

Abstract
miRNAs have recently been implicated in hepatocarcinogenesis, although the actions and mechanisms of individual miRNAs remain incompletely understood. We examined the biological functions and molecular mechanisms of miR-185 in hepatocellular carcinoma (HCC). The expression of miR-185 is decreased in human HCC tissues compared with the nonneoplastic liver parenchyma. Quantitative RT-PCR showed a reduction of miR-185 in human HCC cells compared with primary hepatocytes. miR-185 overexpression in human HCC cells inhibited cell proliferation and invasion in vitro and prevented tumor growth in SCID mice. miR-185 overexpression inhibited DNMT1 3′ untranslated region luciferase reporter activity in HCC cells; this effect was abolished when the miR-185 binding site was mutated. miR-185 mimic or overexpression decreased the level of DNMT1 protein in HCC cells. These findings establish DNMT1 as a bona fide target of miR-185 in HCC cells. The role of DNMT1 in miR-185–induced inhibition of HCC growth was further supported by the fact that DNMT1 overexpression prevented miR-185–induced inhibition of HCC cell proliferation/invasion. miR-185 mimic or overexpression reduced PTEN promoter DNA methylation and enhanced PTEN expression, leading to the inhibition of Akt phosphorylation; these effects were partially reversed by DNMT1 overexpression. These results provide novel evidence that miR-185 inhibits HCC cell growth by targeting DNMT1, leading to PTEN induction and Akt inhibition. Thus, reactivation or induction of miR-185 may represent a novel therapeutic strategy for HCC treatment.
Hepatocellular carcinoma (HCC) is a highly malignant primary liver cancer with a poor prognosis.1–8 HCCs often develop in the background of pre-existing chronic liver diseases, such as chronic viral hepatitis B and C, alcoholic and nonalcoholic steatohepatitis, and cirrhosis. The incidence of HCC is rising in the United States and globally as a result of an epidemic of chronic hepatitis C infection and other risk factors, such as obesity and diabetes. Currently, HCC is the third leading cause of cancer-associated death, with an estimated continued rise in incidence during the next two decades. Despite improvements in surgery and other treatments, the overall 5-year survival rate for patients with HCC remains low. Thus, further studies are needed to define the mechanisms of hepatocarcinogenesis and develop new target therapies.
miRNAs are small noncoding RNA molecules that regulate gene expression post-transcriptionally through complementary base pairing with messenger RNAs.9 Recent studies have shown an important role of miRNAs in the regulation of HCC growth.10–16 Profiling of HCC miRNA expression has identified signatures associated with diagnosis, staging, progression, prognosis, and response to treatment. For example, Budhu et al17 identified a 20-miRNA tumor signature associated with HCC venous metastasis that can predict HCC survival and recurrence in patients with multinodular or solitary tumors; one of these miRNAs is miR-185. A subsequent study by Zhi et al18 found that low miR-185 expression correlates with more tumor recurrence and a lower patient survival rate compared with the high miR-185 group and that miR-185 reduces HCC cell growth and invasion in vitro, suggesting that miR-185 may represent a tumor-suppressive miRNA in HCC. However, to date, the biological functions of miR-185 in hepatocarcinogenesis remain largely unknown, and its mechanisms of action have not yet been defined.
This study was designed to further examine the effect and mechanism of miR-185 in human HCC. We show herein that miR-185 is a tumor suppressor miRNA in HCC and that its expression is down-regulated in human HCC tissues and cells. Our findings indicate that miR-185 inhibits HCC cell growth in vitro and in SCID mice by targeting the DNMT1/PTEN/Akt signaling pathway.
Materials and Methods
Cells, Plasmids, and Reagents
Human hepatocellular cancer cell lines (HepG2, Huh7, and Hep3B cells) were obtained from ATCC (Manassas, VA). All the cell lines were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO). Primary human hepatocytes were purchased from Lonza Inc. (Walkersville, MD) and were cultured in collagen I–coated plates (BD Biosciences, Bedford, MA) with hepatocyte basal medium supplemented with HCM SingleQuots growth factors (Lonza Inc.). The cells were maintained at 37°C and 5% CO2. The plasmids, including pcDNA3.1/DNMT1 and pcDNA3.1, were purchased from Addgene Inc. (Cambridge, MA). DNMT1 3′ untranslated region (3′-UTR) luciferase reporter plasmid was purchased from OriGene Technologies Inc. (Rockville, MD). Lentivirus encoding human miR-185 (L/miR-185) and control lentivirus expressing scramble control miRNA (L/control) were purchased from GeneCopoeia Inc. (Rockville, MD). The antibodies against PTEN, total Akt, and phospho-Akt (p-Akt) were obtained from Cell Signaling Technology Inc. (Beverly, CA). The antibody against β-actin was purchased from Sigma-Aldrich. The anti-DNMT1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 5-Aza-2′-deoxycytidine (5-Aza-CdR) and trichostatin A were obtained from Sigma-Aldrich. Other chemicals were of analytical grade.
