

Abstract
Flufenamic acid has been known since the 1960s to have anti-inflammatory properties attributable to the reduction of prostaglandin synthesis. Thirty years later, flufenamic acid appeared to be an ion channel modulator. Thus, while its use in medicine diminished, its use in ionic channel research expanded. Flufenamic acid commonly affects non-selective cation channels and chloride channels, but also modulates potassium, calcium and sodium channels with effective concentrations ranging from 10-6 M in TRPM4 channel inhibition to 10-3 M in two-pore outwardly rectifying potassium channel activation. Because flufenamic acid effects develop and reverse rapidly, it is a convenient and widely used tool. However, given the broad spectrum of its targets, experimental results have to be interpreted cautiously. Here we provide an overview of ion channels targeted by flufenamic acid to aid in interpreting its effects at the molecular, cellular, and systems levels. If it is used with good practices, flufenamic acid remains a useful tool for ion channel research. Understanding the targets of FFA may help reevaluate its physiological impacts and revive interest in its therapeutic potential.
Keywords: Flufenamic acid, flufenamate, TRP, non-selective cation channel, chloride channels, channel blockers
1. From medicine to widely used ion channel modulator
Flufenamic acid (FFA), namely N-(alpha,alpha,alpha-trifluoro-m-tolyl) anthranilic acid (CI-440), is an aromatic amino acid consisting of anthranilic acid carrying an N-(trifluoromethyl)phenyl substituent (Fig. 1). Its anti-inflammatory and analgesic effects were recognized in the 1960s (Winder et al., 1963) and thus FFA is included in the family of non-steroidal anti-inflammatory drugs (NSAIDs) with mefenamic, meclofenamic (MFA) and niflumic acids (NA). Anti-inflammatory actions occur mainly through reduction of prostaglandin synthesis from arachidonic acid by inhibiting the cyclo-oxygenases (Fig. 1) (Flower et al., 1972).
Figure 1. Anti-inflammatory effect of flufenamic acid.
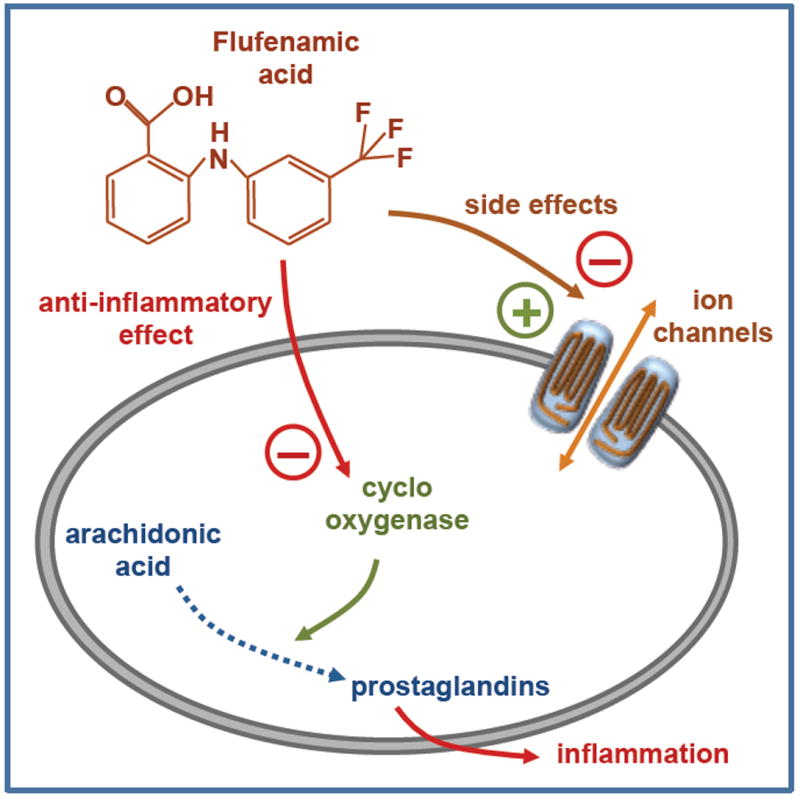
Chemical structure of flufenamic acid and its main targets: cyclooxygenase for anti-inflammatory effect and ion channels for additional effects.
Despite lower effectiveness than other NSAIDs (Flower, 1974), FFA was locally applied for analgesia against pain and inflammation associated with musculoskeletal and joint disorders, peri-articular and soft tissue disorders. Oral administration was discontinued because of large intersubject variability in FFA absorption (Lentjes & van Ginneken, 1987). In addition, the dermal administration reduces first-pass metabolism (Roberts & Walters, 2008). FFA, similar to other NSAIDs, has side effects including gastrointestinal perturbations (which are reduced in dermal application) (Ravi et al., 1986) and renal damage. Due to these deleterious side effects, and because its benefits were weak compared to other NSAIDs, the use of FFA in medicine remained somewhat limited. Nevertheless, human trials in more than 10,000 patients in 1998 re-affirmed NSAIDs effectiveness for acute and chronic pain relief, and particularly emphasized FFA topical application in combination with salicylic acid (Moore et al., 1998).
Interest in FFA revived following the 1976 report of an effect on calcium and sodium uptake in lymphoid cells, which suggested that ion-handling proteins were affected (Famaey & Whitehouse, 1976). Indeed, during the 1990s FFA became recognized as a common regulator of ionic currents in native tissues. The story was elaborated in the 2000s as the molecular identities of the ion channels targeted by FFA were discovered, including Cl-, Na+, K+ and, most notably, non-selective cation channels. Therefore, FFA made a comeback in basic research as a convenient pharmacological tool to study ion channels. However, its broad spectrum of targets may produce complex experimental results that are difficult, or impossible, to interpret unambiguously.
Here, we focus on ion channels modulated by FFA, including native currents and cloned channel proteins. We aim to provide an overview of the currents modulated by FFA to help differentiate its effects in physiological contexts where several ion channel types could be affected pharmacologically (See Table 1 and Figure 2 for specific ion channel targets, permeability, FFA efficiency, and experimental conditions).
Table 1. Information about ion channels and currents affected by FFA.
Depending on the reports, a single FFA concentration was used ([FFA]) or concentration for half maximal effect (EC50) or dissociation constant (KD) was provided.
