantibodies were shown to have inflammatory properties as they can increase the cellular uptake of inflammatory material (such as DNA or RNA) as immune complexes. In fact this inflammatory nature is believed to be part of the pathogenesis of autoantibody mediated inflammation36. Therefore, it is conceivable that autoantibodies to extracellular ASC exist in patients with autoimmune diseases. To test this hypothesis, we assessed the sera of 80 patients with autoimmune diseases and high anti-nuclear antibody (ANA+) titers for the presence of Abs reacting with ASC specks. We first screened the sera with a flow cytometric assay to assess the presence of Abs that reacted with ASC specks (Fig. 7f). Indeed, among the ANA+ patients tested, 18 % showed anti-ASC speck reactive antibodies. The anti-ASC speck positive sera were then confirmed by an ASC speck ELISA (Supplementary Fig. 6a). Next, we assessed whether anti-ASC speck Abs would also develop in mice with experimental lupus37. Similar to what we had observed in autoimmune patient sera, we could also detect anti-ASC speck reactive autoantibodies in the sera of mice with experimental lupus (Supplementary Fig. 6b). These data suggested that ASC specks can be opsonized by autoantibodies and that this may contribute to the inflammatory response. To test this hypothesis, we incubated ASC specks with sera from autoimmune mice before addition to macrophages and found that they boosted IL-1β responses in recipient cells similar to purified commercially available anti-ASC Abs (Fig. 7g). This effect was not observed when ASC specks were incubated with sera from control mice. Together, these data suggest that anti-ASC Abs exist in inflammatory diseases and could potentially exacerbate the effects of the extracellular ASC specks.
DISCUSSION
Inflammasome activation induces the rapid polymerization of ASC and caspase-1 into a fibrillar signaling platform. Similar processes can be found in other signaling pathways38-40 and it is thought that these functional protein fibrils favor digital on-off responses leading to cleavage and release of caspase-1 substrates. Since caspase-1 activation also results in cell death4-6, each activated cell can only respond once. Thus, the inflammatory output of the individual inflammasome-activated cells would thus depend solely on the available cellular pool of caspase-1 substrates and such a one-time response would greatly limit the possible inflammatory output of inflammasome-activated cells. Our study reveals previously unappreciated extracellular activities of ASC specks, which persist even after the demise of the activated cell. The capacity of ASC specks to maturate caspase-1 and its substrates in the extracellular space could contribute to a ‘depot-effect’ of inflammasomes leading to prolonged inflammatory activities. Furthermore, we found that ASC specks can be recognized as a danger signal akin to other aggregated substances and this cell-to-cell transfer of aggregated ASC leading to de novo cellular signaling represents an additional cell-to-cell communication mechanism.
The seeding behavior and fibrillar structure of ASC specks is reminiscent of prion proteins, which propagate from cell-to-cell in an aggregated protein state27,28,31. Prion proteins can induce the conversion of correctly folded proteins to the prion form which accumulate and eventually cause diseases27,28,31. Some prion proteins misfold into a characteristic amyloid fold. However, our extensive attempts to identify amyloid folds in ASC specks were negative suggesting that ASC specks are not bona fide amyloid proteins, but rather have prion-like activities. Another feature of prions is their resistance to proteases. Indeed, we observed that ASC specks remained visible after injection into tissues for prolonged times. Deposition of ASC specks in tissues induced the recruitment of neutrophils. After their recruitment to the endangered tissues, neutrophils can amplify tissue inflammation as they degranulate and release various inflammatory mediators, enzymes and danger signals. Of note, neutrophils can carry ASC and caspase-1 in secretory vesicles and tertiary granule compartments, which are released upon activation41. Since ASC specks can recruit soluble ASC and activate caspase-1, neutrophil-derived ASC or caspase-1 may thereby further contribute to the inflammatory milieu.
It is conceivable that an extracellular function of inflammasomes has evolved to enhance and to focus the inflammatory tissue reaction to the site of injury. On the other hand, the failure to appropriately clear ASC specks in inflamed tissues could potentially lead to their accumulation and subsequent perpetuation of immune responses. In agreement with this hypothesis, the presence of ASC and other inflammasome components in cerebrospinal fluid correlated with poor outcomes in patients with brain injury42.