In Situ Hybridization for miR-185 in Human HCC Tissue Specimens
In situ hybridization for miR-185 was performed in human HCC tissue microarray (serial # LV8013; US Biomax Inc., Rockville, MD). The tissue microarray consisted of 80 tissue core samples, including 20 cases of HCC, 10 cases of liver cirrhosis, and 10 cases of normal liver tissue (each case with duplicate samples) (more detailed information is available from US Biomax Inc.). The tissue microarray was prepared using the UVP HB-1000 Hybridizer hybridization oven (UVP LLC, Upland, CA), designed to create the ideal environment for nucleic acid hybridization. Human miR-185 miRCURY LNA probes were purchased from Exiqon Inc. (Woburn, MA), and the in situ hybridization procedure was performed according to the manufacturer's instruction. A scramble miRNA control probe having no homology with sequences in the miRBase database was used as a negative control. A U6 small nuclear RNA control probe was used as a positive control. The U6 positive control and the scramble negative control were used to ensure a functional working system and for optimization and troubleshooting. The staining score was measured by values from 0 to 4+ and was categorized as no staining = 0, minimal staining = 1+, weak staining = 2+, moderate staining = 3+, or strong staining = 4+.
Quantitative RT-PCR
Total RNA was extracted using a TRIzol plus RNA purification kit (Life Technologies Inc., Gaithersburg, MD) and were reverse transcribed to cDNA using SuperScript II reverse transcriptase (Life Technologies Inc.). The cDNA samples were used for quantitative RT-PCR (RT-qPCR) analysis in triplicate to determine the expression level of miR-185 using a QuantiFast SYBR Green PCR kit (Qiagen Inc., Valencia, CA) on a Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA). Experiments were followed according to the manufacturer's instructions.
Western Blot Analysis
The logarithmically growing cells were washed twice with ice-cold phosphate-buffered saline and lyzed in a lysis buffer (50 mmol/L Tris-HCl, pH 8.0; 150 mmol/L NaCl; 1% Nonidet P-40 (Caledon Laboratories Ltd., Georgetown, ON, Canada); 5 mmol/L EDTA, pH 8.0), with protease inhibitor mixtures and phosphatase inhibitor from Roche Applied Science (Indianapolis, IN). Cell lysates were centrifuged at 12,000 × g for 20 minutes at 4°C after sonication on ice, and supernatants were collected to measure protein concentrations. After boiling for 10 minutes in the presence of 10% β-mercaptoethanol, samples containing cells or tissue lysate proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane (Bio-Rad Laboratories), and then blocked in 10% dry milk/phosphate-buffered saline with 0.1% Tween 20 for 1 hour at room temperature. The blots were incubated with primary antibody (dilution 1:1000) overnight at 4°C. After three washes, the membranes were incubated with secondary antibody (IRDye 680LT/IRDye 800CW secondary antibodies; Li-Cor Biosciences, Lincoln, NE) in phosphate-buffered saline with 0.1% Tween 20 (dilution 1:10,000) for 60 minutes at room temperature or 4°C overnight. Signals were visualized using the ODYSSEY infrared imaging system (Li-Cor Biosciences). The antibodies used include the following: DNMT1, PTEN, p-Akt, total Akt, β-actin, and IRDye 680LT/800CW secondary antibodies from Li-Cor Biosciences.
Tumor Cell Invasion Assay
Invasion studies were performed using the 24-well (8-μm), colorimetric QCM ECMatrix cell invasion assay (EMD Millipore, Billerica, MA) for the evaluation of invasive tumor cells. Warm serum-free medium was added to the interior of the inserts to rehydrate the extracellular matrix layer for 1 to 2 hours. A Huh7 cell suspension of 1.0 × 106 cells/mL in serum-free medium was prepared. Medium was removed from the insert, and 300 μL of prepared cell suspension was added to the insert. In addition, 500 μL of medium containing 10% fetal bovine serum was added to the lower chamber. This 24-well plate was incubated for 24 hours in a tissue culture incubator. Using a cotton-tipped swab, noninvaded cells and ECMatrix gel (EMD Millipore) were removed from the inserts. The invasive cells on the lower surface of membrane were stained in staining solution for 20 minutes. The membranes were dipped in water several times and allowed to air-dry. Cells were counted by photographing the membrane through the microscope.
Cell Proliferation
Cells were seeded in 6-well plates in quadruplicate in standard growth medium. On the indicated days, the cells were detached from the wells using 0.05% trypsin-EDTA and were resuspended in 500 μL of standard growth medium. Aliquots were mixed 1:1 with trypan blue stain and were counted using a Countess automated cell counter (Invitrogen). Cell numbers were counted at 24-, 48-, and 72-hour time points. Triplicate measurement was performed for each period, and the results are expressed as means ± SD.
Selection of HCC Cells with Stable Overexpression of miR-185
Huh7 cells were infected with L/miR-185 or L/control to establish the stable cell lines in the presence of 6 μg/mL of puromycin (Invitrogen). We monitored transfection efficiency by green fluorescent protein signaling under a fluorescent microscope. Cells with stable overexpression of miR-185 were confirmed by real-time PCR.
Transient Transfection of miR-185 Mimic Oligonucleotide
Using a 24-well plate, 1.0 × 105 Huh7 cells were seeded per well in 500 μL of Dulbeco's modified Eagle's medium containing 10% fetal bovine serum and antibiotics. These Huh7 cells were transfected with double-stranded miR-185 mimic or its relative negative control RNA (Qiagen Inc.), with a final concentration of 5 nmol/L using Lipofectamine 2000 reagent in Opti-Mem I medium (both from Invitrogen) for 24 hours. After transfection, these cells were collected for Western blot analysis and real-time PCR experiments.
Overexpression of DNMT1 Devoid of the 3′-UTR
Huh7 cells stably infected with miR-185 or control lentivirus were seeded at 1.5 × 105 cells per well in a 24-well plate and were transfected with pcDNA3.1 or pcDNA3.1-DNMT1 using Lipofectamine 2000 for 24 hours. The resulting four cell groups were used for DNMT1 rescue experiments.