| Channel name |
Perme- ability |
Current | Cell | Configuration | FFA effect | [FFA] in 10-6 M |
EC50 in 10-6 M |
KD in 10-6 M | Other fenamates |
mechanisms | references |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
|
Chloride channels
| |||||||||||
| CFTR | Cl- | cAMP-activated Cl- current | Xenopus oocyte | Whole-cell | inhibition | 200 to 1000 | direct interaction in the open state | Mc Carty et al. 1993 | |||
| Single channel | |||||||||||
|
| |||||||||||
| CIC-Ka | Cl- | Voltage-gated Cl- current | Xenopus oocyte | Whole-cell | inhibition | 57 to 121 | MFA>FFA | direct interaction in the vestibule | Liantonio et al. 2006 | ||
|
| |||||||||||
| CIC-Kb | Cl- | Voltage-gated Cl- current | Xenopus oocyte | Whole-cell | activation | 200 | NA>FFA | Liantonio et al. 2006 | |||
|
| |||||||||||
| CIC-1 | Cl- | Voltage-gated Cl- current | Xenopus oocyte | Whole-cell | inhibition | 4.5 | FFA>NA | direct interaction | Liantonio et al. 2007 | ||
|
| |||||||||||
| GABAA-R | Cl- | GABA-inducced Cl- current | Xenopus oocyte | Two electrodes voltage-clamp | inhibition | 16 | Woodward et al. 1993 | ||||
| HEK-293 | Whole-cell | inhibition | 2 | ||||||||
|
| |||||||||||
| PanX-1 | Cl- | HEK-293 | Whole-cell | inhibition | >1000 | FFA=NA | Ma et al. 2009 | ||||
|
| |||||||||||
|
Non-selective cation channels
| |||||||||||
| TRPC3 | Na+, K+, Ca2+ | Redox-sensitive NSC current | HEK-293 | Whole-cell | inhibition | 100 | Inoue et al. 2001 | ||||
|
| |||||||||||
| TRPC4 | Na+, K+, Ca2+ | Redox-sensitive NSC current | HEK-293 | Whole-cell | inhibition | 55 | FFA>NA>MFA | direct interaction | Jiang et al. 2012 | ||
|
| |||||||||||
| TRPC5 | Na+, K+, Ca2+ | HEK-293 | Whole-cell | inhibition | 37 | FFA>MFA>NA | direct interaction | Jiang et al. 2012 | |||
|
| |||||||||||
| TRPC6 | Na+, K+, Ca2+ | α-adrenorec-activated NSC current | HEK-293 | Whole-cell | inhibition | 17 | FFA>MFA>NA | Klose et al. 2011 | |||
|
| |||||||||||
| TRPC6 | Na+, K+, Ca2+ | α-adrenorec-activated NSC current | HEK-293 | Whole-cell | activation | 100 | FFA≫NA | direct interaction | Inoue et al. 2001; Foster et al. 2009 | ||
|
| |||||||||||
| TRPC7 | Na+, K+, Ca2+ | HEK-293 | Whole-cell | inhibition | 100 | Inoue et al. 2001 | |||||
|
| |||||||||||
| TRPM2 | Na+, K+, Ca2+ | Hydrogen peroxide-activated NSC | HEK-293 | Whole-cell | inhibition | 155.1 | FFA>NA=MFA | Klose et al. 2011 | |||
|
| |||||||||||
| TRPM3 | Na+, K+, Ca2+ | Hypoosmolarity -activated NSC | HEK-293 | Whole-cell | inhibition | 33.1 | MFA>FFA>NA | Klose et al. 2011 | |||
|
| |||||||||||
| TRPM4 | Na+, K+ | NSCca | HEK-293 | Whole-cell | inhibition | 2.8 | Ullrich et al. 2005 | ||||
|
| |||||||||||
| TRPM5 | Na+, K+ | NSCca in taste cells | HEK-293 | Whole-cell | inhibition | 24.5 | Ullrich et al. 2005 | ||||
|
| |||||||||||
| TRPV1 | Na+, K+, Ca2+ | Capsaicin-activated NSC current | Xenopus oocyte | Two electrodes voltage-clamp | inhibition | 100 | Hu et al. 2010 | ||||
|
| |||||||||||
| TRPV3 | Na+, K+, Ca2+ | Thermo-sensitive NSC current | Xenopus oocyte | Two electrodes voltage-clamp | inhibition | 100 | Hu et al. 2010 | ||||
|
| |||||||||||
| TRPV4 | Na+, K+, Ca2+ | Thermo-sensitive NSC current | HEK-293 | Whole-cell | inhibition | 40.7 | FFA>NA>MFA | Klose et al. 2011 | |||
|
| |||||||||||
| TRPA1 | Na+, K+, Ca2+ | Heat-activated NSC current | HEK-293 | Whole-cell | activation | 57 | Hu et al. 2010 | ||||
|
| |||||||||||
| α3–β2 nAch-R | Na+, K+, Ca2+ | Neuronal-nicotinic Ach-receptor | Xenopus oocyte | Two electrodes voltage-clamp | inhibition | 90 | FFA>NFA | Direct interaction | Zwart et al. 1995 | ||
|
| |||||||||||
| α3–β4 nAch-R | Na+, K+, Ca2+ | Neuronal-nicotinic Ach-receptor | Xenopus oocyte | Two electrodes voltage-clamp | activation | 30 | FFA>NFA | Direct interaction | Zwart et al. 1995 | ||
|
| |||||||||||
| Cx 43 | Na+, K+, Ca2+ | Gap junction | Rat kidney fibroblast | Dye measurements | inhibition | 40 | MFA>FFA | Harks et al. 2001 | |||
|
| |||||||||||
| Cx 50 | Na+, K+, Ca2+ | Gap junction | N2A neuroblast oma cells | Two electrodes voltage-clamp | inhibition | 47 | NA>FFA=MFA | Reduction of open probability. Binding in a modulatory site within membrane | Srinivas et al. 2003 | ||
|
| |||||||||||
|
Potassium channels
| |||||||||||
| KCa 1.1 | K+ | Ca2+-activated K+ current (BKCa) | Xenopus oocyte | Two electrodes voltage-clamp | activation | >300 | FFA=NA | Gribkoff et al. 1996 | |||
|
| |||||||||||
| KV 11.1 | K+ | Human ether à gogo related current (HERG) | Xenopus oocyte | Two electrodes voltage-clamp | activation | 100 | FFA>NA | Malykhina et al. 2002 | |||
|
| |||||||||||
| KV 7.1 | K+ | Delayed-rectifier K+ current | Xenopus oocyte | Two electrodes voltage-clamp | activation | 100 | Slowing of channel deactivation | Busch et al. 1994 | |||
|
| |||||||||||
| KCa 4.2 | K+ | Two pores outward rectifyer K+ current | Xenopus oocyte | Two electrodes voltage-clamp | activation | 1100 | MFA>FFA>NA | Binding in the pore region | Garg and Sanguinetti, 2012 | ||
|
| |||||||||||
| K2p 2.1 | K+ | Lipid-sensitive mechano-gated 2P domain K+ channel | Cos-7 | Perforated patch-clamp | activation | 100 | FFA>NA=MFA | Takahira et al. 2005 | |||
|
| |||||||||||
| K2p 4.1 | K+ | TWIK-related arachidonic acid-stimulated K+ channel | Cos-7 | Perforated patch-clamp | activation | >500 | FFA=NA>MFA | Takahira et al. 2005 | |||
|
| |||||||||||
| K2p 10.1 | K+ | Inward rectifier K+ channel | Cos-7 | Perforated patch-clamp | activation | >100 | FFA>NA=MFA | Takahira et al. 2005 | |||
|
| |||||||||||
|
Sodium channels
| |||||||||||
| BLINaC | Na+ | Brain liver intestine Na+ channel | Xenopus oocyte | Two electrodes voltage-clamp | activation | >1000 | FFA>NA | Increase of Na+ selectivity | Wiemuth and Grunder, 2011 | ||
| Current | Permeability | Cell | Configuration | FFA effect | EC50 in 10-6 M | KD in 10-6 M | Other fenamates | mechanisms | references |
|
| |||||||||
| CaCCs | Cl- | Xenopus oocyte | Two electrodes voltage-clamp | inhibition | 35.4 | 28 | F=NA>MFA | Direct interaction in the open state | White al. 1990 |
| Oh et al.2008 | |||||||||
|
| |||||||||
| ICl, swell | Cl- | Human gastric epithelial cells | Whole-cell | inhibition | 50<IC50<200 | Jin et al. 2003 | |||
|
| |||||||||
| NMDA-R current | Na+, K+, Ca2+ | Spinal cord neurons | Ask publication to Christopher | Independent from NMDA | Lerma et al. 1992 | ||||
|
| |||||||||
| Voltage-gated INa | Na+ | Rat Hippocampal pyramidal neurons | Whole-cell | inhibition | 189 | Modification of inactivation kinetic | Yau et al. 2012 | ||
|
| |||||||||
| Voltage-gated ICa | Ca2+ | Smooth muscle cells of rat carotid artery | Whole-cell | inhibition | 100 | Shimamura et al. 2002 | |||
Figure 2. Ion channels targeted by flufenamic acid.