Typically fairly large doses of aggregated or crystalline substances are required for NRLP3 activation. We thus hypothesize that the NLRP3 activation threshold combined with the ability of macrophages to degrade sub-threshold amounts of ASC specks will limit the spread of inflammation leading to a concentration-dependent effect. Additionally, the activity of caspase-1 is sensitive to oxidation, which could block the enzymatic function of extracellular specks43. Consistent with an activation threshold by which extracellular ASC specks are recognized by cells, antibody-coated ASC specks were phagocytosed more efficiently resulting in higher IL-1β production from cells in vitro and increased inflammatory response to inflammasome activation in vivo.
Our finding that autoantibodies against ASC specks exist in a high percentage of patients with autoimmune diseases warrants further studies in larger populations. Additionally, whether autoantibodies against ASC specks are associated with inflammatory pathologies remains to be investigated. The fact that treatment with anti-ASC Abs altered the in vivo inflammatory response to crystals suggests that extracellular ASC specks appear after inflammasome activation and opens the possibility that therapies directed against ASC specks could modulate inflammation. So far only the inflammasome effector molecules, i.e., IL-1β and IL-18, or their receptors have been targeted for pharmaceutical intervention. Indeed, this idea is supported by studies in traumatic brain and spinal cord injury models in which anti-ASC therapy was able to reduce caspase-1 and IL-1β activation, resulting in significant reduction in tissue damage44,45. However, as shown by our in vivo studies with anti-ASC Abs, different Abs can have opposite effects warranting careful design and selection of the Abs against ASC. Modern antibody engineering techniques allow for production of Abs, which do not interact with Fc receptors for example. In addition, Abs that interfere with the ASC speck-mediated maturation of extracellular caspase-1 or IL-1β could be generated, and hence different activities of ASC specks could be targeted therapeutically.
ONLINE METHODS
Reagents
Ultrapure LPS (E. coli 0111:B4), was from Invivogen; nigericin was from Invitrogen and ATP was from Sigma-Aldrich. Silica crystals were from US Silica. Antibodies to ASC were from BioLegend (mAb, 653902, clone TMS-1, 1:500 dilution), Millipore (mAb, 04-147, clone 2EI-7, 1:1,000 dilution), Adipogen (pAb, AG-25B-0006, clone AL177, 1:1,000 dilution) and Santa Cruz (pAb, sc-22514-R, clone N-15R, 1:200 dilution). Mouse monoclonal Abs to NLRP3 (Cryo-2 AG-20B-0014-C100, 1:1,000 dilution) and caspase-1 p20 (casper-1, AG-20B-0042-C100, 1:1,000 dilution) were from Adipogen. Anti-caspase-1 p45/p10 was from Santa Cruz (pAb, sc-514, 1:200 dilution). Biotin-conjugated anti-IL-1β (BAF401, 1:1,000 dilution) was from R&D Systems. The enzyme-linked immunosorbent assay ELISA kit for mouse IL-1β was from R&D Systems. The Invitrogen Zenon Tri-Colour kit (Z-25080) was used to directly label 1 μg of anti-ASC IgG1 mAbs or same amount of a purified IgG1 isotype (Invitrogen, 02-6100). Purified rabbit IgG (Invitrogen, 02-6102, 100 μg) was used as a control for injection into mice for comparison with rabbit anti-ASC pAbs from Adipogen (AL177). Anti-GFP antibody was from Invitrogen (A11122, 1:2,000). The combination of anti-mouse Abs: CD11b-PE (clone M1/70, eBioscience), Ly-6G (Gr-1) APC (clone RB6-8C5, eBioscience), Ly-6C-A450 (clone HK1.4, eBioscience) and the isotype controls Rat IgG2b-PE (clone eB149/10H5, eBioscience), Rat IgG2b-APC (clone eB149/10H5, eBioscience), and Rat IgG2a K-A488 (clone eBR2a, eBioscience) were used for the identification of neutrophils, inflammatory monocytes by flow cytometry. Dead cells that stained positively for 7AAD (BD Pharmingen, 559925) were gated out and excluded from the analysis. The anti-mouse Ly-6GA647 Abs (clone 1A8, BioLegend); F4/80 (clone BM8, eBiosciences), anti-CD31 Abs (Clone MEC 13.3, BD Biosciences) and Collagen IV (clone ab19808, abcam) were used to identify neutrophils, macrophages and endothelial/stroma cells (ECs) respectively in confocal experiments. PE-labelled calibrite beads (BD Biosciences #349502), and polystyrene particles of 0.7 - 0.9 μm (Spherotech Inc. PP-08-10) were used as reference in flow cytometry experiments. PE-beads were also used as control for injections ear skin whole mounts experiments.