DNMT1 3′-UTR Luciferase Reporter Assay
DNMT1 3′-UTR luciferase reporter plasmid was obtained from OriGene Technologies Inc. Mutant DNMT1 3′-UTR luciferase reporter plasmid was constructed by cloning synthetic DNMT1 3′-UTR nucleotide (with deletion of the 7 bp of hsa–miR-185 seed sequence) into the pMirTarget vector. For transfection, Huh7 stable cells were seeded at 1.5 × 105 cells per well in a 24-well plate; 24 hours later, the cells were transfected with the wild-type or mutant DNMT1 3′-UTR luciferase reporter plasmid using Lipofectamine 2000. The cells were harvested 48 hours after transfection, and the luciferase reporter activities were analyzed by using the Dual-Luciferase reporter assay system (Promega Corp., Madison, WI) according to the manufacturer's protocol.
PTEN Promoter Methylation Assay
PTEN tumor suppressor gene at +1 transcription start site has a CpG island region located 250 Bp upstream from the transcription start site of PTEN and overlaps in the PTEN promoter region by 989 Bp, with a total length of 1239 Bp. The CpG island region in the PTEN promoter was amplified by methylation-specific PCR using primers that amplified a 186-Bp fragment (this region has been shown to allow satisfactory detection of PTEN promoter hypermethylation19). The methylation status was assessed by using the EpiTect II DNA methylation enzyme kit along with the EpiTect methyl qPCR primer assay PTEN human (CpG island 28526) (both from Qiagen Inc.) (the primers are located in the PTEN promoter region with a central position on chromosome 10: 89613408 of CpG island location Chr10: 89611752-89614108). The method used was based on detection of the remaining input DNA after cleavage with a methylation-sensitive and/or a methylation-dependent restriction enzyme (these enzymes digest unmethylated and methylated DNA, respectively). The relative fractions of methylated and unmethylated DNA were determined by comparing the amount in each digest with that of a mock (no enzymes added) digest using a ΔCT method.
HCC Xenograft Study in SCID Mice
Four-week-old SCID mice from The Jackson Laboratory (Bar Harbor, ME) were used for in vivo studies analyzing tumor growth. A suspension of 1 × 107 live cells/mL was mixed with BD Matrigel matrix high concentration (BD Biosciences) in a 0.5-mL total volume for a 1:1 ratio. Five microliters of cell suspension mixture was used for direct liver injection using a Hamilton syringe (cemented NDL, 26s gauge, 2 in, point style 2) (model 75 N SYR; Hamilton Co, Reno, NV). The high protein concentration (18 to 22 mg/mL) allowed the BD Matrigel matrix plug to maintain its integrity after direct liver injections into the mice. This allowed the injected tumor cells to remain localized for future excision. The xenograft tumors were recovered 30 days after inoculation. Approximately 0.5 cm of tumor samples were fixed in 10% buffered formalin for paraffin embedding. Tissue sections (4 μm thick) were processed for hematoxylin and eosin staining. Immunostaining for the proliferation marker Ki-67 was also performed. Portions of the liver tumor samples were frozen in liquid nitrogen, followed by −80°C storage. Data analyses were performed using a paired t-test. The animal experiments were performed according to the protocol approved by the Tulane University Institutional Animal Care and Use Committee.
Statistical Analysis
Statistical analyses were performed using SPSS for Windows software, version 13.0 (SPSS Inc., Chicago, IL) and Microsoft Excel spreadsheet software (Microsoft Corp., Redmond, WA). Xenograft tumors were analyzed using the paired t-test. In situ hybridization results were analyzed using the U-test. P ≤ 0.05 was considered statistically significant.
Results
Decreased Expression of miR-185 in Human HCC Cells
We performed in situ hybridization for miR-185 in the human liver tissue microarray consisting of 80 tissue samples (20 cases of HCC, 10 cases of cirrhotic liver, and 10 cases of normal liver; duplicate samples for each case). Although miR-185 is readily detectable in nonneoplastic hepatocytes, its level of expression is significantly reduced in HCC cells (Figure 1A). U-Test analysis revealed a significant reduction of miR-185 in HCCs arising from chronic hepatitis C and/or chronic hepatitis B compared with the normal liver tissues (there was no statistical significance between normal and cirrhotic liver tissues) (Figure 1B). This observation is corroborated by the results of RT-qPCR analysis showing decreased miR-185 expression in cultured human HCC cells (Huh7, HepG2, and Hep3B) compared with human primary hepatocytes (Figure 1C).
Figure 1.

Down-regulation of miR-185 in human HCC. A: Representative in situ hybridization for miR-185 in human HCC and normal liver tissues. The expression of miR-185 was decreased in HCC cells compared with nonneoplastic hepatocytes. Boxed areas on the left correspond to higher-magnification images on the right. B: miR-185 staining intensity in different types of tissue specimens [normal liver, cirrhotic liver, hepatitis B virus (HBV)–associated HCC, and hepatitis C virus (HCV)–associated HCC]. Staining intensities were compared and are depicted in a box plot. C: RT-qPCR showed a lower level of miR-185 expression in human HCC cells (Huh7, HepG2, and Hep3B) compared with human primary hepatocytes (HHs). Data are given as means ± SD. ∗P < 0.05, ∗∗P < 0.01. Original magnification: ×40 (A, left panels); ×400 (A, right panels).
