

Abstract
We have previously reported that members of the NR4A family of orphan nuclear receptors can augment insulin’s ability to stimulate glucose transport in adipocytes. In the current study, we endeavored to test for an insulin-sensitizing effect in muscle cells and to identify a potential transactivator. Lentiviral constructs were used to engineer both hyperexpression and shRNA silencing of NR4A3 in C2C12 myocytes. The NR4A3 hyper-expression construct led to a significant increase in glucose transport rates in the presence of maximal insulin while the NR4A3 knock-down exhibited a significant reduction in insulin-stimulated glucose transport rates. Consistently, insulin-mediated AKT phosphorylation was increased by NR4A3 hyperexpression and decreased following shRNA NR4A3 suppression. Then, we examined effects of prostaglandin A2 (PGA2) on insulin action and NR4A3 transactivation. PGA2 augmented insulin-stimulated glucose uptake in C2C12 myocytes and AKT phosphorylation after 12-h treatment, without significant effects on basal transport or basal AKT phosphorylation. More importantly, we demonstrated that PGA2 led to a greater improvement in insulin-stimulated glucose rates in NR4A3 overexpressing C2C12 myocytes, when compared with Lac-Z controls stimulated with insulin and PGA2. Moreover, the sensitizing effect of PGA2 was significantly diminished in NR4A3 knockdown myocytes compared to scramble controls. These results show for the first time that: (i) PGA2 augments insulin action in myocytes as manifested by enhanced stimulation of glucose transport and AKT phosphorylation; and (ii) the insulin sensitizing effect is dependent upon the orphan nuclear receptor NR4A3.
Keywords: NR4A3, MINOR, insulin sensitivity, prostaglandin A2, glucose transport, skeletal muscle, C2C12 cells
Introduction
For the past several decades, the global prevalence of Type 2 Diabetes Mellitus (T2DM) and the Metabolic Syndrome has been increasing. Insulin resistance is central to the pathogenesis of these diseases, and involves impaired insulin action in skeletal muscle. Even so, the mechanisms causing insulin resistance have not been fully elucidated, and optimal strategies for pharmacological intervention have not been developed.
Recently, there has been a growing interest in NR4A orphan nuclear receptors as potential targets for metabolic diseases. There are 3 members in NR4A family: NR4A1 (TR3, NGFI-B, N10, DHR38, NAK-1, TIS1 or Nur77), NR4A2 (HZF-3, RNR-3, TINUR, NOT or Nurr1), and NR4A3 (NOR-1, TEC, CHN or MINOR). NR4A family members differ from other nuclear receptors in that they lack the classic hydrophobic cleft for recruitment of coactivators and corepressors on their C-terminal ligand-binding domain [1]. They bind the NGFI-B response element (NBRE) and putatively activate gene expression in a ligand-independent manner [2]. NR4As are immediate early stress response genes that can be induced by a wide range of physiological signals such as inflammatory cytokines [3], prostaglandins [4], and growth factors [5]. This subgroup has also been shown to be involved in various pathological conditions, including Parkinson’s disease [6], inflammation [7], atherosclerosis [8] and cancer [9]. Furthermore, all 3 NR4A members are inducible by chronic caloric restriction in rat liver and skeletal muscle [10]. This finding is particularly interesting since both chronic and intermittent caloric restriction have been shown to enhance insulin sensitivity in numerous species [11]. Finally, NR4A1 null mice exhibit insulin resistance in skeletal muscle and liver together with an impaired fat metabolism [12].
Our laboratory has reported that NR4A3, a member of NR4A orphan nuclear receptor family, can enhance insulin sensitivity and glucose transport stimulation in 3T3-L1 adipocytes, and is downregulated in insulin resistant or diabetic rodent models [13]. While these results suggest a potential role for NR4A3 as a therapeutic target for modulation of insulin action, it will be important to demonstrate that NR4A3 enhances insulin sensitivity in skeletal muscle, which is responsible for the bulk of insulin-mediated glucose uptake in human metabolism. It would also be imperative to show that a small molecular agonist of the orphan receptor could produce an increase in insulin action.
In our current study, we have examined whether alterations in NR4A3 expression affect insulin sensitivity in C2C12 muscle cells. Furthermore, we have studied prostaglandin A2 (PGA2) as a potential transactivator of NR4A3 in muscle cells following the report by Kagaya et al. [4] showing that PGA2 enhances NR4A3-dependent transcriptional activity. PGA2 belongs to the cyclopentanone prostaglandin family, which was thought to exert potent and specific regulatory effects on protein activities, as extensively reviewed by Strauss et al. [14]. This transactivation was largely dependent on a direct physical interaction between PGA2 and the ligand binding domain of NR4A3. We have shown for the first time that PGA2 exerts an insulin-sensitizing effect, and that this action is dependent upon NR4A3. These results are consistent with our previous report in adipocytes [13], and confirmed the importance of NR4A3 in the regulation of insulin action in skeletal muscle cells. Further, our findings highlight the potential role of PGA2 or other small molecule agonists of NR4A3 as therapeutic modulators of insulin action.
Materials and Methods
Reagents
Mouse C2C12 myoblast cells were purchased from American Type Culture Collection (Manassas, VA, USA). Tissue culture media were products of Invitrogen. NR4A3 antibody (Catalogue No: PP-H7833-00) was purchased from R & D Systems (MN, USA), and actin antibody (Catalog No: sc-130065) was from Santa Cruz (CA, USA), 2-deoxy-D-[3H]- and L-[1-3H]glucose were purchased from Amersham Biosciences. All other reagents were purchased from Sigma, unless specified. Unless specified, all other antibodies were purchased from Cell Signaling.