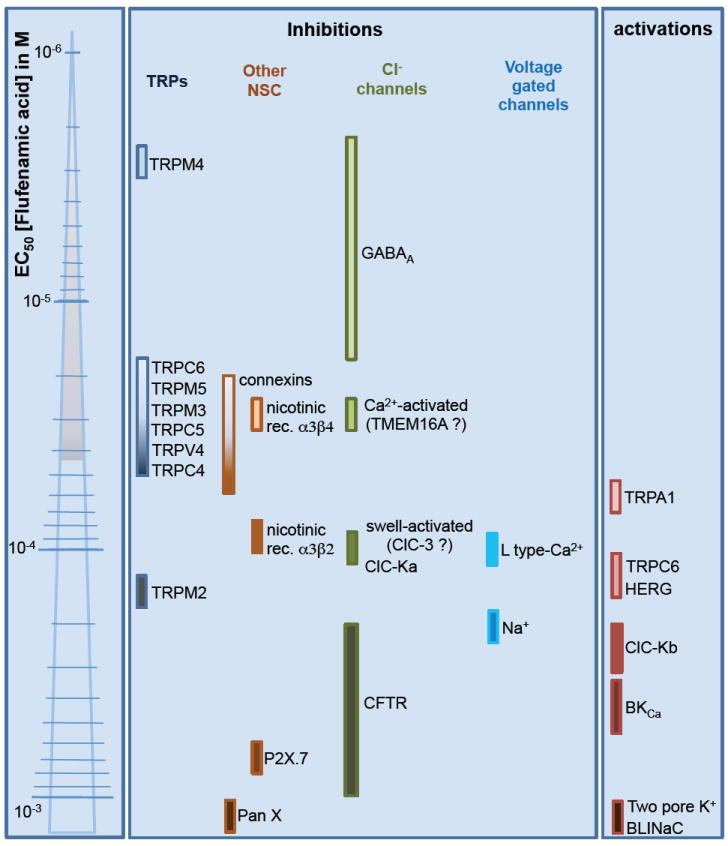
Flufenamic acid produces inhibition or activation of ion channels. Coloured bars near ionic channel name correspond to the estimated EC50 for flufenamic effect. References are provided within the text.
2. Flufenamic acid as an ion channel modulator
2.1. Chloride channels
Anion channels poorly differentiate between anions but because Cl- is most abundant, the channels are referred to as chloride channels. These channels are implicated in a variety of physiological processes, depending on their regulatory properties, including sensitivity to voltage, cell volume, internal Ca2+, cAMP, pH, and ligand binding (see Duran et al., 2010 for review). Chloride channels were the first family shown to be affected by FFA, which is considered to be a classical chloride channel blocker, along with disulfonic stilbenes (DIDS, SITS, DNDS), anthracene carboxylates (9-AC), arylaminobenzoates (DPC), indanylalkanoic acids (IAA-94), chlofibric acid derivatives (CPP) and other fenamates such as NPPB and niflumic acid (see Suzuki et al., 2006 for review).
2.1.1. Cystic fibrosis transmembrane conductance (CFTR)
A cAMP-dependent chloride current, later recognized as the cystic fibrosis transmembrane conductance regulator (CFTR), was the first identified FFA target among ion channels (McCarty et al., 1993). CFTR is an ATP-binding cassette (ABC) protein containing 1480 amino acids divided into two domains, each composed of six transmembrane domains. CFTR forms a PKA and PKC-activated chloride channel mediating chloride transport in a variety of tissues, with a major role in airway epithelia. Altering its activity or expression leads to cystic fibrosis and secretory diarrhea. CFTR is intensively studied because of its role in pathology (Welsh et al., 1992; Duran et al., 2010 for review).
CFTR modulators were sought to correct chloride transport in cystic fibrosis (Becq & Mettey, 2004). FFA, which is membrane permeable, inhibits CFTR heterologously expressed in Xenopus oocytes (McCarty et al., 1993). FFA inhibition is stronger at positive voltages. In addition, since the effect is observed in inside-out patches, FFA inhibits CFTR by direct interaction with the channel, producing an open-channel block. However, high concentrations are necessary to inhibit the channel; indeed, the CFTR currents are reduced by only 20-30 % by 200 μM FFA (McCarty et al., 1993).
Probably due to its low efficiency, FFA has rarely been used to study CFTR in physiological preparations (Liu et al., 2006). However, because CFTR is expressed in apical membranes of epithelia, its inhibition has to be considered when using FFA at high concentrations in tissues such as airway epithelia, intestine, pancreas, kidney, sweat duct and testis, as well as cardiac cells that express CFTR (Duan, 2009 for review).
2.1.2. Ca2+-activated chloride currents (CaCCs) and Bestrophins
A Ca2+-activated chloride current (CaCC) first described in Xenopus oocytes (Barish, 1983) has been similarly recorded in excitable and non-excitable cells (Huang et al., 2012a for review). For example, CaCC is present in Cl- secretory epithelia and in tissues expressing cAMP-activated Cl- current attributed to CFTR, where both channel types co-localize in the apical membrane (Cliff & Frizzell, 1990). Also, CaCC is present in cardiomyocytes; its cytosolic Ca2+ activation profile, outward rectification, and time-dependent inactivation contribute to cardiac action potential repolarization (Duan, 2009).
FFA inhibits the archetypal CaCC from Xenopus oocytes with an IC50 ranging from 28-35 μM (White & Aylwin, 1990; Oh et al., 2008). Inhibition by FFA has also been observed in other native CaCCs in rabbit portal vein, pig ventricular cardiomyocytes, as well as olfactory receptors neurons from moth Spodoptera littoralis (Greenwood & Large, 1995; Gwanyanya et al., 2010; Pezier et al., 2010). FFA appears to exert an open-channel block like in CFTR (Greenwood & Large, 1995). Despite its lack of specificity for CaCC, FFA remains a useful tool to study these currents because its IC50 is comparably low and, until now, no other CaCC-specific inhibitors have been identified.
The molecular identity of CaCCs remains unknown. Three major protein families have been proposed (Huang et al., 2012a). The first candidate comes from the Ca2+ activated chloride channel (CLCA) protein family, initially shown to produce chloride currents. However, its identity as an ion channel has been strongly debated and it is now considered as a secreted non-integral membrane protein (Winpenny et al., 2009). Moreover, the CaCC endogenous expression levels do not match the expression levels that characterize CLCA. The second candidate comes from the Bestrophin family, so called because mutations of the prototypic member Best-1 causes Best disease, an inherited form of retinal macular dystrophy (Xiao et al., 2010). The Bestrophin family is composed of four members found in the human genome. The expression of some of these four transmembrane domain proteins produces a Cl- current activated by physiological levels of internal Ca2+. In hippocampal astrocytes, 100 μM FFA inhibits a Ca2+-activated anionic current by 75% (Park et al., 2009). This endogenous current is reduced by the expression of mBest-1-specific short hairpin RNA, which suggests that the Ca2+-activated anion current corresponds to Best-1 and, thus, indirectly demonstrates Best-1 sensitivity to FFA. At present, to the best of our knowledge, there are no reports demonstrating direct effects of FFA on bestrophins. The most recent candidate for the molecular identity of CaCCs is the transmembrane protein 16A (TMEM16A), which forms a CaCC channel subunit (Huang et al., 2012b). This eight transmembrane segment protein may form a functional channel as a homodimer. No existing data show TMEM16A modulation by FFA, even though the effects of FFA have been reported on TMEM16A-expressing cells such as pulmonary artery smooth muscle cells and human airway gland cells (Fischer et al., 2010; Yamamura et al., 2011).