DNMT1 Is a Direct Target of miR-185
We performed in silico analyses for target gene prediction by using Target Scan Human software version 5.2 and the resources from http://www.microRNA.org (last accessed August 24, 2011), which led to the identification of DNA (cytosine-5)-methyltransferase 1 (DNMT1) as one of the high-scoring candidate genes of miR-185 targets. Consequently, we performed further experiments to evaluate the effect of miR-185 on DNMT1 in HCC cells. Transient transfection of miR-185 mimic significantly decreased the level of DNMT1 in Huh7 and HepG2 cells (Figure 2). Similarly, stable transduction of Huh7 cells with an L/miR-185 also significantly decreased DNMT1 protein levels (successful overexpression of miR-185 was confirmed by RT-qPCR) (Figure 3, A and B). Moreover, Huh7 cells stably overexpressing miR-185 showed a significant reduction of cell proliferation and invasion (an approximately fourfold reduction of cell proliferation; an approximately 50% decrease of cell invasion) (Figure 3, C and D). These results demonstrate that miR-185 targets DNMT1 and inhibits HCC cell growth in vitro.
Figure 2.

miR-185 targets DNMT1 in HCC cells. Huh7 (A) and HepG2 (B) cells were transiently transfected with miR-185 mimic and miRNA scramble control oligonucleotide. The cell lysates were obtained for Western blot analysis to measure DNMT1 protein levels. DNMT1 protein expression was analyzed with β-actin as an internal control. The experiments were repeated five times (N = 5). Normalized fold expression of DNMT1 in Huh7 cells transfected with miR-185 mimic was compared with scramble control as depicted in bar graphs. Data are given as means ± SD. ∗P < 0.05.
Figure 3.

miR-185 targets DNMT1 and inhibits HCC proliferation and invasion in vitro. A: Real-time PCR quantification of miR-185 showed a significant increase in Huh7 cells stably transduced with L/miR-185 compared with cells stably transduced with L/Control. B: Western blot analysis for DNMT1 in Huh7 cells with or without stable overexpression of miR-185. Representative Western blots and average densitometry data. The level of DNMT1 protein is decreased in Huh7 cells stably expressing miR-185 (gray bars) compared with control cells (white bars). C: Cell proliferation was measured at 24, 48, and 72 hours in Huh7 stable cells after plating 1 × 103 per well in 6-well plates. miR-185 overexpression decreased cell proliferation at all the time points. D: A cell invasion assay was performed in Huh7 stable cells. Huh7 miR-185 stable cells showed an approximately 50% reduction in the number of invaded cells on the matrix membrane surface compared with control after 24 hours. The images depict invaded cells. Data are given as means ± SD. N = 5 (B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
We identified a putative miR-185 binding site in the 3′-UTR of DNMT1 (Figure 4A); this binding site is located at positions 306 to 312 of DNMT1 3′-UTR. To determine whether this predicated site is involved in miR-185 targeting of DNMT1 in HCC cells, we transfected luciferase reporter constructs containing DNMT1 3′-UTR (with or without miR-185 binding site mutation) into Huh7 cells (with or without miR-185 overexpression). miR-185 overexpression significantly reduced the DNMT1 3′-UTR luciferase reporter activity (by 2.3-fold), and the effect was abolished when the miR-185 binding site was mutated (Figure 4B). Thus, DNMT1 is a bona fide target of miR-185 in HCC cells.
Figure 4.

miR-185 decreases DNMT 3′-UTR luciferase reporter activity in HCC cells. A: The putative miR-185 binding site in DNMT1 3′-UTR is depicted in the top sequence pairs. The original strand of DNMT1 3′-UTR contains the 7-Bp seed sequences that bind to miR-185; the DNMT1 3′-UTR mutant strand shows deletion of the 7-Bp seed sequence. B:DNMT1 3′-UTR luciferase reporter acidity assay in Huh7 stable cells transfected with wild-type or mutant DNMT1 3′-UTR reporter construct. Data are given as means ± SD. N = 3. ∗P < 0.05.
Restoration of DNMT1 Prevents miR-185–Induced Inhibition of HCC Cell Proliferation and Invasion
To further determine the role of DNMT1 in miR-185–induced inhibition of HCC growth, we transfected miR-185 overexpressed cells with a DNMT1 expression vector devoid of the 3′-UTR. Specifically, Huh7 stable cells (with or without miR-185 overexpression) were transfected with a DNMT1 expression vector (DNMT1 in pcDNA3) or a control vector (pcDNA3), and the cells were analyzed for proliferation and invasion. DNMT1 overexpression prevented miR-185–induced inhibition of cell proliferation and invasion (Figure 5).
Figure 5.

Restoration of DNMT1 prevents miR-185–induced inhibition of HCC cell proliferation and invasion. A: Western blot analysis for DNMT1, PTEN, p-Akt, and Akt in Huh7 stable cells with or without DNMT1 overexpression. B: Cell proliferation analysis of Huh7 stable cells with or without DNMT1 overexpression. The experiments were performed 72 hours after plating 1 × 103 per well in 6-well plates. The experiments were repeated three times. C: Cell invasion assay of Huh7 stable cells with or without DNMT1 overexpression. The numbers of invaded cells were counted 24 hours after cell plating. The images depict invaded cells. Data are given as means ± SD. N = 5 (A); N = 3 (B and C). ∗P < 0.05.