Cell culture and stimulation
The C2C12 are maintained as growing myoblasts in Dulbecco’s minimal essential medium (DMEM, Mediatech) containing 1 g/l glucose, and 10 % fetal bovine serum (GIBCO). Mass cultures reached 70–80 % confluence within 48 h. Differentiation of C2C12 myoblasts was induced by reducing the serum concentration (2 % horse serum, Hy-Clone). The cultures were supplied with 2 % horse serum DMEM daily thereafter, and then serum-starved overnight preceding experiments.
Recombinant lentiviruses and lentiviral-transduced cell lines
Lentiviral-transduced NR4A3 overexpression and shRNA construct cloning procedures have been described in our previous report [13]. Briefly, the full-length human NR4A3 cDNA coding sequence and a V5 epitope tag were cloned into a Vira-Power-CMV vector (Invitrogen). The NR4A3 construct and a control LacZ gene plasmid were transfected into HEK293 cells. Western blots were performed to confirm successful transfection, and infectious virus particles were produced according to the manufacturer’s protocol (Invitrogen). To establish stable C2C12 myocytes that overexpress NR4A3 or LacZ genes, recombinant NR4A3 or LacZ lentiviral stocks were used to infect C2C12 myoblasts with Polybrene (Specialty Media, Phillipsburg, NJ, USA) at a final concentration of 6μg/ml. Seventy two h after transfection, cells were placed under blasticidin selection (30μg/ml) for 20 days. Western blot analyses were performed to test for stable NR4A3 or LacZ gene expression after antibiotic selection.
Lentiviral based endogenous NR4A3 gene hypoexpression
Three shRNA hairpin oligonucleotides (sense 5′-CAC CGC TGT TTG TCC TCA GAC TTT CCG AAG AAA GTC TGA GGA CAA ACA GC-3′, antisense 5′-AAA AGC TGT TTG TCC TCA GAC TTT CTT CGG AAA GTC TGA GGA CAA ACA GC-3′; sense 5′-CAC CGC TGA GCA TGT GCA ACA ATT CCG AAG AAT TGT TGC ACA TGC TCA GC-3′, antisense 5′-AAA AGC TGA GCA TGT GCA ACA ATT CTT CGG AAT TGT TGC ACA TGC TCA GC-3′; sense 5′-CAC CGC TCT TGT CCG AGC TTT AAC ACG AAT GTT AAA GCT CGG ACA AGA GC-3′, antisense 5′-AAA AGC TCT TGT CCG AGC TTT AAC ATT CGT GTT AAA GCT CGG ACA AGA GC-3′) that are complementary to the mouse NR4A3 gene coding sequences were synthesized by Integrated DNA Technologies (Coralville, IA, USA), and subcloned into the pENTR™/U6 lentiviral vectors (Invitrogen) to create the NR4A3 shRNA constructs following the manufacturer’s instructions. Three specific target sequences in the gene coding region for the knockdown experiments were 5′-GCT GTT TGT CCT CAG ACT TTC-3′, 5′-GCT GAG CAT GTG CAA CAA TTC-3′, and 5′-GCT CTT GTC CGA GCT TTA ACA-3′ for the 3 synthesized short oligonucleotides. The recombinant shRNA-NR4A3 lentiviral plasmid or control scramble construct was transfected into HEK293 cells to generate lentiviruses. Thereafter, shRNA lentiviruses were transfected into C2C12 myoblasts to generate cell lines exhibiting NR4A3 suppression in parallel with the scramble control cell lines. Stable knockdown cell lines were selected under the same conditions as the NR4A3 overexpression cell lines described above.
Westernblot analyses
Myocytes were treated with cell lysis buffer (1 × phosphate-buffered saline, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS) containing freshly added protease inhibitor mixture (Sigma). Fifty micrograms of protein per lane and known molecular weight markers from Bio-Rad were separated by SDS-polyacrylamide gel electrophoresis. Proteins were electrophoretically transferred onto nitrocellulose membranes and incubated overnight at 4 °C with blocking solution (5 % nonfat milk in Tris-buffered saline). The blocked membranes were incubated with Akt or phospho-Akt antibodies (Zee Biologic) and then peroxidase-conjugated second antibody (1:1 000 or 1:5 000 dilution with 1 % nonfat milk in Tris-buffered saline (TBS)) for 1 h for each at room temperature and washed 3 times each with TBS buffer containing 0.1 % Tween 20 for 15 min at room temperature with rocking. Immunodetection analyses were performed using the Enhance Chemiluminescence Kit (PerkinElmer Life Sciences). Typical data were shown after similar results were obtained in 3 or more independent experiments.