Patients
This study was performed in compliance with the Declaration of Helsinki (1989) of the World Medical Association. Bronchoalveolar lavages (BAL) specimens were obtained from patients undergoing bronchoscopy and BAL for routine medical diagnostics. Written informed consent was obtained from each patient prior to enrolment. A total of twelve Caucasian patients were sampled in this study; chronic obstructive lung disease (COPD) (n = 4), Pneumonia (n = 4), Pulmonary Hypertension (n = 2) and healthy donors (n = 2). Patients were recruited from the Department of Internal Medicine, University of Bonn, Germany. COPD was diagnosed in accordance with the current ATS/ERS/WASOG guidelines.
Serum samples from a total of 80 patients with autoimmune disease that tested highly (≥1:2,560) positive for antinuclear antibodies (ANA+) were obtained from the Department of Immunology and Rheumatology, Hannover Medical School, Germany. Written informed consent was obtained from all patients. The sera were screened for their reactivity to purified ASC specks based on flow cytometry analysis as shown in Fig. 7f. Serum samples from a total of 7 healthy donors served as controls.
Mice
Mice were housed pathogen-free and mice were treated in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. Pycard−/− and Il-1r−/−, mice have previously been described17,46. C57BL/6J mice (6 - 8 weeks old, females) were purchased from Jackson Laboratories.
Animal model of COPD
The animal model of COPD was described previously35. Briefly, wild type BALB/c mice were exposed to cigarette smoke (twice per day and 5 times per week for 8 weeks) by using a custom-designed and purpose-built nose-only, directed flow inhalation and smoke-exposure system (CH Technologies, Westwood, NJ) housed in a fume and laminar flow hood. Each exposure lasted 75 minutes. At the end of the treatment mice BALF samples were harvested by lavage. The quantification of ASC specks in cell-free supernatants or BALF were carried out on a MACSQuant analyzer (Miltenyi Biotec), after gating on debris-sized events using micro sized beads of 0.7 - 0.9 μm (Spherotech Inc.) or 6.0 μm (BD Biosciences) as reference for their distribution on a FSC vs. SSC scatter. Data was analysed in FlowJo X 10.0.7 software.
Cells
The inflammasome reporter macrophages stably transduced with constructs for the expression of mCerulean or mCherry-tagged ASC have been described14. Bone-marrow derived macrophages (BMDMs) were obtained by culturing bone marrow cells from 6- to 8-week old C57BL/6 mice in DMEM supplemented with 10% FBS, 10 μg/ml Ciprobay-500 and 40 ng/ml M-CSF (R&D Systems). Six days later, BMDMs were collected and plated. Immortalized BMDMs were cultured in DMEM supplemented with 10% FBS and 10 μg/ml Ciprobay-500. Human peripheral blood mononuclear cells (PBMCs) were purified from whole blood over Ficoll density gradients (GE Healthcare); erythrocytes were lysed in red cell lysis buffer (Miltenyi Biotec) and seeded in RPMI supplemented with 10% FBS and 10 μg/ml Ciprobay-500. The monocytic cell line THP1 was cultured in RPMI 1640 supplemented with 10% FBS and 10 μg/ml Ciprobay-500. For stimulation assays, cells were treated with 100 nM of phorbol 12-myristate 13-acetate (PMA) overnight, primed with 1 μg/ml of LPS for 2 h and further activated with 10 μM of nigericin for the time indicated in figure legends.