miR-185 Regulates the PTEN/Akt Signaling Pathway in HCC Cells
DNMT1 is essential for maintaining DNA methylation patterns during carcinogenesis and is important for cancer cell survival and proliferation.20–22 The effect of DNMT is mediated, in part, through methylation of 5′-CpGs of target gene promoters, thus silencing the expression of tumor suppressor genes (including PTEN).23,24 We performed a methylation-specific PCR assay to determine the extent of DNA methylation in the PTEN promoter region. Stable overexpression of miR-185 in Huh7 cells significantly reduced PTEN promoter DNA methylation (Figure 6A). miR-185 mimic treatment enhanced the expression of PTEN protein in Huh7 cells (Figure 6B) and HepG2 cells (Supplemental Figure S1). Consistent with the documented inhibition of Akt phosphorylation by PTEN, we observed decreased levels of p-Akt in miR-185 mimic–treated cells. Furthermore, stable overexpression of miR-185 also enhanced PTEN protein and inhibited Akt phosphorylation; these effects were partially reversed by overexpression of DNMT1 (devoid of the 3′-UTR) (Figure 5A). We observed that inhibition of DNMT by 5-Aza-CdR also decreased PTEN promoter methylation, enhanced PTEN protein, and reduced Akt phosphorylation (Supplemental Figure S2), which are comparable with those induced by miR-185 mimic or overexpression. Taken together, these findings suggest that miR-185 inhibits HCC cell growth by targeting DNMT1, which leads to induction of PTEN expression and subsequent inhibition of Akt phosphorylation. This assertion is further corroborated by the documented role of PTEN and Akt in hepatocarcinogenesis.25–29
Figure 6.
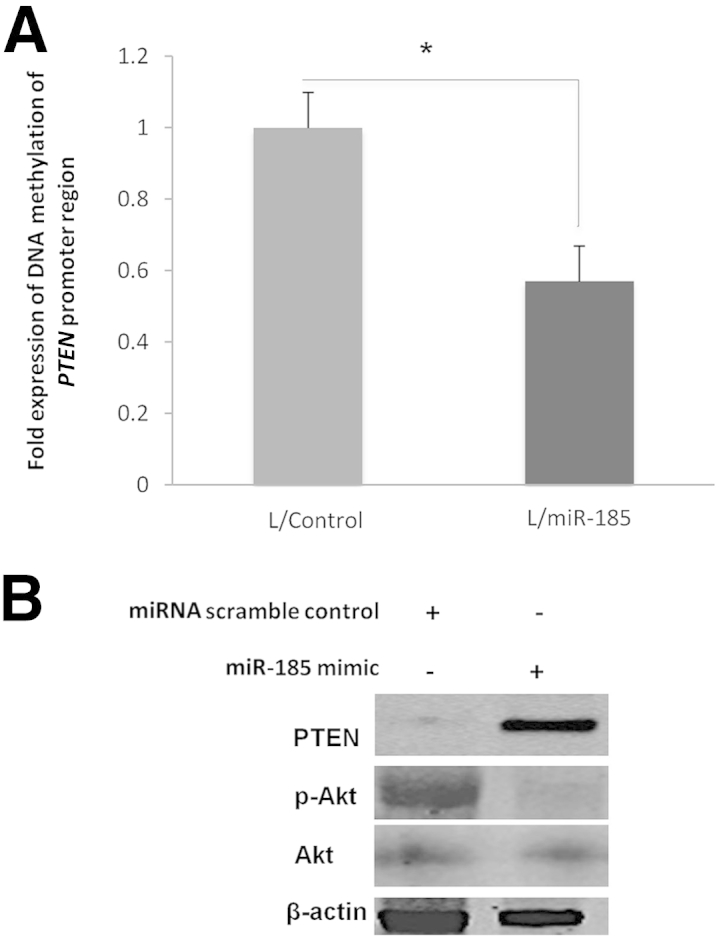
miR-185 decreases PTEN promoter methylation and enhances PTEN expression in Huh7 cells. A: Huh7 cells stably infected with miR-185 or control lentivirus were subjected to methylation-specific PCR assay. miR-185 overexpression decreased methylation in the PTEN CpG island promoter region. B: Huh7 cells were transiently transfected with miR-185 mimic and scramble control oligonucleotide, and the cell lysates were obtained for Western blot analysis to determine the levels of PTEN, p-Akt, and Akt. Data are given as means ± SD. N = 5. ∗P < 0.05.
Because the established epigenetic/global methylation–regulated tumor suppressor genes in patients with HCC include p14ARF, p16INK4A, and SOCS-1,30 we further examined the levels of these proteins in our systems. We observed that the protein levels of p14ARF, p16INK4A, and SOCS-1 were significantly higher in Huh7 cells treated with 5-Azad-CdR compared with cells treated with vehicle control (Supplemental Figure S3). However, lentiviral viral transduction of miR-185 in Huh7 cells did not significantly alter the protein levels of p14, p16, and SOCS-1. Similarly, the protein levels of p14ARF, p16INK4A, and SOCS-1 in miR-185 overexpressed Huh7 xenograft tumors were not significantly different compared with the control tumors. Thus, miR-185 does not seem to influence the levels of p14ARF, p16INK4A, and SOCS-1 in our system, despite that miR-185 targets DNMT1. The explanation for the latter phenomenon is unknown and remains speculative. We cannot exclude the possibility that miR-185 might influence these molecules through indirect mechanisms that are yet to be identified. It is also possible that the observed phenomenon may reflect cell type–specific changes. Nonetheless, the findings in this study support the involvement of DNMT1-regulated PTEN in miR-185–mediated inhibition of HCC cell growth.
miRNA-185 Inhibits HCC Growth in Vivo
To determine the effect of miR-185 on HCC growth in vivo, we inoculated Huh7 cells stably expressing miR-185 or scramble control miRNA into the livers of SCID mice [direct intrahepatic inoculation was achieved by injecting cell suspension mixed with Matrigel solution (BD Biosciences)]; the animals were observed for 30 days to monitor for tumor growth in the liver. The cells overexpressing miR-185 formed smaller tumor nodules in the liver compared with the controls (an approximately 11-fold decrease in tumor volume) (Figure 7A). Immunostaining for the cell proliferation marker Ki-67 showed a reduction of Ki-67–positive cells in miR-185 overexpressed tumors (Figure 7B). Western blot analysis confirmed a reduction of DNMT with up-regulation of PTEN and a decrease of p-Akt in miR-185 overexpressed tumor tissues (Figure 7C). These results demonstrate that miR-185 inhibits HCC growth in vivo.