Modified glucose uptake assay
Glucose transport was assayed in monolayers as initial rates of 2-deoxy-[3H]glucose uptake as we have described previously [15]. Modification was made in order to enhance insulin response in C2C12 myotubes as described [16]. Fully differentiated C2C12 myotubes were starved using low glucose (1 g/l), serum-free DMEM for 4 h. The cells were then washed 3 times with glucose-free KRPH buffer (20 mM HEPES, 120 mM NaCl, 1.2 mM MgSO4, 2 mM CaCl2, 2.5 mM KCl, 1 mM NaH2PO4, and 1 mM sodium pyruvate) and stimulated with or without 100 nM insulin. Fifteen minutes following insulin stimulation, glucose was added to KRPH buffer to a final concentration of 25 mM (4.5 g/l, equivalent to “High Glucose Culture Medium” concentration). Cells were allowed to incubate for additional 45 min. Glucose transport was determined by the incubation of 2-deoxy-[3H]glucose for 3 min (0.1 mM, 0.2μCi/ml; PerkinElmer Life and Analytical Science). Cells were then placed on ice and washed 3 times with ice-cold KRPH buffer to remove any 2-deoxy-[3H] glucose remaining in the buffer. Finally, 2-deoxy-[3H]glucose uptake was assessed by counting β-emissions from the cells.
Statistics
Experimental results are shown as the mean ± S.D. Statistical analyses were performed by unpaired Students t -test or 2-way ANOVA assuming unequal variance unless otherwise indicated. Significance was defined as *p < 0.05, or **p < 0.01.
Results
Our group has shown that NR4A3 is induced during differentiation of 3T3-L1 adipocytes [13]. In C2C12 myocytes, we now similarly observed that NR4A3 expression is induced during differentiation (Fig. 1). The increments in NR4A3 protein expression accompanied both the histological evidence of muscle cell differentiation (Fig. S1A, S1B) and the induction of myosin heavy chain protein (Fig. S1C). In order to elucidate the role of NR4A3 in insulin action in C2C12 myocytes, we made lentiviral constructs for both NR4A3 stable hyperexpression and knockdown, together with transduced LacZ and scramble control cells. The hyperexpression construct results in approximately doubled NR4A3 protein expression compared to LacZ control (Fig. 2A, Fig. S2A), and the knockdown construct decreases NR4A3 protein by approximately 75% compared to scramble control (Fig. 2B, Fig. S2B, S2C). It is important to note that NR4A1 and NR4A2 protein expression was not changed by NR4A3 overexpression or knockdown (Fig. S2D). In addition, our initial observation suggested that the extent and temporal progression of muscle cell differentiation were not affected by lentivirus-mediated NR4A3 hyperexpression, nor by shRNA-mediated NR4A3 repression and this result was further confirmed with unchanged myosin heavy chain expression or cell morphology upon NR4A3 overexpression or knockdown (Fig. S2E, S2F). Thus, NR4A3 does not appear to influence differentiation from a histological or biochemical perspective.
Fig. 1.
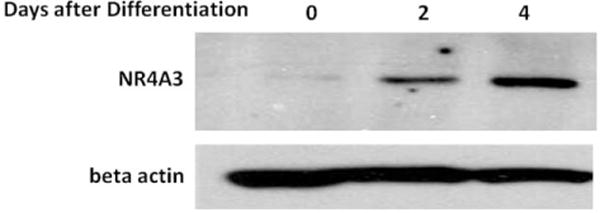
NR4A3 Protein is induced during differentiation of C2C12 cells. Wild type C2C12 myoblasts were allowed to differentiate as described in Materials and Methods. A sequential increase of NR4A3 protein expression was observed after 2 days and 4 days of differentiation. The results are representatives of 3 independent experiments.
Fig. 2.
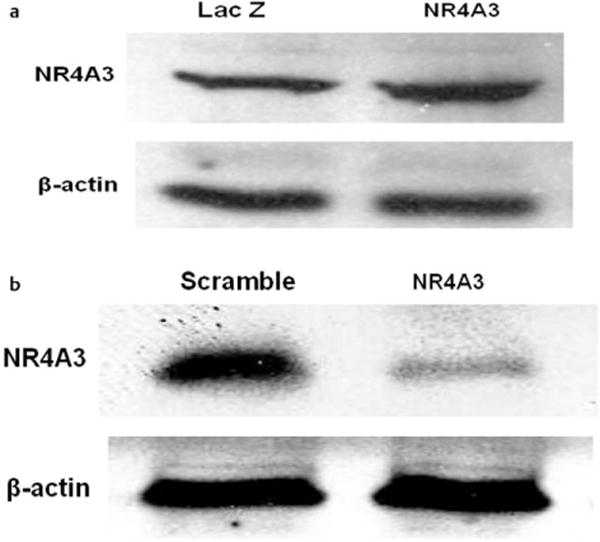
Characterization of lentiviral transduced cell lines. a Lentiviral-mediated NR4A3 overexpression in C2C12 myocytes. A one-fold increase of NR4A3 protein expression was observed in NR4A3 overexpression cell line compared to LacZ control cell line. b shRNA mediated silencing of NR4A3 in C2C12 myocytes. A 75 % decrease of NR4A3 protein expression was observed in NR4A3 knockdown cell line compared to scramble control cell line.
Even so, our data did indicate that NR4A3 was involved in achieving a fully insulin-responsive functional phenotype in myocytes as we had previously demonstrated in adipocytes [13]. As a consequence of NR4A3 hyperexpression, we observed a significant elevation in insulin-stimulated glucose transport in C2C12 myocytes compared to LacZ controls in a concentration dependent manner (Fig. 3A, S3A), while no significant effects on basal glucose transport were observed. We also found that NR4A3 hyperexpression significantly enhanced insulin-mediated Akt/PKB ser473 phosphorylation (Figs. 3b, c, S3B, S3C), without affecting GSK-3β ser9 phosphorylation (Fig. S3D, S3E). Conversely, NR4A3 knockdown led to a state of relative insulin resistance with a significant decline in insulin’s ability to stimulate glucose transport activity (Fig. 4a, S4A) and Akt/PKB phosphorylation (Fig. 4b, c, S4B, S4C). Again, no significant basal glucose transport change was observed after NR4A3 silencing.