2.1.3. Swelling-activated chloride currents (IClswell) and ClC-3
Chloride channels that activate under hypo-osmotic conditions can prevent cellular injuries associated with swelling. In conjunction with K+ channels, they allow KCl leakage, leading to intracellular dilution, net water loss, and volume decrease. A swelling-activated Cl- current named IClswell has been characterized in virtually every cell yet examined, including in the heart where ICIswell may combat arrhythmias (Baumgarten & Clemo, 2003; Duran et al., 2010). Tissue-specific differences in biophysics and pharmacology suggest that different channel proteins give rise to IClswell in different cells. IClswell is supported by an outwardly rectifying Cl- channel in rabbit and human myocytes (Duan et al., 1997a; Demion et al., 2006). Open probability is not voltage-dependent but the single-channel conductance increases from 10 to 80 pS as voltage ascends, resulting in a pronounced outward rectification (Duan et al., 1997a; Demion et al., 2006).
In human gastric epithelial cells, 100 μM FFA reduced IClswell by 82% (Jin et al., 2003). More recently, the same concentration has been shown to inhibit IClswell in microglia (Schlichter et al., 2011) and reduce regulatory volume decrease in bovine ciliary epithelium (Do et al., 2006).
The molecular identity of IClswell is a subject of debate. The confusion is probably due to several underlying channel proteins whose expression differs with tissue type. One of the strongest candidates belongs to the chloride channel (ClC) family initially identified by the cloning of the voltage-gated Cl- channel from the electric organ of the torpedo electric ray. Nine members comprise the ClC family in mammals (Duran et al., 2010). The constituent molecules, composed of 10 to 12 transmembrane domains, have two conducting pores. Among ClCs, ClC-3, cloned in 1997, is broadly distribution among tissues and its expression gives rise to an outwardly rectifying chloride channel activated by cell swelling (Duan et al., 1997b). Following the cloning of ClC-3, competing studies putatively demonstrated or, alternatively, invalidated the idea that ClC-3 mediated the endogenous swelling-activated chloride-current involved in cell volume regulation (Duan et al., 2001; Weylandt et al., 2001; Duran et al., 2010). It is unfortunate for our purposes that none of these studies directly evaluated the effects of FFA on the ClC-3 cloned protein. Nonetheless, FFA and anti-ClC-3 antibodies attenuated IClswell in human gastric epithelial cells and disrupted the attendant regulatory volume decrease, which suggests that ClC-3 is sensitive to FFA (Jin et al., 2003).
2.1.4. Renal transepithelial Cl- transport and ClC-K
Within the ClC family, the expression of ClC-K channels is restricted to the basolateral membrane of kidney cells (from the thin ascending limb to the collecting duct), where they play a major role in urine concentration. ClC-K is also expressed in the inner ear where these channels participate in endolymph production (Fahlke & Fischer, 2010). Two human ClC-K isoforms (ClC-Ka and ClC-Kb) correspond to ClC-K1 and ClC-K2 orthologs in rat. Unlike other ClCs, ClC-K channels require the presence of an additional β-subunit called barttin (Estevez et al., 2001), which produces a chloride current with moderate outward rectification (Estevez et al., 2001; Waldegger et al., 2002). Mutations in ClC-Kb that reduce channel activity cause type III Bartter’s syndrome, a renal disease characterized by severe salt wasting (Simon et al., 1997; Seyberth & Schlingmann, 2011). Mutations in barttin cause Bartter’s syndrome type IV, which is characterized by renal failure and sensorineural deafness (Birkenhager et al., 2001).
Experiments sought to identify ClC-K ligands to discover pharmacological interventions for Bartter’s diseases. FFA inhibits ClC-Ka in Xenopus oocytes with a binding constant ranging from 57 μM at -140 mV to 121 μM at +60 mV (Liantonio et al., 2006). The authors predicted 1:1 binding based on the dose response curve. Therefore, KD might be equivalent to the EC50 for ClC-Ka. This FFA inhibition is abolished by the N68D mutation, a residue putatively located on the extracellular vestibule (Liantonio et al., 2006). A non-coplanar conformation in the aromatic group of FFA is necessary for the inhibitory binding site (Liantonio et al., 2008); FFA derivatives with coplanar aromatic groups are too rigid to enter the narrow part of the extracellular vestibule. Nonetheless, coplanar ligands bind to an activating site and activate ClC-Ka (Gradogna & Pusch, 2010).
ClC-Kb does not exhibit the same FFA sensitivity: 200 μM FFA increases ClC-Kb current by two-fold while, at the same dose, it reduces ClC-Ka current by half (Liantonio et al., 2006). At present there are no clear explanations for these discrepancies.
To our knowledge, the effects of FFA on renal and inner ear trans-epithelial salt transport systems remain unknown even though other ClC-K blockers were recently shown to increase water dieresis in rat (Liantonio et al., 2012).
2.1.5. Skeletal muscle voltage-gated chloride current and ClC-1
In a study designed to evaluate skeletal muscle chloride currents sensitivity to niflumic acid, the authors also observed that 100 μM FFA abolished about all the endogenous chloride current from native rat muscle fibers (Liantonio et al., 2007). Because skeletal muscle chloride conductance is mainly attributable to the ClC-1 chloride channel protein (Steinmeyer et al., 1991), they subsequently tested the effect of FFA on ClC-1 expressed in Xenopus oocytes. The blocking potency of FFA, with a KD value of 4.5 μM, was enhanced compared to niflumic acid (Liantonio et al., 2007). This unique report of FFA-sensitive ClC-1 awaits further confirmation since it is based on only five recordings. However, if confirmed, the inhibition of ClC-1 by FFA might have physiological importance because it occurs at low concentrations and dysfunction of this channel causes congenital myotonia from both autosomal dominant (Thomsen type) and autosomal recessive (Becker type) inherited patterns (Tang & Chen, 2011 for review).
2.1.6. Synaptic inhibition and GABAA-Receptor
The γ-aminobutyric acid (GABA) receptor mediates fast inhibitory neurotransmission in the central nervous system by opening anion channels. GABA channels share a five-subunit structure with other ligand-gated ion channels, in which each subunit is composed of four transmembrane domains. Of the three types of GABA receptors, GABAA and GABAC form Cl- channels (GABAB receptors are G-protein coupled and linked to K+ channels). The single-channel conductance of GABAA and GABAC ranges from 10-30 pS. Its current-voltage relationship is linear at the single channel level, yet exhibits weak outward rectification at the macroscopic level (Bormann et al., 1987; Macdonald et al., 1989). GABAA receptor subunit mutations that reduce GABA-activated currents are associated with epilepsy (Baulac et al., 2001; Wallace et al., 2001; Macdonald et al., 2010).
FFA, like other NSAIDs, modulates GABAA receptors. Nevertheless, whereas most NSAIDs exert a potentiating effect, FFA reduces the GABA-induced current with an IC50 of 16 μM in a model of GABAA receptors expressed in Xenopus oocytes (Woodward et al., 1994) and 2 μM in a model of GABAA receptors expressed in HEK-293 cells (Rae et al., 2012). FFA effects on GABAA receptors may depend on the subunit composition in mammalian brain, because FFA exerts a potentiating effect on several GABAA subunits, while inhibiting others (Smith et al., 2004). Interestingly, FFA is specific for the GABAA isoform because it does not exert any effect on GABAC (Jones & Palmer, 2011).