Production and purification of ASC specks
ASC specks were assembled in vitro by incubating cytosolic contents at 37 °C for 45 min (Supplementary Fig. 2c) or purified from LPS-primed, ATP or nigericin activated cells as described previously4,47. The ASC specks were further runned through a 50% percoll cushion in CHAPS buffer (20 mM Hepes-KOH, pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 0.1 mM PMSF, 0.1 % CHAPS) for purification as described (Supplementary Fig. 2d)47. For EM imaging fluorescent ASC specks were further FACS sorted prior to analysis. To investigate oligomerization of ASC, ASC specks were washed with 0.5 mL CHAPS buffer and chemically cross-linked with 2 mM of DSS (Pierce) for 30 min at room temperature before being subjected to immunoblotting.
Immunoblotting
Proteins were separated on 4–12% precast SDS-PAGE gels (Novex, Invitrogen) with MES or MOPS buffer (Novex, Invitrogen). Proteins were then transferred onto PVDF membranes (Millipore) and blocked in 3% BSA in Tris-buffered saline containing 0.1% Tween-20 before overnight incubation with specific primary Abs. Membranes were then washed and incubated with appropriate secondary Abs (IRDye 800CW or IrDye 680RD coupled, LI-COR Biosciences, 1:15,000), and immunoreactivity was observed by near-infrared detection and analysed with a Odyssey CLx imaging system (LI-COR Biosciences).
Confocal Imaging
Confocal Laser Scanning Microscopy (CLSM) was performed with a Leica TCS SP5 SMD confocal system (Leica Microsystems). For live imaging, temperature was maintained at 37°C with 5% CO2 using an environmental control chamber (Life Imaging Services and Solent Scientific). Images were acquired, using a 63X objective, at time points noted in the figures or figure legends and analyzed using the LAS AF version 2.2.1 (Leica Microsystems) or Volocity 6.01 software.
Analysis of crystal or ASC speck-induced lysosomal damage
Assessment of lysosomal stability was performed as described21. Cells were seeded in 8 well glass-bottom dishes and were allowed to adhere overnight. Cells were then treated with 40 μg/ml of Alexa Fluor 647–coupled dextran (10 kDa; Invitrogen) alongside with inflammasome activators or fluorescent ASC specks for 4 h. Cells were washed twice with PBS, fixed with 4% formaldehyde and imaged in the LAS AF Confocal Microscope as described above.
Bacterial infection
Mice were injected in the footpad with 30 μl of PBS containing 107 colony-forming units (CFU) P. aeruginosa48. Four hours post infection the draining popliteal LNs were harvested and fixed with PLP buffer (0.05 M phosphate buffer containing 0.1 M L-lysine [pH 7.4], 2 mg/ml NaIO4, and 10 mg/ml paraformaldehyde) for 12 h, then dehydrated in 30% sucrose prior to embedding in OCT freezing media (Sakura Finetek). 30 μm sections were cut on a CM3050S cryostat (Leica), adhered to Superfrost Plus slides (VWR), stained, mounted with Fluormount G (Southern Biotech), and imaged on a LSM 710 confocal microscope (Carl Zeiss Microimaging).
Recruitment of neutrophils in ear skin whole mounts
Mice were subcutaneously injected in the ear with 10 μl of PE-labelled calibrite beads (BD Biosciences) or 5 μg of fluorescent ASC specks in 10 μl of PBS. Four hours after injections, mice were euthanized, ears excised, divided into dorsal and ventral halves, ventral halves fixed in 1% paraformaldehyde (Electron Microscopy Sciences) and stained with Abs diluted in washing buffer consisting of 1× PBS, 1% BSA and 0.025% (v/v) Triton X-100 (Sigma-Aldrich). For identification of neutrophils, ventral ear whole mounts were stained with Ly-6G-A647 Abs; macrophages were stained with F4/80-A488 and endothelial/stroma cells (ECs) were stained with anti-CD31 Abs. Confocal microscopy of skin whole mounts was performed with a LSM 710 confocal microscope equipped with a 20×/0.8 NA Plan Apochromat objective (Carl Zeiss Microimaging) using 1 μm optical slices.