Fig. 3.
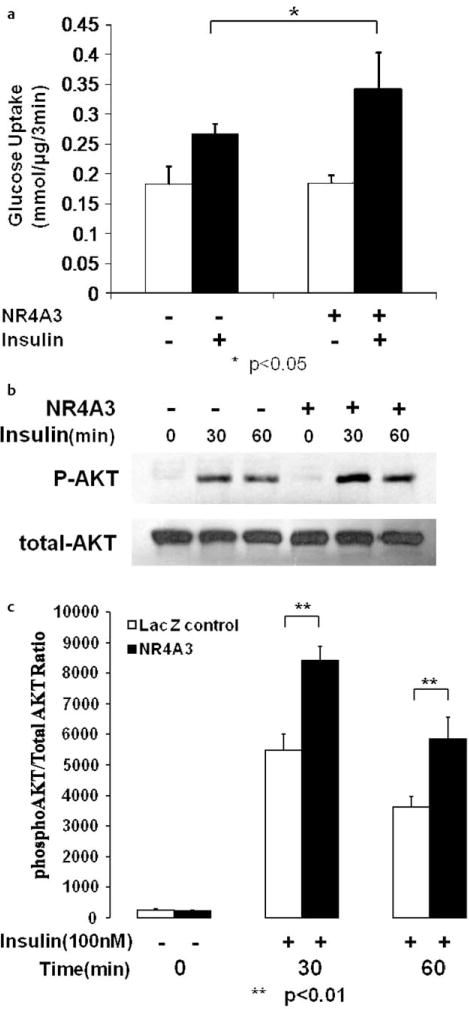
NR4A3 hyperexpression augments insulin sensitivity in C2C12 myocytes. a NR4A3 hyperexpression enhances insulin stimulated glucose transport. Glucose transport was measured as 3H radioactivity intake in the initial 3 min as described in Materials and Methods. A 30 % (p < 0.05) increase in glucose transport rates in the presence of 100 nM insulin was observed without significant effects on basal transport, when compared to control cells. The results were obtained from 4 independent experiments. b NR4A3 hyperexpression enhances insulin stimulated AKT Ser473 phosphorylation in C2C12 myocytes. A typical Western blot data was shown after similar results were obtained in 3 independent experiments. c The quantification of insulin stimulated AKT phosphorylation enhancement after NR4A3 overexpression. The results were obtained from 3 independent experiments.
Fig. 4.
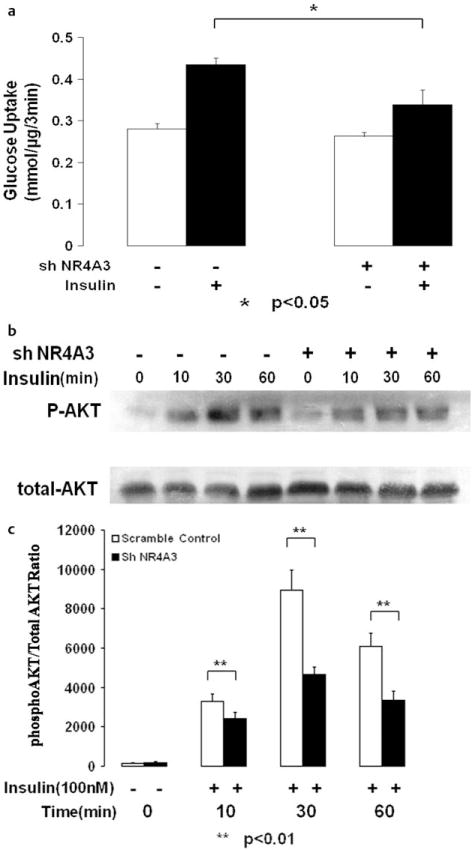
NR4A3 silencing reduces insulin sensitivity in C2C12 myocytes. a NR4A3 knockdown suppresses insulin stimulated glucose transport activity. A 35 % (p < 0.05) decrease in glucose transport rates in the presence of 100 nM insulin was observed without significant effects on basal transport, when compared to control cells. The results were obtained from 3 independent experiments. b NR4A3 knockdown reduces insulin mediated AKT phosphorylation. C2C12 myoblasts were allowed to differentiate for 5–6 days before cellular protein was harvested. A typical blot data was shown after similar results were obtained in 3 independent experiments. c The quantification of insulin stimulated AKT phosphorylation impairment after NR4A3 knockdown. The results were obtained from 3 independent experiments.