The impact of GABAA modulation by FFA on neurophysiology is incompletely understood. It has been shown that FFA suppresses epileptiform activity (Schiller, 2004; Fernandez et al., 2010). However, at least in hippocampus, this effect is more likely due to NMDA receptor modulation than effects on GABAA receptors.
2.1.7. Pannexins
The recently identified mammalian pannexins (PanX) are molecules that bear amino acid sequence homologies with innexins, the gap junction-forming invertebrate proteins. PanX was considered to have a structure similar to connexins (see paragraph 2.2.3) and thus suspected to form non-selective transmembrane pores. However, the three known isoforms form typical anion channels and do not form gap junctions but most likely function as hemi-channels when expressed in HEK-293 cells (Ma et al., 2012). PanX1 is inhibited by FFA at very high concentrations; the IC50 is estimated to exceed 1 mM (Ma et al., 2009).
2.2. Non-selective cation channels
Following its description as a Cl- channel blocker, FFA was shown to modulate non-selective cation channels (NSC). NSC channels are a heterogeneous family whose members do not strongly differentiate between permeable cations. Initially characterized at the current level in native cells, now a large number of cloned genes are known to code for NSC channels. They are usually classified as ligand-gated NSC channels (e.g., nicotinic acetylcholine receptors, glutamate receptors, P2X purinergic receptors), cyclic nucleotide-gated channels (cGMP-gated or cAMP-gated channels), connexins and, the large group of transient receptor potential (TRP) channels. Despite their heterogeneity in structure, FFA modulates members in all subfamilies of NSCs except cyclic nucleotide-gated channels.
2.2.1. Transient Receptor Potential channels
TRP channels, first characterized in tissue from the Drosophila eye (Minke, 1977; Montell & Rubin, 1989), are classified for mammals into six sub-families: TRPC (Canonical, seven members), TRPV (Vanilloid, six members), TRPM (Melastatin, eight members), TRPP (Polycystin, three members), TRPML (Mucolipin, three members) and TRPA (Ankyrin, one member) (Gees et al., 2010). Most TRPs are permeable to Ca2+ as well as monovalent cations. However, some are strictly Ca2+ selective (TRPV5, TRPV6), whereas others are Ca2+ impermeable (TRPM4, TRPM5). Major physiological functions of TRP channels include Ca2+ signaling, sensory detection in peripheral neurons, as well as burst-generating functions in central neurons (Gees et al., 2010). TRP proteins are composed of subunits containing six transmembrane domains that assemble as tetramers. A large variety of TRP modulators have been described, including intracellular or extracellular messengers (e.g., ATP, Ca2+, phosphatidylinositol 4,5-bisphosphate), as well as biophysical modulators such as voltage and temperature. FFA inhibits a wide spectrum of TRP channels, including: C3, C7, M2, M3, M4, M5, M7, M8, V1, V3, and V4; but FFA activates at least two TRP channels (C6 and A1), as described below.
TRPCs
An α-adrenoreceptor-activated and Ca2+-permeable NSC channel is activated by FFA in rabbit portal vein smooth muscle (Yamada et al., 1996). TRPC6 is responsible for this current, and, when the protein is expressed in HEK-293 cells, its amplitude doubles in the presence of 100 μM FFA (Inoue et al., 2001). Interestingly, the FFA activating effect is not reproduced by niflumic acid, which suggests that TRPC6-activation is not a general property of the fenamate family (Foster et al., 2009). In addition, cyclo-oxygenase inhibitors do not affect this activating effect, which favors a direct interaction of FFA with the channel (Foster et al., 2009). Surprisingly, a recent paper reported an inhibitory effect of FFA (IC50 = 17 μM) on TRPC6 heterologously expressed in HEK-293 cells (Klose et al., 2011). The effect of FFA has also been evaluated on the closely related channels TRPC3 and TRPC7 that share, with TRPC6, activation by diacylglycerol, thus forming a subgroup of TRPCs. 100 μM FFA inhibits TRPC3 and TRPC7 by 60 and 90%, respectively (Inoue et al., 2001). This inhibitory action of FFA was reproduced in TRPC3-like native currents from rabbit ear arterial myocytes (Albert et al., 2006). The fact that FFA exerts opposite effects on TRPC6 vs. TRPC3/7 channels indicates that FFA and diacylglycerol may act through different mechanisms on channel activity.
In the other TRPC subgroup (TRPC1/4/5), mouse TRPC5 current is reduced by 92% by 100 μM FFA (Lee et al., 2003). A more recent study reports the inhibition by FFA of human TRPC4 and TRPC5 heterologously expressed in HEK-293 cells with IC50 of 55 and 37 μM respectively (Jiang et al., 2012).
TRPMs
TRPM4 and TRPM5 are unique among TRPs because they do not conduct Ca2+ but instead are activated by internal Ca2+ (Guinamard et al., 2011 for review). TRPM4/5 support one of the major NSC currents often called the Ca2+-activated non-selective cation current (NSCCa) and sometimes Ca2+-activated non-specific cation current (ICAN). NSCCa has been recorded in a wide variety of tissues, and is inhibited by FFA in, for example, pancreatic acinar cells (IC50 < 10 μM), rat liver cells (Simon et al., 2002), cardiomyocytes (Gogelein et al., 1990; Guinamard et al., 2002), and neurons (Partridge & Valenzuela, 2000; Pace et al., 2007). It is now well established that TRPM5 is responsible for NSCCa in taste receptor cells (Liman, 2007a; b). In contrast, insulin secretion, immune response, constriction of cerebral arteries, neural burst discharge in breathing-related neurons, and cardiac dysfunctions are associated with TRPM4 function (or dysfunction) (Guinamard et al., 2011). TRPM4 occupies a special position, particularly in the present review, because of its high sensitivity to FFA. Indeed, TRPM4 is inhibited with an IC50 of 2.8 μM when expressed in HEK-293 cells. Interestingly, in native tissue, our group measured a similar IC50 of 5.5 μM for the inhibition of an endogenous TRPM4 current in rat cardiomyocytes (Guinamard et al., 2006b). The closest relative, TRPM5, is inhibited with 10 fold higher doses, the IC50 for TRPM5 being 24.5 μM (Ullrich et al., 2005). Low concentrations of FFA (~10 μM) may be appropriate to evaluate the physiological role of TRPM4 in situ, which would be expected to have little to no effect on other ion currents whose FFA sensitivity is much lower. Consistent with this idea, 10 μM FFA was used to differentiate breathing-related neurons that depend putatively on TRPM4 for ICAN-mediated neural bursts in the respiratory oscillator preBötzinger complex in mice (Del Negro et al., 2005). TRPM4 modulation may represent a major common explanation for the physiological effects of FFA given its ubiquitous expression profile and high sensitivity to FFA. This is particularly important because plasma concentrations of 4-12 μM, measured in conditions of FFA clinical use, are sufficient to strongly inhibit TRPM4 (Aly et al., 2000).