Activity assay of ASC specks
Pycard−/− immortalized macrophages were primed with LPS as indicated to induce NLRP3 and pro-IL-1β. Cells were then lysed in CHAPS buffer (see above) by syringing 25X through a 20G needle. Lysates were centrifuged at 19,000 × g for 8 min to obtain crude lysates before ultra-centrifugation at 100,000 × g for 30 min at 4 °C to obtain S100 cytosols. The cytosolic contents were incubated with ASC specks assembled in vitro or a corresponding volume of PBS for 1 h at 37°C. For the activity assay of ASC specks in supernatants of activated WT BMDMs, cell-free supernatants (40 μl) of LPS-primed (500 ng/ml, 3 h) and ATP (2.5 mM, 40 min), or nigericin (5 μM, 40 min) activated BMDMs were incubated with 10 μl of serum free DMEM, or 10 μl of in vitro-assembled fluorescent ASC specks (1 mg/ml generated from ASC-mCerulean expressing WT or Casp1−/− iMøs) for 1 h. The reaction mixtures were then fractionated by SDS-PAGE and analyzed by immunoblotting with anti-caspase-1 (Adipogen) or anti-IL-1β (R&D Systems) Abs.
Assessment of pro-caspase-1 recruitment to ASC specks
ASC-mCerulean specks (50 μg) generated from WT or Casp1−/− iMøs expressing ASC-mCerulean were incubated with cell-free supernatants (180 μl) generated as described above, or serum free DMEM for 1 h. The ASC specks were pelleted (2,500 × g, 8 min), washed twice with PBS, fractionated by SDS-PAGE and analyzed by immunoblotting for caspase-1 and GFP.
Flow cytometry
For the assessment of recruitment of inflammatory cells to peritoneal cavity, mice (C57BL/6, female, 6 - 8 weeks old) were injected intraperitoneally with 200 μg of silica crystals, or 20 - 200 μg of ASC specks in 200 μl of PBS, or with PBS alone. After 16 h, peritoneal lavage cells were harvested and pre-incubated with 10 μl of monoclonal Abs 2.4G2 (FcgRIIB/III receptor blocker, Miltenyi Biotech) before staining with fluorescently-conjugated monoclonal Abs against CD11b, Ly-6G, Ly6C, F4/80 or fluorescently-conjugated isotype control Abs (see reagents). Dead cells were excluded based on their staining with 7AAD. The absolute number of neutrophils (CD11b+, Ly-6G+, Ly6C+) and inflammatory monocytes (CD11b+, Ly6C+, Ly6G+, F4/80 − ) was determined by flow cytometry as described elsewhere21. Data was analysed in FlowJo X 10.0.7 software.
Transmission electron microscopy of cytosolic lysates of WT or Pycard−/− iMøs
10 μl of ASC specks assembled in vitro from S100 cytosolic contents of WT or same volume of a mock speck preparation from Pycard−/− iMøs were placed on carbon-coated copper grids and incubated for 1 min. After removing excess liquid, grids were washed three times with water prior to staining with 2% aqueous uranyl acetate for 1 min. Samples were analyzed on a Philips CM12 electron microscope (transmission electron microscope with an acceleration voltage of 80 keV).
Transmission electron microscopy of purified ASC specks
5 μl of purified ASC specks from supernatants of ATP activated ASC-mCerulean expressing iMøs were placed on formvar/carbon coated copper grids. After removing excess liquid, grids were washed twice with water prior to staining with 2% aqueous uranyl acetate for 20 seconds. Samples were analyzed on a JEM-2200FS transmission electron microscope (JEOL, Tokyo) operated at acceleration voltage of 200 keV and equipped with a Tietz Tem-Cam-F416 (TVIPS, Gauting). Magnification ranged from 8000× to 20000× corresponding to pixel size at the specimen level of 1.47 to 0.58nm.