A previous report indicated that PGA2 is able to bind NR4A3 and activate NR4A3-dependent transcription [4]. Based on this discovery and the fact that NR4A3 regulates insulin action and downstream signaling, we hypothesized that PGA2 and other cyclopentenone prostaglandins would also increase insulin sensitivity through NR4A3 agonism. PGA2 did significantly enhance insulin sensitivity without altering basal glucose uptake in wild-type C2C12 myocytes (Fig. 5a, p < 0.01). We also observed increased Akt/PKB ser473 phosphorylation (Fig. 5b, c) when cells were incubated in the presence of both insulin and PGA2, with no changes in GSK3β phosphorylation (Fig. S4D, S4E). However, GLUT4 protein expression seemed to be unchanged after NR4A3 hyperexpression, knockdown or PGA2 treatment, which indicates that GLUT4 expression change was not involved in NR4A3- or PGA2 -mediated insulin sensitization (Fig. S5A, S5B). Interestingly, the insulin sensitization effect of PGA2 could not be attributed to decreased insulin EC50. Our data suggested that PGA2 enhances maximal insulin action (Fig. 5d). Additional data indicated that the insulin sensitization effect of PGA2 is both time and concentration related, with a 12-h incubation in 10 μM PGA2 producing the maximal increase in insulin-stimulated glucose transport (Fig. 6a, b, respectively).
Fig. 5.
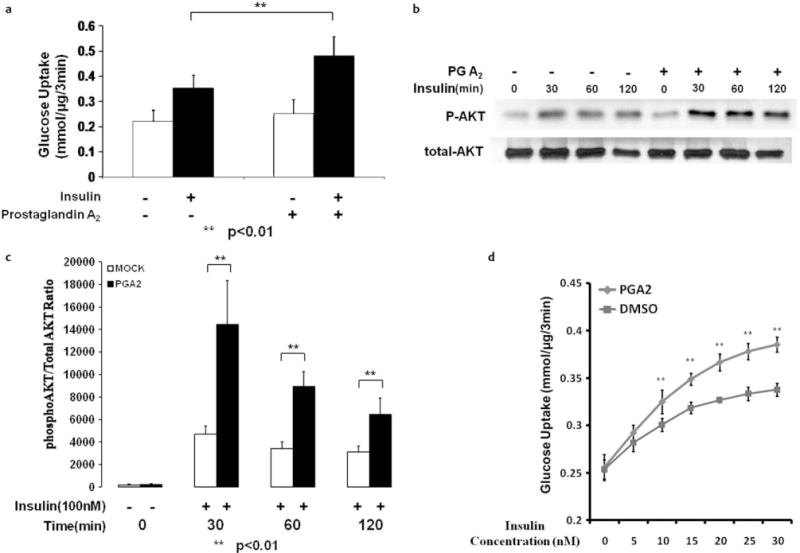
PGA2 augments insulin sensitivity in C2C12 myocytes. a 10 μM PGA2 12-h treatment significantly enhances insulin stimulated glucose transport. A 30 % (p < 0.05) increase in glucose transport rates in the presence of 100 nM insulin was observed without significant effects on basal transport, when compared to control cells. The results were obtained from 3 independent experiments. b 10 μM PGA2 12 h treatment enhances insulin stimulated AKT Ser473 phosphorylation. A typical blot data was shown after similar results were obtained in 3 independent experiments. c The quantification of insulin stimulated AKT phosphorylation enhancement after PGA2 treatment. The results were obtained from 3 independent experiments. d 10 μM PGA2 12-h treatment did not change insulin EC50 but increases maximal insulin stimulated glucose transport. PGA2 treatment significantly enhances insulin (with concentrations > 5 nM) mediated glucose transport activity in C2C12 cells at different insulin concentrations. The results were obtained from 3 independent experiments (**p < 0.01 at each insulin concentration, PGA2 treatment vs. DMSO control).
Fig. 6.
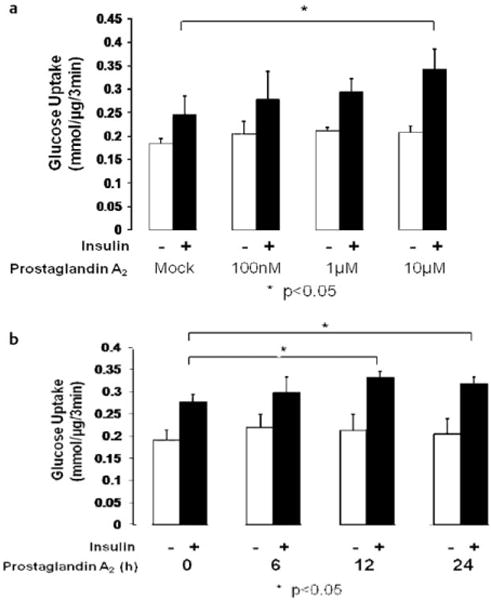
PGA2 enhances insulin action in C2C12 myocytes in a concentration and time related manner. The results were obtained from 3 independent experiments. a PGA2 treatment enhances insulin stimulated glucose transport in a concentration related manner. 10 μM treatment significantly enhances insulin stimulated glucose transport (30 %, p < 0.05) without a significant impact on basal glucose transport. b 10 μM PGA2 treatment of different time length enhances insulin-mediated glucose transport. 12-h treatment enhances insulin stimulated glucose transport by 20 % (p < 0.05) and 24-h treatment by 15 % (p < 0.05).
In wild-type C2C12 cells, we did not observe any effects of PGE2 or PGA2-isoprostanes on insulin-stimulated glucose transport (data not shown). The PGA2 metabolite 15-deoxy-12,14-PGA2 increased mean insulin-stimulated glucose transport, but this effect did not reach statistical significance (data not shown).