Figure 7.

miR-185 overexpression inhibits HCC growth in SCID mice. An Huh7 stable cell suspension mixed in Matrigel solution was inoculated into SCID mice via direct liver injections. A: Gross images depict liver tumors that originated from miR-185 overexpressed or control cells. Local liver tumors that originated from miR-185–transduced Huh7 cells are significantly smaller (11-fold decrease) in tumor volume compared with control. B: Ki-67 immunostaining showed a lower proliferative rate in miR-185 overexpressed tumors compared with the control. C: Western blot analysis for DNMT1, PTEN, and p-Akt in miR-185 overexpressed or control tumors recovered from the livers of SCID mice. Normalized fold expressions for DNMT1, PTEN, and p-Akt are depicted in bar graphs. D: Schematic illustration of miR-185–induced inhibition of HCC growth. Data are given as means ± SD. N = 12 (A); N = 5 (C). ∗P < 0.05.
Discussion
This study provides novel evidence that miR-185 is a tumor-suppressive miRNA that inhibits HCC cell growth by targeting DNMT1. This assertion is based on several observations: i) the expression of miR-185 is decreased in human HCC tissues and cell lines compared with the nonneoplastic hepatocytes; ii) forced overexpression of miR-185 in human HCC cells inhibits tumor cell growth in vitro and in SCID mice; iii) miR-185 directly targets DNMT1, a key enzyme that silences the expression of tumor suppressor genes (including PTEN); iv) overexpression of DNMT1 devoid of the 3′-UTR prevents miR-185–induced inhibition of HCC cell proliferation and invasion; v) miR-185 mimic or overexpression reduces PTEN promoter DNA methylation and restores PTEN expression (leading to Akt inhibition), and these effects are partially reversed by forced overexpression of DNMT1; and vi) the effects of miR-185 mimic or overexpression on PTEN in HCC cells are similar to those induced by the DNMT inhibitor 5-Aza-CdR. These findings demonstrate that miR-185 inhibits HCC cell growth by targeting DNMT1, which leads to induction of PTEN and inhibition of Akt (Figure 7D).
Budhu et al17 reported that miR-185 is one of the 20 miRNAs associated with HCC venous metastasis that can predict HCC survival and recurrence. They identified miR-185 as one of the four miRNAs that are up-regulated in HCCs with venous metastasis compared with HCCs without venous metastasis (16 miRNAs were found to be down-regulated in HCCs with venous metastasis compared with HCCs without venous metastasis); however, whether these miRNAs differ between HCC and nontumorous liver tissues was not addressed. Zhi and colleagures18 found that low miR-185 expression in early-stage HCC correlated with more tumor recurrence and worse patient survival compared with the high miR-185 group; the latter study also did not address whether miR-185 differed between HCC and nontumorous liver tissues. Therefore, the role of miR-185 in venous metastasis and in HCC recurrence seems conflicting, with high miR-185 expression found to be associated with venous metastasis but less tumor recurrence and better patient survival. Although it is possible that multiple factors might have contributed to the discrepancy (including different patient populations, different tumor stages, different parameters analyzed, and different study designs), an important pitfall is that in both of the previous studies, the levels of miR-185 in HCC tissues were measured by RT-qPCR, which prevents comparison of their levels in different cell populations in the tumor microenvironment. This is an important issue given that miRNAs in different cell populations in the tumor microenvironment may mediate distinct functions.15,16 In this context, the in situ hybridization technique used in the present study allows direct comparison of miR-185 in HCC and nonneoplastic liver parenchymal tissues. These data indicate that in nonneoplastic liver parenchyma, miR-185 is predominantly expressed in hepatocytes, whereas its expression is significantly reduced in neoplastic hepatocytes. This observation is further corroborated by the RT-qPCR analysis showing decreased miR-185 expression in cultured human HCC cell lines compared with primary human hepatocytes. These results, along with the functional and mechanistic studies described herein, demonstrate that miR-185 is a tumor suppressor miRNA that inhibits HCC growth.
Another important aspect of the present study is the identification of DNMT1 as the direct target of miR-185 in human HCC cells. DNMT1 is the most abundant DNMT in mammalian cells (including hepatocytes) that catalyzes the methylation of cytosine at the 5′ position, forming 5′-methylcytosine; it is responsible for de novo and maintenance methylation of tumor suppressor genes in cancer cells.31,32 The notion that miR-185 directly targets DNMT1 is supported by the observations that i) a putative miR-185 binding site is present in the 3′-UTR of DNMT mRNA; ii) miR-185 mimic or overexpression decreased DNMT1 protein levels in HCC cells; iii) miR-185 overexpression significantly reduced the DNMT1 3′-UTR luciferase reporter activity in HCC cells, and the effect was abolished when the miR-185 binding site was mutated; and iv) restoration of DNMT1 prevented miR-185–induced inhibition of HCC cell proliferation and invasion.