Cryo- transmission electron microscopy of purified ASC specks
5 μl of FACS sorted ASC specks assembled in vitro from ASC-mCerulean expressing iMøs were applied on glow discharged (Pelco easiGlow, Ted Pella, CA) Quantifoil grids (R 3.5/1). The sample was frozen in liquid ethane using an automated plunge freezing machine (Vitrobot™ Mark IV, FEI Company). Samples were stored in liquid nitrogen until imaging. Plunge frozen samples of ASC specks were imaged under low-dose conditions in a cryo- transmission electron microscope (Titan Krios, FEI Company, Eindhoven) operated at accelerating voltage of 300kV. Images were recorded on a direct electron detector, Falcon II (FEI Company, Eindhoven). Magnification ranged from 6500× to 14000× corresponding to pixel size at the specimen level of 1-0.5nm.
Statistics
Statistical analysis was performed with Prism 6.0 (GraphPad Software Inc.) software. Unpaired two-tailed Student's t-tests were performed after data were confirmed to fulfill the criteria of normal distribution and equal variance; otherwise Mann–Whitney U-tests were applied. The suitable test for each experiment that required statistical analysis is indicated in individual figure legends. Data are typically presented as mean ± SD, or mean ± s.e.m., when multiple experiments were combined. A P value of < 0.05 was considered significant. All cell culture experiments requiring statistical analysis were performed at least 3 times. For animal experiments requiring statistical analysis we used at least 4 mice per group. No specific method of randomization was used for generation of samples or in animal experiments. Animal models were not performed in a blinded fashion, and the mice were assigned arbitrary numbers during experimentation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors would like to thank Cinzia Tiberi and Ping-I Chiang for technical support. This work was supported by grants from the DFG SFB670 (to EL), the NIH (to EL), the Alexander von Humboldt Foundation (to BSF) and the intramural BONFOR research support at the University of Bonn (to BSF). EL is a member of the ImmunoSensation cluster of Excellence in Bonn and a member of German Center for Infection Research (DZIF). AA was supported by an ERC Advanced Grant, the EU Neurinox Consortium, the Novartis Research Foundation and the Swiss National Science Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
BSF, LB and WK designed and performed experiments and analyzed data. JMR, AS, BM, GE, MM, PB and SH performed experiments. CB, MN, SRM, AA-A, SH and AA performed and analyzed EM and Cryo-EM data. AA, TE, PB, AMR, and WK analyzed data and provided critical suggestions and discussions throughout the study. SZ provided BALF samples from COPD patients. LB, MF and RES provided sera samples from autoimmune patients. BJ, AGJ and PH provided the mouse COPD model. BSF, and EL designed the study. BSF, DD, and EL wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests.
REFERENCES
- 1.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Latz E, et al. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masumoto J, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes-Alnemri T, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu A, et al. Unified PolymerizationMechanism for the Assemblyof ASC-Dependent Inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai X, et al. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell. 2014;156:1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsushita K, et al. A splice variant of ASC regulates IL-1beta release and aggregates differently from intact ASC. Mediators of Inflammation. 2009;2009:287387. doi: 10.1155/2009/287387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H. Higher-order assemblies in a new paradigm of signal transduction. Cell. 2013;153:287–292. doi: 10.1016/j.cell.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ. The CARD plays a critical role in ASC foci formation and inflammasome signalling. J Biol Chem. 2009;284:32932–32941. doi: 10.1042/BJ20121198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faustin B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 11.Bergsbaken T, et al. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller M, Ruegg A, Werner S, Beer H-D. Active Caspase-1 Is a Regulator of Unconventional Protein Secretion. Cell. 2008;132:14–14. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 13.Balci-Peynircioglu B, et al. Expression of ASC in renal tissues of familial mediterranean fever patients with amyloidosis: postulating a role for ASC in AA type amyloid deposition. Exp Biol Med (Maywood) 2008;233:1324–1333. doi: 10.3181/0803-RM-106. [DOI] [PubMed] [Google Scholar]