We hypothesized that the insulin-sensitizing effect of PGA2 was mediated via NR4A3 in muscle cells. To address this question, NR4A3 knockdown and scramble control C2C12 myocyte lines were pretreated with or without PGA2, and then acutely stimulated by insulin. As expected, NR4A3 knockdown not only reduced insulin-mediated glucose transport but also abrogated the insulin sensitizing effect of PGA2 (Fig. 7a) Likewise, NR4A3 hyperexpression significantly augmented both insulin-mediated glucose transport in wild type cells and the effect of with PGA2 to improve insulin sensitivity (Fig. 7b). Taken together, we were able to conclude that NR4A3 is required for the insulin sensitizing effect of PGA2.
Fig. 7.
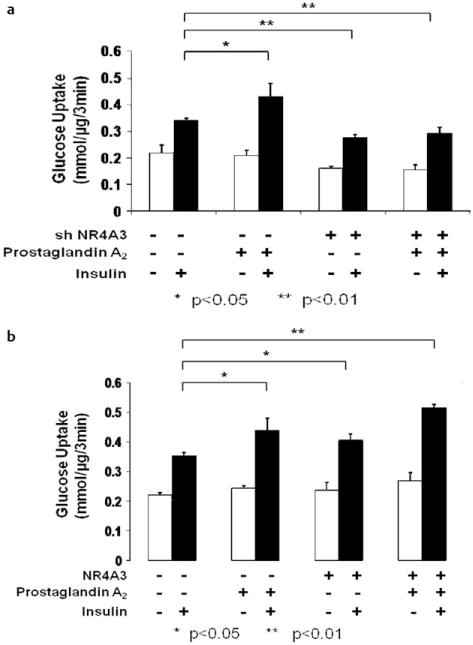
The insulin sensitization effect of PGA2 is dependent on the full presence of NR4A3. C2C12 myoblasts differentiation was induced and glucose transport was measured as previously described. The results were obtained from 3 independent experiments. a PGA2 (10 μM, 12 h) enhances insulin stimulated glucose transport by 30 % (p < 0.05) in control cells but not in NR4A3 knockdown cell lines (p > 0.05). b PGA2 (10 μM, 12 h) and NR4A3 hyperexpression enhances insulin stimulated glucose transport additively. PGA2 (10 μM, 12 h) treatment enhances insulin stimulated glucose transport by 28% (p < 0.05) and NR4A3 hyperexpression enhances by 17 % (p < 0.05). PGA2 treatment and NR4A3 hyperexpression together enhances insulin stimulated glucose transport by 42 % (p < 0.05), which is significantly higher than PGA2 treatment alone (p < 0.05) and NR4A3 hyperexpression alone (p < 0.05).
Discussion
NR4A nuclear receptors appear to exert diverse influences on cell physiology. In cultured adipocytes, we had previously shown that NR4A3 overexpression increased insulin-stimulated glucose transport rates by increasing GLUT4 translocation, with no change in total cellular GLUT4 content. The data presented in this report extend our previous findings to skeletal muscle. As observed in adipocytes, NR4A3 overexpression significantly enhances insulin-stimulated glucose transport in C2C12 muscle cells, while shRNA suppression of NR4A3 reduces insulin-stimulated glucose transport.
NR4A family members can be functionally redundant in specific instances. NR4A3 and NR4A1 have been implicated in metabolic regulation in liver, and adenoviral overexpression of NR4A3 or NR4A1 in primary mouse hepatocytes increases expression of both fructose bisphosphatase-2 (a.k.a. phosphofructokinase 2) and enolase-3 and increases pyruvate-derived glucose production [17]. With respect to metabolism in skeletal muscle, NR4A3 has not been as well studied; however, when the current data are examined in light of previous publications concerning NR4A1, it is clear that both NR4A family members can overlap in their metabolic effects. Importantly, overexpression of NR4A1 in C2C12 cells enhances basal glucose transport, with no change in GLUT1 expression [18]. It is also interesting to note that overexpression of NR4A1 increases expression of GLUT4, phosphofructokinase, and glycogen phosphorylase in C2C12 cells and in rat muscle [18]. Furthermore, NR4A1 null mice are no different from wild-type mice when fed with standard diet, but are more susceptible to metabolic dysfunction when maintained on high fat diet; thus, when fed with high fat diet, NR4A1−/− mice become more obese and insulin resistant, with impaired insulin signaling in skeletal muscle driving these changes. Interestingly, NR4A1 knockout mice also displayed increased intramyocellular lipid content and hepatic steatosis, but maintained normal hepatic insulin sensitivity [12]. These data, along with ours, suggest that NR4A3 and NR4A1 modulate cell-type specific metabolic functions, and enhance metabolic functions and insulin actions in muscle. The current effects on insulin action occurred as a result of specific modulation of NR4A3 expression, without changes in NR4A1, indicating that the induced hyperexpression and suppression of NR4A3 predominate in the modulation of insulin sensitivity under these experimental conditions.