DNMT catalyzes the methylation of cytosine at the CpG islands of tumor suppressor genes (including PTEN).33 Although CpG islands are usually present in an unmethylated state, DNMT enzyme activity leads to conversion to the methylated status, resulting in suppression of gene transcription.34 Accordingly, hypermethylation of tumor suppressor gene promoters has been recognized as an important mechanism in hepatocarcinogenesis.35,36 In the present study, we found that miR-185 mimic or overexpression reduced DNA methylation in the PTEN promoter and enhanced PTEN protein expression, and these effects were reversed by forced overexpression of DNMT1. In addition, we note that the effects of miR-185 mimic or overexpression on PTEN promoter DNA methylation and protein expression in HCC cells are comparable with the effect induced by the DNMT inhibitor 5-Aza-CdR. These findings suggest that miR-185 prevents HCC growth through inhibition of DNMT protein synthesis, thus restoring PTEN expression. Consistent with the documented role of PTEN in the suppression of Akt phosphorylation, we observed that miR-185 mimic or overexpression decreased Akt phosphorylation in HCC cells. These findings are consistent with the well-documented suppression of PTEN and activation of Akt in HCC.25–29
The relationship between epigenetic and miRNA regulation has been increasingly providing a better understanding of the development and progression of human cancers.37,38 Epigenetic modifications include DNA methylation, histone modifications, and RNA-based mechanisms in which miRNAs are critically implicated. In this context, it is noteworthy that the present study provides novel evidence for an important tumor-suppressive role of miR-185 in HCC through targeting DNMT, a key enzyme in cancer epigenetics.
In summary, this study provides novel evidence that miR-185 is a tumor suppressor miRNA that inhibits HCC cell growth by targeting DNMT1 and up-regulating PTEN expression. Therefore, reactivation or induction of miR-185 may represent a novel therapeutic strategy for HCC treatment that warrants further investigation. Furthermore, it remains to be determined whether miR-185 and DNMT can be concomitantly targeted for effective anti-HCC therapy.
Footnotes
Supported by NIH grants CA106280, CA102325, CA134568, and DK077776 (T.W.).
Disclosures: None declared.
Supplemental Data
miR-185 increases PTEN protein expression and reduces Akt phosphorylation in HepG2 cells. HepG2 cells were transiently transfected with miR-185 mimic or scramble control miRNA, and the cell lysates were obtained for Western blot analysis to determine the levels of PTEN, p-Akt, and Akt. The experiments were repeated five times (N = 5).
The effect of 5-Aza-CdR/trichostatin A (TSA) on PTEN promoter methylation and PTEN protein expression. A: Western blot analysis for PTEN, p-Akt, and Akt in Huh7 cells treated with 1 μmol/L 5-Aza-CdR/10 μmol/L TSA and the densitometry data. The experiments were repeated five times (N = 5). B: Methylation-specific PCR assay. Huh7 cells treated with 1 μmol/L 5-Aza-CdR/10 μmol/L TSA showed decreased methylation in the PTEN CpG island promoter region. Data are expressed as means ± SD. ∗P < 0.05.
Western blot analyses for p14ARF, p16INK4A, and SOCS-1 in Huh7 cells. Huh7 cells were treated with or without 1 μmol/L 5-Aza-CdR/10 μmol/L trichostatin A or were transfected with or without miR-185 mimic. The cell lysates were collected and subjected to SDS-PAGE and Western blot analysis for p14ARF, p16INK4A, and SOCS-1. The experiments were repeated five times (N = 5).
References
- 1.Farazi P.A., DePinho R.A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 2.Seeff L.B., Hoofnagle J.H. Epidemiology of hepatocellular carcinoma in areas of low hepatitis B and hepatitis C endemicity. Oncogene. 2006;25:3771–3777. doi: 10.1038/sj.onc.1209560. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag H.B. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 4.Llovet J.M., Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altekruse S.F., McGlynn K.A., Reichman M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forner A., Llovet J.M., Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 7.Yang J.D., Roberts L.R. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villanueva A., Llovet J.M. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valencia-Sanchez M.A., Liu J., Hannon G.J., Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 10.Thorgeirsson S.S., Lee J.S., Grisham J.W. Functional genomics of hepatocellular carcinoma. Hepatology. 2006;43:S145–S150. doi: 10.1002/hep.21063. [DOI] [PubMed] [Google Scholar]
- 11.Aravalli R.N., Steer C.J., Cressman E.N. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–2063. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
- 12.Giordano S., Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57:840–847. doi: 10.1002/hep.26095. [DOI] [PubMed] [Google Scholar]