FFA also inhibits TRPM2, the most abundant TRP in the brain, which is implicated in cell death resulting from oxidative stress (Hill et al., 2004). FFA inhibits 90% of the TRPM2 current in HEK-293 cells at a dose of 50 μM (Hill et al., 2004) or 200 μM (Togashi et al., 2008). Interestingly, the inhibitory effects of FFA increase in response to extracellular acidification. This phenomenon can be explained by the fact that FFA assumes its uncharged form at acidic pH, which favors membrane crossing to the cytosolic face of TRPM2. It can also be also explained by a modification of the channel itself, which favors FFA interaction (Hill et al., 2004). A more recent study in the same preparation reports an IC50 of 155 μM for TRPM2 inhibition and an IC50 of 33 μM for TRPM3 inhibition (Klose et al., 2011). The inhibitory effect of FFA has been further established using peroxide-stimulated endogenous TRPM2 currents from CR1-G1 insulinoma cells and CHO cells (Hill et al., 2004; Naziroglu et al., 2007) or endogenous currents from hippocampal neurons (Olah et al., 2009) and dorsal root ganglion from rat (Naziroglu et al., 2011).
Three recent publications report a 50% reduction of TRPM7-like currents by 10-4 M FFA in rat brain microglia, the human breast cancer cell line MCF-7, and in mouse renal tubule (Jiang et al., 2003; Guilbert et al., 2009; Guinamard et al., 2012). Nevertheless, the direct inhibition of TRPM7 by FFA remains to be clearly demonstrated. In addition, a tiny inhibition of 16 to 30 % by 10-4 M FFA has been also reported for TRPM8 heterologously expressed in Xenopus oocyte (Hu et al., 2010).
TRPVs
Sensitivity to vanilloid characterizes TRPV1, which became the founding member of the thermo-sensitive TRP channels (Xia et al., 2011). Subsequently, this channel was shown to be modulated by capsaicin (Cortright et al., 2001) and has been implicated in somatic pain sensing. As a consequence, TRPV1 became an attractive target for pharmaceutical research in order to identify new analgesic drugs. Human TRPV1 is mainly expressed in dorsal root ganglia (and trigeminal root ganglia) but also in the central nervous system, kidney and liver (Cortright et al., 2001). TRPV1 is expressed in the plasma membrane but also in intracellular organelles such as the endoplasmic reticulum membrane (Wisnoskey et al., 2003). Therefore, TRPV1 is a target for molecules that are membrane permeable such as FFA, as previously shown (McCarty et al., 1993).
Unfortunately, only one study reports the FFA sensitivity of TRPV1; 10-4 M FFA reduces the TRPV1 current by 57-75% when heterologously expressed in Xenopus oocytes (Hu et al., 2010). TRPV3, in the same TRPV family, is inhibited to the same extent (57-67%) by 10-4 M FFA, as measured in Xenopus oocytes (Hu et al., 2010).
The mechanosensitive TRPV4 channel is inhibited by FFA with an IC50 of 41 μM when stably expressed in HEK-293 cells (Klose et al., 2011), which must be considered when investigating the effects of FFA in cell swelling.
TRPA
Among the most recently cloned TRP channels, TRPA1 is expressed in sensory neurons and is implicated in inflammatory pain as well as nociception (Gees et al., 2010). Given the anti-inflammatory properties of fenamates, TRPA1 seemed to be an obvious target to study in detail. A variety of fenamates including niflumic, mefenamic and flufenamic acids were shown to activate TRPA1 current following expression in HEK-293 cells, with an EC50 of 57 μM for FFA (Hu et al., 2010). This activation effect has also been observed for the TRPA1 endogenous current from WI-38 fibroblasts (Hu et al., 2010). Nevertheless, warming (from 23 to 39 °C) prevents TRPA1 activation by FFA (300 μM) (Wang et al., 2012).
2.2.2. Ligand-gated non-selective cation channels
FFA effects have been described for three types of ligand-gated non-selective cation channels activated by acetylcholine, glutamate or ATP. However, the physiological significance of these FFA effects remains incompletely understood.
An inhibitory, non-competitive effect of FFA has been described for the N-methyl-D-aspartate (NMDA) glutamate receptors in spinal cord neurons (Lerma & Martin del Rio, 1992). NMDA receptors form non-selective cation channels that flux Ca2+, which can subsequently activate an NSCCa. Because NSCCa are inhibited by FFA, as described above, the effects of FFA on NMDA-induced responses must be interpreted with caution. NMDA receptors are implicated in epilepsy and their inhibition by 100 μM FFA has been shown to suppress epileptiform activity in the hippocampus (Fernandez et al., 2010). Nevertheless, this effect of FFA may involve the inhibition of NSCCa subsequently activated by NMDA receptor-mediated Ca2+ current (Schiller, 2004). Interestingly, FFA does not affect other types of glutamate receptors (Lerma & Martin del Rio, 1992).
Neuronal nicotinic acetylcholine receptors (nAChRs) form pentameric non-selective cation channels. FFA exerts differential effects on nAChRs in Xenopus oocytes, depending on the β subunit that is expressed. FFA inhibits the α3β2 nAChR current with an IC50 of 90 μM, whereas FFA activates the α3β4 nAChR current with an EC50 of 30 μM (Zwart et al., 1995). Once again, interpreting FFA effects is problematic because nAChRs are Ca2+ permeable, and their activation can elevate intracellular Ca2+ and subsequently evoke FFA-sensitive NSCCa, as shown in mesencephalic dopamine neurons (Zwart et al., 1995).
ATP induced Ca2+-entry is reduced by FFA with a low EC50 of 655 nM in the 1321N1 astrocytoma cell line stably transfected with the purinergic receptor P2X7R, which also forms a non-selective cation channel (Suadicani et al., 2006). The authors attributed this reduction to the inhibition of the P2X7R. However this interpretation is now controversial since it was observed that 100 μM FFA had no effect on P2X7R currents in HEK-293 transfected cells (Ma et al., 2009).
2.2.3. Gap junction channels
FFA inhibits gap junctions, channels that electrically connect adjacent cells. Gap junctions are composed of two hemichannels that associate in series and can span the plasma membrane of neighboring cells. Hemichannels are composed of six connexin subunits, wherein each connexin is composed of four transmembrane segments. There are 21 connexin (Cx) isoforms in human; nomenclature depends on molecular weight, from Cx26 to Cx62 (Maeda & Tsukihara, 2011 for review). The single-channel conductance of homomeric connexin channels spans 20-300 pS. These channels are permeable to most cations, sometimes anions, and several intracellular signaling molecules. The principal characteristic that influences permeability is size, which has to be under 1kDa. A wide variety of tissues express connexins, which can synchronize intracellular Ca2+ signaling and membrane potential trajectory among cells. Gap junction modifications perturb the development of cerebral, cardiac, and auditory functions (Kar et al., 2012). Consequently, connexins represent important targets for pharmacological research (Bodendiek & Raman, 2010).
A variety of fenamates inhibit gap junctions in rat kidney fibroblasts, a result reproduced in SKHep1 cells overexpressing Cx43 (Harks et al., 2001). In this model, FFA inhibits intercellular communication with an IC50 of 40 μM. This inhibitory effect was later described for Cx46 and Cx50 expressed in Xenopus oocytes (Eskandari et al., 2002). The effect was further investigated at the current level after overexpressing a variety of connexins in N2A neuroblastoma cells; Cx23, 32, 40, 43, 46, and 50 are inhibited by FFA with an IC50 ranging from 20 to 60 μM (Srinivas & Spray, 2003). Interestingly, FFA does not appear to affect single-channel conductance. The molecule does not bind connexin within the conduction pore but rather in a modulatory site, presumably within the membrane, inducing channel closure (Srinivas & Spray, 2003).
2.3. Potassium channels
K+ channels form the largest ion channel family with close to one hundred genes that encode such channels that have an extensive array of physiological functions. There are only a few noteworthy effects of FFA on these channels. K+ channels are subdivided according to biophysics as voltage-gated K+ channels (Kv), Ca2+-activated K+ channels (KCa), inward rectifier K+ channels (Kir), and two-pore K+ channels (K2P). In contrast to its effect on most others channels, FFA exerts an activating effect on K+ channels in nearly all cases.