Cyclopentenone prostaglandins are known to modulate the transcriptional activity of other nuclear receptors, including PPAR-γ, NF-κB, AP-1, Nrf2, HIF1-α [19], and estrogen receptor-α [20]. These prostaglandins include PGA2, PGA1, and PGJ2, and are formed by dehydration of the cyclopentane rings of PGE2, PGE1, and PGD2, respectively, resulting in an unsaturated carbonyl group that is electrophilic and reactive [14,21]. Unlike receptor-binding prostaglandins, cyclopentenone prostaglandins covalently bind specific target proteins, and these adducts result from covalent interaction between protein cysteine thiols and the α,β-unsaturated carbonyl moiety in the cyclopentenones [20]. The reaction between the electrophilic carbons and their target proteins is nonpromiscuous, highly specific, and can alter protein activities [14,21–24]. Because the ligand-binding domain of NR4A proteins lacks a hydrophobic cleft, it was thought that these receptors may function constitutively in the absence of ligand. However, Kagaya et al. have identified PGA2 as a specific transactivator of NR4A3 [4]. This group showed that PGA2 activated NR4A3-dependent transcription, and that this PGA2 action was dependent upon interaction with the NR4A3 ligand binding domain [4]. Based on these results, we hypothesized that PGA2 would improve skeletal muscle insulin sensitivity through transactivation of NR4A3.
The improvement in insulin action due to the interaction between a prostanoid (PGA2) and a nuclear transcription factor (NR4A3) is analogous to the effects of PGJ2 metabolites interacting with PPARγ. PGJ2 was discovered as a dehydration product of PGD2 in studies assessing their antineoplastic properties [25], and can undergo albumin-assisted catalysis to A12-PGJ2 and 15-deoxy-Δ12,14-PGJ2 [26]. In 1995, 15-deoxy-Δ12,14-PGJ2 was found to enhance insulin action as a high affinity ligand and agonist for PPARγ [27,28]. In light of the current results, it is important to note that PGA2 is not known to interact with PPARγ [14]. Kagaya et al. have comprehensively screened a large number of naturally occurring arachidonic acid and glycerolipid metabolites from the KEGG database, and found that only PGA2 and PGA1 have the unique ability to activate NR4A3-dependent transcription using a GAL4-based reporter system [4]. Furthermore, using mutational analyses and a Biacore resonance sensor, they demonstrated that PGA2 action was dependent upon binding of PGA2 to the ligand binding domain of NR4A3. Even so, the possible biochemical mechanism by which PGA2 interacts with NR4A3 in muscle cells remains unclear even though our data indicate that NR4A3 is necessary for the insulin-sensitizing effect of PGA2.
We should admit that it still remains unclear how NR4A3 and PGA2 treatment augment insulin action in C2C12 myocyte culture system. Our previous data from 3T3-L1 adipocytes suggested that GLUT4 translocation but not expression may be involved in NR4A3 mediated insulin sensitivity augment in 3T3-L1 cells. However, in C2C12 myocytes it may be different since Tortorella et al. reported that C2C12 myocytes lacks GLUT4 translocation machinery which exists in adipose tissue [29]. Further studies showed that GLUT4 translocation may not play a major role in insulin action in C2C12 myocytes and a modified glucose uptake assay protocol should be used in this system [16]. These observations, together with our data, suggested that an underlying mechanism other than GLUT4 translocation may be involved in NR4A3- and PGA2 -mediated insulin sensitization in C2C12 myocytes.
In the future animal models such as skeletal muscle-specific NR4A3 knockout and transgenic mice would likely provide valuable insight regarding the in vivo significance of NR4A3 as a modulator of glucose metabolism and insulin sensitivity.
In closing, our results demonstrate that NR4A3 mediates an increase in insulin-stimulated glucose transport and AKT phosphorylation in muscle cells. It seems to be an attractive pharmacological target for the modulation of insulin sensitivity, and diseases characterized by insulin resistance such as Type 2 Diabetes and the Metabolic Syndrome. We have also shown for the first time that PGA2 enhances insulin sensitivity, and this occurs via a mechanism involving NR4A3. This provides proof-of-principle that a small molecule mediator can have an insulin-sensitizing effect through an interaction with NR4A3.
Supplementary Material
Acknowledgments
This work was supported from grants from the National Institutes of Health (DK-083562, DK-038764, HL-055782 to W.T.G.), the Merit Review program of the Department of Veterans Affairs (W.T.G.), and the American Diabetes Association (Y.F.). We gratefully acknowledge the support of the research core facilities of the UAB Diabetes Research and Training Center (P60-DK079626) and the UAB Nutrition and Obesity Research Center (P30-DK56336).
Footnotes
Supporting Information available online at http:/www.thieme-connect.de/ejournals/toc/hmr
Conflict of Interest
The authors have no conflict of interest.