- 13.Borel F., Konstantinova P., Jansen P.L. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J Hepatol. 2012;56:1371–1383. doi: 10.1016/j.jhep.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Kogure T., Lin W.L., Yan I.K., Braconi C., Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237–1248. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo G., Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542–552. doi: 10.1038/nrgastro.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabo G., Sarnow P., Bala S. MicroRNA silencing and the development of novel therapies for liver disease. J Hepatol. 2012;57:462–466. doi: 10.1016/j.jhep.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Budhu A., Jia H.L., Forgues M., Liu C.G., Goldstein D., Lam A., Zanetti K.A., Ye Q.H., Qin L.X., Croce C.M., Tang Z.Y., Wang X.W. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- 18.Zhi Q., Zhu J., Guo X., He S., Xue X., Zhou J., Hu B., Li H., Chen S., Zhao H., Kuang Y. Metastasis-related miR-185 is a potential prognostic biomarker for hepatocellular carcinoma in early stage. Biomed Pharmacother. 2013;67:393–398. doi: 10.1016/j.biopha.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 19.Yin L., Cai W.J., Liu C.X., Chen Y.Z., Hu J.M., Jiang J.F., Li H.A., Cui X.B., Chang X.Y., Zhang W.J., Sun K., Li F. Analysis of PTEN methylation patterns in soft tissue sarcomas by MassARRAY spectrometry. PLoS One. 2013;8:e62971. doi: 10.1371/journal.pone.0062971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen T., Hevi S., Gay F., Tsujimoto N., He T., Zhang B., Ueda Y., Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 21.Park I.Y., Sohn B.H., Yu E., Suh D.J., Chung Y.H., Lee J.H., Surzycki S.J., Lee Y.I. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology. 2007;132:1476–1494. doi: 10.1053/j.gastro.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 22.You H., Ding W., Rountree C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology. 2010;51:1635–1644. doi: 10.1002/hep.23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hino R., Uozaki H., Murakami N., Ushiku T., Shinozaki A., Ishikawa S., Morikawa T., Nakaya T., Sakatani T., Takada K., Fukayama M. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009;69:2766–2774. doi: 10.1158/0008-5472.CAN-08-3070. [DOI] [PubMed] [Google Scholar]
- 24.Phuong N.T., Kim S.K., Lim S.C., Kim H.S., Kim T.H., Lee K.Y., Ahn S.G., Yoon J.H., Kang K.W. Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Res Treat. 2011;130:73–83. doi: 10.1007/s10549-010-1304-2. [DOI] [PubMed] [Google Scholar]
- 25.Peyrou M., Bourgoin L., Foti M. PTEN in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig Dis. 2010;28:236–246. doi: 10.1159/000282095. [DOI] [PubMed] [Google Scholar]
- 26.Sze K.M., Wong K.L., Chu G.K., Lee J.M., Yau T.O., Ng I.O. Loss of phosphatase and tensin homolog enhances cell invasion and migration through AKT/Sp-1 transcription factor/matrix metalloproteinase 2 activation in hepatocellular carcinoma and has clinicopathologic significance. Hepatology. 2011;53:1558–1569. doi: 10.1002/hep.24232. [DOI] [PubMed] [Google Scholar]
- 27.Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S.T., Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian T., Nan K.J., Guo H., Wang W.J., Ruan Z.P., Wang S.H., Liang X., Lu C.X. PTEN inhibits the migration and invasion of HepG2 cells by coordinately decreasing MMP expression via the PI3K/Akt pathway. Oncol Rep. 2010;23:1593–1600. doi: 10.3892/or_00000800. [DOI] [PubMed] [Google Scholar]
- 29.Chen J.S., Wang Q., Fu X.H., Huang X.H., Chen X.L., Cao L.Q., Chen L.Z., Tan H.X., Li W., Bi J., Zhang L.J. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP-9. Hepatol Res. 2009;39:177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 30.Herath N.I., Leggett B.A., MacDonald G.A. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006;21:15–21. doi: 10.1111/j.1440-1746.2005.04043.x. [DOI] [PubMed] [Google Scholar]
- 31.Gravina G.L., Festuccia C., Marampon F., Popov V.M., Pestell R.G., Zani B.M., Tombolini V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol Cancer. 2010;9:305. doi: 10.1186/1476-4598-9-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., Lay F., Han H., Jones P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–546. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanai Y., Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28:2434–2442. doi: 10.1093/carcin/bgm206. [DOI] [PubMed] [Google Scholar]
- 34.Deaton A.M., Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schagdarsurengin U., Wilkens L., Steinemann D., Flemming P., Kreipe H.H., Pfeifer G.P., Schlegelberger B., Dammann R. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene. 2003;22:1866–1871. doi: 10.1038/sj.onc.1206338. [DOI] [PubMed] [Google Scholar]
- 36.Yu J., Ni M., Xu J., Zhang H., Gao B., Gu J., Chen J., Zhang L., Wu M., Zhen S., Zhu J. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer. 2002;2:29. doi: 10.1186/1471-2407-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berdasco M., Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Fabbri M., Calin G.A. Epigenetics and miRNAs in human cancer. Adv Genet. 2010;70:87–99. doi: 10.1016/B978-0-12-380866-0.60004-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miR-185 increases PTEN protein expression and reduces Akt phosphorylation in HepG2 cells. HepG2 cells were transiently transfected with miR-185 mimic or scramble control miRNA, and the cell lysates were obtained for Western blot analysis to determine the levels of PTEN, p-Akt, and Akt. The experiments were repeated five times (N = 5).
The effect of 5-Aza-CdR/trichostatin A (TSA) on PTEN promoter methylation and PTEN protein expression. A: Western blot analysis for PTEN, p-Akt, and Akt in Huh7 cells treated with 1 μmol/L 5-Aza-CdR/10 μmol/L TSA and the densitometry data. The experiments were repeated five times (N = 5). B: Methylation-specific PCR assay. Huh7 cells treated with 1 μmol/L 5-Aza-CdR/10 μmol/L TSA showed decreased methylation in the PTEN CpG island promoter region. Data are expressed as means ± SD. ∗P < 0.05.
Western blot analyses for p14ARF, p16INK4A, and SOCS-1 in Huh7 cells. Huh7 cells were treated with or without 1 μmol/L 5-Aza-CdR/10 μmol/L trichostatin A or were transfected with or without miR-185 mimic. The cell lysates were collected and subjected to SDS-PAGE and Western blot analysis for p14ARF, p16INK4A, and SOCS-1. The experiments were repeated five times (N = 5).