- 14.Stutz A, Horvath GL, Monks BG, Latz E. ASC Speck Formation as a Readout for Inflammasome Activation. Methods Mol Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- 15.Jakobs C, Bartok E, Kubarenko A, Bauernfeind F, Hornung V. Immunoblotting for active caspase-1. Methods Mol Biol. 2012;1040:103–115. doi: 10.1007/978-1-62703-523-1_9. [DOI] [PubMed] [Google Scholar]
- 16.Lima-Junior DS, et al. Inflammasome-derived IL-1. Nat Med. 2013:1–9. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 17.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheedy FJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013:1–11. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westwell-Roper C, Dunne A, Kim ML, Verchere CB, Masters SL. Activating the NLRP3 inflammasome using the amyloidogenic peptide IAPP. Methods Mol Biol. 2013;1040:9–18. doi: 10.1007/978-1-62703-523-1_2. [DOI] [PubMed] [Google Scholar]
- 20.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinon F, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nat Cell Biol. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 23.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masters SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duewell P, Duewell P, Latz E, Latz E. Assessment and Quantification of Crystal-Induced Lysosomal Damage. Methods Mol Biol. 2013;1040:19–27. doi: 10.1007/978-1-62703-523-1_3. [DOI] [PubMed] [Google Scholar]
- 26.Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459:924–925. doi: 10.1038/459924a. [DOI] [PubMed] [Google Scholar]
- 27.Hofmann JP, et al. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5951–5956. doi: 10.1073/pnas.1217321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. Nature Publishing Group. 2013:1–15. doi: 10.1038/nri3553. [DOI] [PubMed] [Google Scholar]
- 29.Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nature Neuroscience. 2012;15:936–939. doi: 10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, et al. The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem. 2008;283:21530–21539. doi: 10.1074/jbc.M800466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmes BB, Diamond MI. Cellular mechanisms of protein aggregate propagation. Current Opinion in Neurology. 2012;25:721–726. doi: 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutterwala FS, et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Nardo D, De Nardo CM, Latz E. New Insights into Mechanisms Controlling the NLRP3 Inflammasome and Its Role in Lung Disease. The American Journal of Pathology. 2013 doi: 10.1016/j.ajpath.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Botelho FM, et al. IL-1α/IL-1R1 Expression in Chronic Obstructive Pulmonary Disease and Mechanistic Relevance to Smoke-Induced Neutrophilia in Mice. PLoS ONE. 2011;6:e28457. doi: 10.1371/journal.pone.0028457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sci E, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. Journal of Allergy and Clinical Immunology. 2013;131:752–762. e7. doi: 10.1016/j.jaci.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leiss H, et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. CORD Conference Proceedings. 2012;22:778–792. doi: 10.1177/0961203313492869. [DOI] [PubMed] [Google Scholar]
- 38.Hou F, et al. MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu, et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Bin, et al. Structural Basis for dsRNA Recognition, Filament Formation, and Antiviral Signal Activation by MDA5. Cell. 2012;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 41.Bakele M, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. 2014;289:5320–5329. doi: 10.1074/jbc.M113.505636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adamczak S, et al. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome. Journal of Neurosurgery. 2012:1–7. doi: 10.3171/2012.9.JNS12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008;9:866–872. doi: 10.1038/ni.1633. [DOI] [PubMed] [Google Scholar]
- 44.de Rivero Vaccari JP, et al. A Molecular Platform in Neurons Regulates Inflammation after Spinal Cord Injury. Journal of Neuroscience. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Rivero Vaccari JP, et al. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart CR, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2009:1–8. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandes-Alnemri T, Fernandes-Alnemri T, Alnemri ES, Alnemri ES. Assembly, purification, and assay of the activity of the ASC pyroptosome. Meth Enzymol. 2008;442:251–270. doi: 10.1016/S0076-6879(08)01413-4. [DOI] [PubMed] [Google Scholar]
- 48.Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T, Germain RN. A Spatially-Organized Multicellular Innate Immune Response in Lymph Nodes Limits Systemic Pathogen Spread. Cell. 2012;150:1235–1248. doi: 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.