References
- 1.Wilson TE, Fahrner TJ, Johnston M, Milbrand J. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science. 1991;252:1296–1300. doi: 10.1126/science.1925541. [DOI] [PubMed] [Google Scholar]
- 2.Davis IJ, Lau LF. Endocrine and neurogenic regulation of the orphan nuclear receptors Nur77 and Nurr-1 in the adrenal glands. Mol Cell Biol. 1994;14:3469–3483. doi: 10.1128/mcb.14.5.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borghaei RC, Rawlings PL, Jr, Mochan E. Interleukin-4 suppression of interleukin-1-induced transcription of collagenase (MMP-1) and stromelysin 1 (MMP-3) in human synovial fibroblasts. Arthritis Rheum. 1998;41:1398–1406. doi: 10.1002/1529-0131(199808)41:8<1398::AID-ART8>3.0.CO;2-B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kagaya S, Ohkura N, Tsukada T, Miyagawa M, Sugita Y, Tsujimoto G, Matsumoto K, Saito H, Hashida R. Prostaglandin A2 acts as a transactivator for NOR1 (NR4A3) within the nuclear receptor superfamily. Biol Pharm Bull. 2005;28:1603–1607. doi: 10.1248/bpb.28.1603. [DOI] [PubMed] [Google Scholar]
- 5.Hazel TG, Nathans D, Lau LF. A gene inducible by serum growth factors encodes a member of the steroid and thyroid hormone receptor superfamily. Proc Natl Acad Sci USA. 1988;85:8444–8448. doi: 10.1073/pnas.85.22.8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 7.Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol. 2006;20:786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 8.Rius J, Martinez-Gonzalez J, Crespo J, Badimon L. NOR-1 is involved in VEGF-induced endothelial cell growth. Atherosclerosis. 2006;184:276–282. doi: 10.1016/j.atherosclerosis.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Ohkubo T, Ohkura N, Maruyama K, Sasaki K, Nagasaki K, Hanzawa H, Tsukada T, Yamaguchi K. Early induction of the orphan nuclear receptor NOR-1 during cell death of the human breast cancer cell line MCF-7. Mol Cell Endocrinol. 2000;162:151–156. doi: 10.1016/s0303-7207(00)00222-7. [DOI] [PubMed] [Google Scholar]
- 10.Oita RC, Mazzatti DJ, Lim FL, Powell JR, Merry BJ. Whole-genome microarray analysis identifies up-regulation of Nr4a nuclear receptors in muscle and liver from diet-restricted rats. Mech Ageing Dev. 2009;130:240–247. doi: 10.1016/j.mad.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr) 2010;32:97–108. doi: 10.1007/s11357-009-9118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, Ding SY, Reue K, Watt MJ, Newgard CB, Pilch PF, Hevener AL, Tontonoz P. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes. 2009;58:2788–2796. doi: 10.2337/db09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y, Luo L, Luo N, Zhu X, Garvey T. NR4A orphan nuclear receptors modulate insulin action and the glucose transport system: potential role in insulin resistance. J Biol Chem. 2007;282:31525–31533. doi: 10.1074/jbc.M701132200. [DOI] [PubMed] [Google Scholar]
- 14.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 15.Mayor P, Maianu L, Garvey WT. Glucose and insulin chronically regulate insulin action via different mechanisms in BC3H1 myocytes. Effects on glucose transporter gene expression. Diabetes. 1992;41:274–285. doi: 10.2337/diab.41.3.274. [DOI] [PubMed] [Google Scholar]
- 16.Nedachi T, Kanzaki M. Regulation of glucose transporters by insulin and extracellular glucose in C2C12 myotubes. Am J Physiol Endocrinol Metab. 2006;291:E817–E828. doi: 10.1152/ajpendo.00194.2006. [DOI] [PubMed] [Google Scholar]
- 17.Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med. 2006;12:1048–1055. doi: 10.1038/nm1471. [DOI] [PubMed] [Google Scholar]
- 18.Chao LC, Zhang Z, Pei L, Saito T, Tontonoz P, Pilch PF. Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol Endocrinol. 2007;21:2152–2163. doi: 10.1210/me.2007-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim EH, Surh YJ. 15-deoxy-Delta12,14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochem Pharmacol. 2006;72:1516–1528. doi: 10.1016/j.bcp.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 20.Kim HJ, Kim JY, Meng Z, Wang LH, Liu F, Conrads TP, Burke TR, Veenstra TD, Farrar WL. 15-deoxy-Delta12,14-prostaglandin J2 inhibits transcriptional activity of estrogen receptor-alpha via covalent modification of DNA-binding domain. Cancer Res. 2007;67:2595–2602. doi: 10.1158/0008-5472.CAN-06-3043. [DOI] [PubMed] [Google Scholar]
- 21.Milne GL, Musiek ES, Morrow JD. The cyclopentenone (A2/J2) isoprostanes – unique, highly reactive products of arachidonate peroxidation. Antioxid Redox Signal. 2005;7:210–220. doi: 10.1089/ars.2005.7.210. [DOI] [PubMed] [Google Scholar]
- 22.Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levonen AL, Dickinson DA, Moellering DR, Mulcahy RT, Forman HJ, Darley-Usmar VM. Biphasic effects of 15-deoxy-delta-(12,14)-prostaglandin J(2) on glutathione induction and apoptosis in human endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21:1846–1851. doi: 10.1161/hq1101.098488. [DOI] [PubMed] [Google Scholar]
- 24.Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, Zanoni G, Vidari G, Morrow JD, Darley-Usmar VM. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2006;290:H1777–H1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- 25.Fukushima M, Kato T, Ota K, Arai Y, Narumiya S, Hayaishi O. 9-deoxy-delta 9-prostaglandin D2, a prostaglandin D2 derivative with potent antineoplastic and weak smooth muscle-contracting activities. Biochem Biophys Res Commun. 1982;109:626–633. doi: 10.1016/0006-291x(82)91986-6. [DOI] [PubMed] [Google Scholar]
- 26.Fitzpatrick FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2. Identification of products formed in vitro. J Biol Chem. 1983;258:11713–11718. [PubMed] [Google Scholar]
- 27.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 28.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 29.Tortorella LL, Pilch PF. C2C12 myocytes lack an insulin-responsive vesicular compartment despite dexamethasone-induced GLUT4 expression. Am J Physiol Endocrinol Metab. 2002;283:E514–E524. doi: 10.1152/ajpendo.00092.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.