FFA affects a large conductance Ca2+-activated K+ channels, known as the Ca2+-activated big K+ channels (BKCa), as shown in coronary smooth muscle membrane vesicles incorporated in lipid bilayer for electrophysiological recordings (Ottolia & Toro, 1994), rabbit portal vein smooth muscle cells (Greenwood & Large, 1995), and cultured Vero kidney cells (Kochetkov et al., 2000), among others. The KCa 1.1 gene (or Slo1) encodes BKCa current. Expression of mouse or human KCa 1.1 in Xenopus oocytes results in a K+ current activated by FFA with an EC50 that exceeds 0.3 mM (Gribkoff et al., 1996). FFA may be more efficient in native KCa channels, because the activation of BK currents in coronary and portal vein smooth muscle cells was on the order of 50 μM (Ottolia & Toro, 1994; Greenwood & Large, 1995). Moreover, in human trabecular meshwork 10-5 M FFA stimulated BKCa current by 400% (Stumpff et al., 2001).
FFA has also been shown to activate the channel encoded by the human ether-a-gogo related gene (HERG), also called Kv 11.1. This gene encodes for the pore forming subunit of the rapid component of the delayed rectifier K+ channel participating in action potential repolarization in cardiac myocytes. When heterologously expressed in Xenopus oocytes, Kv 11.1 produces a current enhanced by 20% in the presence of 10-4 M FFA (Malykhina et al., 2002). Interestingly, 10-4 M FFA also enhances the slow component of the delayed rectifier K+ current encoded by Kv 7.1 by slowing its deactivation (Busch et al., 1994).
Recently FFA was shown to stimulate the two-pore outwardly rectifying K+ channel KCa 4.2 (or Slo 2.1) expressed heterologously in Xenopus oocytes, although at a high dose (EC50 of 1.1 - 1.4 mM) (Dai et al., 2010; Garg & Sanguinetti, 2012). Interestingly, the mutant A278R, which substitutes a residue in the transmembrane domain six segment flanking the pore, is 19-times more sensitive to FFA, indicating that FFA binding might occur in this region (Garg & Sanguinetti, 2012). KCa 4.2 encodes a K+ channel gated by voltage as well as internal Na+ and Cl-, which is also inhibited by ATP. The physiological functions of Slo 2.1 are not yet established, but its relative “slack” (or Slo 2.2) may be involved in neural burst generation and termination in particular in central pattern generating neural circuits (Wallen et al., 2007; Krey et al., 2010). FFA also activates the lipid-sensitive mechano-gated two-pore channels encoded by K2P 4.1, K2P 10.1 and K2P 2.1 with EC50 in the range of 1 mM (Takahira et al., 2005).
2.4. Sodium channels
Action potentials in all excitable cells depend on voltage-activated Na+ channels. After an initial depolarization reaches the threshold of activation, Na+ channels open and produce the rapid upstroke of the action potential. Repolarization is achieved, in part, by time-dependent channel inactivation. The Na+ channel protein is composed of one α subunit (four major repeat units, each of which is composed of six transmembrane domains) and two β subunits (each is comprised of one transmembrane segment) encoded by genes SCNXA (or Nav) and SCNXB (Catterall, 2010). A recent paper describes the inhibition of the voltage-activated Na+ channel in hippocampal pyramidal neurons by FFA with an IC50 of approximately 0.2 mM (Yau et al., 2010). FFA affects inactivation by shifting the steady-state inactivation curve to more hyperpolarized membrane potentials.
FFA activates another Na+ conductance in ventricular cardiomyocytes with an EC50 that exceeds 0.2 mM (Macianskiene et al., 2010). The underlying channel remains unknown but may correspond to the brain liver intestine Na+ channel (BLINaC) that is activated by high levels of FFA (EC50 > 1 mM) when heterologously expressed in Xenopus oocytes (Wiemuth & Grunder, 2011). BLINaC belongs to the degenerin/epithelial Na+ channel superfamily. It is predominantly expressed in non-neuronal tissues, in particular epithelia, and weak expression has been observed in heart (Sakai et al., 1999). Its physiological function was unknown until the recent demonstration that the BLINaC channel is expressed in cholangiocytes and is activated by bile acids, suggesting its role in bile duct sensing of bile acids concentrations (Wiemuth et al., 2012).
2.5. Calcium channels
Voltage-gated Ca2+ channels activate in response to depolarization and participate in Ca2+ transients that induce muscle cell contraction as well as a variety of excitable responses in neurons including, notably, chemical synaptic transmission. Ca2+ channels are composed of a central α subunit (organized according to four repeat units of six transmembrane segments each, similar to Na+ channels) encoded by the Cav genes and four additional regulatory subunits (α2, β, γ, δ) (Catterall, 2010). The channels are divided in L, P/Q, N, R and T subtypes. FFA inhibits smooth muscle tone in carotid arteries by directly inhibiting L-type Ca2+ channels with an IC50 of ~0.1 mM (Shimamura et al., 2002). No experiments have been reported to identify the subunit targeted by FFA.
3. Mechanisms involved in current modulation by FFA
The activating or inhibiting effects of FFA are well described. However, the underlying mechanisms remain largely unknown. Because FFA targets numerous ion channels with different structures, biophysics, and regulatory properties, the underlying mechanisms might be different from one to the other.
As illustrated in Fig. 3 and in most studies reported in this review, modulation of ion currents by FFA is not likely to occur via gene expression since the effect develops within minutes. While indirect effects on ion channels through modulation of intracellular pathways may occur, the major accepted mechanism is a direct interaction between FFA and channel proteins. That is particularly evident when FFA is used in excised patch-clamp configurations, as example for CFTR (McCarty et al., 1993), TRPM4 and TRPM5 (Ullrich et al., 2005; Guinamard et al., 2006b) or Cx50 (Srinivas & Spray, 2003). The FFA effect can be abolished by channel mutation such as in ClC-Ka (Liantonio et al., 2006), which also suggests a direct interaction between FFA and channel proteins. This direct effect assumes a binding site within the channel itself. Such a site was suspected for Cl- channels (CFTR and ClC-K) within the narrow part of the protein vestibule since NA is not able to reach the site in ClC-Ka (Liantonio et al., 2006) and FFA showed an apparent binding site at 40-50% of the electrical distance from the cytoplasmic face in CFTR (McCarty et al., 1993). This binding site may be different in non-selective cation channels, at least in Cx50, where it may be a modulatory site comprised within the membrane but not in the pore (Srinivas & Spray, 2003). Although the binding site was not described, FFA interacts directly with TRPC4, C5, and C6 (Jiang et al., 2003; Foster et al., 2009).
Figure 3. Effects of Flufenamic acid on several preparations.
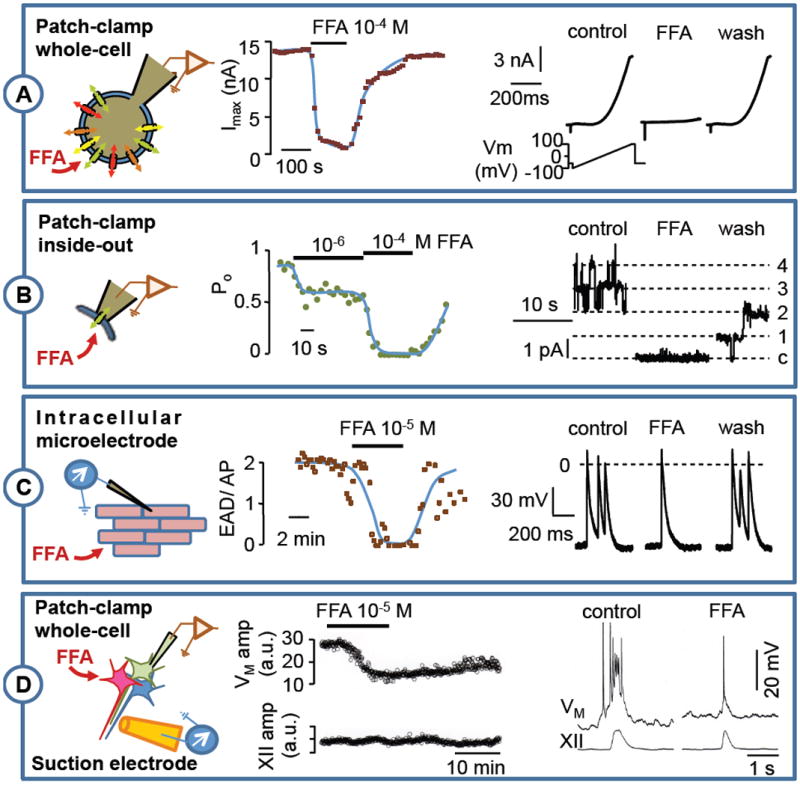
A: Inside-out patch-clamp recording of TRPM4 current on rat ventricular isolated myocyte (Vm = +40 mV). FFA produced a dose-dependent and reversible channel inhibition (see Guinamard et al. 2006-b for protocol). B: Action potential recorded by an intracellular microelectrode on isolated mouse ventricle submitted to a hypoxia and reoxygenation protocol (see Simard et al. 2012 for protocol). FFA superfusion reversibly reduced the number of early after depolarization by action potential (EAD/AP). C: Respiratory bursts recorded in rhythmogenic neurons of the preBötzinger complex (preBötC) as well as hypoglossal nerve root (XII) from neonatal mouse brainstem-slice preparations. Whole-cell patch-clamp recordings in preBötC neurons show that 100 μM FFA attenuates respiratory bursts at the whole-cell level by attenuating ICAN, but has a relatively mild affect motor output from the XII nerve root output (see Picardo et al. 2012 for protocol).
The insights above regarding FFA binding sites cannot be extended to other channels because of large variations in channel structure despite their (sometimes) common sensitivity to FFA.
4. Impact of ion channels modulation by FFA on physiological processes
The effect of FFA has been observed in a wide variety of physiological processes; too many to cover thoroughly in one review. Here, we focus on a few representative examples to illustrate the large spectrum of targets.
FFA affects neurons, smooth muscle cells, and cardiomyocytes. FFA reduces firing rates in neurons, and in particular reduces the rhythmic burst-generating capabilities of inspiratory neurons from the respiratory pre-Bötzinger complex, studied in thin medullary slices from neonatal rodents at concentrations from 10 to 500 μM (Pena et al., 2004; Del Negro et al., 2005). This effect occurs through inhibition of a Ca2+-activated non-selective cation current (ICAN, see above) that was later attributed to the TRPM4 or TRPM5 proteins, both expressed in this tissue (Crowder et al., 2007; Del Negro et al., 2010). The effective dose of FFA was later determined to be ~100 μM (Pace et al., 2007). FFA (100 μM) has been also shown to suppress epileptiform activity in rat CA1 pyramidal neurons of the hippocampus through diminution of glutamatergic excitatory synaptic transmission (Fernandez et al., 2010) and by blocking ICAN (Schiller, 2004). Therefore, FFA was proposed as a potentially effective agent for the treatment of epilepsy. FFA (30 μM) reduces the peptide-induced intra-cardiac neuron firing rate (Merriam et al., 2012), which may involve the TRPC channel inhibition. A reduction of firing rate by FFA (20 μM) was also reported in GABAergic neurons, possibly through TRP current inhibition (Lee et al., 2011b). Finally, FFA (3 μM) reduced dopamine-induced oscillations in pyloric pacemaker neurons of the spiny lobster (Kadiri et al., 2011).
FFA modulates gastrointestinal tract motility by reducing pacemaker potentials of intestinal cells of Cajal in mice (Han et al., 2012; Lee et al., 2012). This effect has also been observed at 50 μM in human intestinal cells of Cajal and attributed to the inhibition of the TRPM7 channel (Kim et al., 2009).
In neuroendocrinology, FFA (100 μM) inhibits pacemaker activity in rat pituitary lactotrophs through non-selective cation channel modulation, leading to decrease in prolactin secretion (Kucka et al., 2012).
Our group recently reported a cardioprotective effect of 10 μM FFA in a model of hypoxia reoxygenation-induced arrhythmia in mouse (Simard et al., 2012). The mechanism is related to the fact that FFA abolishes TRPM4-mediated early after depolarizations observed following reoxygenation. This FFA effect mimics the specific TRPM4 antagonist 9-phenanthrol, suggesting that FFA effect occurs through TRPM4 inhibition. Therefore, FFA may be regarded as a cardiac anti-arrhythmic agent. FFA (25 μM) may also modulate Ca2+ signaling by inhibiting Cx43 in rat ventricular myocytes (Li et al., 2012). A similar result was observed in the murine fibroblast cell line L929, where FFA (100 μM) inhibits ATP release and Ca2+ transients that polarize the actin/myosin complex via inhibition of connexins (Marimuthu et al., 2012). FFA (50 μM) also modulates vascular endothelial growth factor secretion in human retinal pigment epithelial cells by inhibiting Cx43 (Pocrnich et al., 2012).
The impact of FFA is not restricted to excitable cells. Partial reduction of Cl- secretion in human airway gland cells occurs in response to 100 μM FFA, an effect that might be attributed to inhibition of the chloride channel TMEM16A (Fischer et al., 2010). FFA also regulates cell volume in hypotonic as well as hypertonic conditions. Regulatory volume decreases in hypotonic conditions are reduced by FFA, due to a FFA-inhibited swelling-activated Cl- channel (Jin et al., 2003; Do et al., 2006). A regulatory volume increase under hypertonic conditions that protects against apoptosis is reduced by FFA with an EC50 of 300 μM, which occurs through FFA-mediated inhibition of cation current (Wehner et al., 2003). Alpha-subunit of the epithelial Na+ channel (ENaC) was shown to participate in this hypertonicity-inducced current in the human hepatocellular liver carcinoma cell line HepG2 (Bondarava et al., 2009) whereas the current was recently shown to be supported by the TRPM2 channel in the HeLa cells (Numata et al., 2012).
5. Using flufenamic acid in research
In the following section we evaluate the advantages and caveats of using FFA in research. The caveats pertain to several FFA targets in the same preparation and FFA exerting opposite effects on a target channel in a dose-dependent fashion. We discuss the use of FFA in comparison to other NSAIDs and finally identify assets of FFA.
5.1. Multiples targets in the same preparation
As described above, FFA modulates a wide spectrum of ion channels. The same cell can express several FFA-sensitive channels. For example, gonadotropin-releasing hormone neuroendocrine neurons express non-selective cation channels and BK channels (Wang & Kuehl-Kovarik, 2010). Mammalian cardiomyocytes express ion channels inhibited by FFA including TRPC3, TRPC6, TRPM4, TRPM7, and ICl,swell, but also channels that are activated by FFA such as HERG (Malykhina et al., 2002; Demion et al., 2006; Guinamard et al., 2006a; Guinamard et al., 2006b; Inoue et al., 2006). Similarly, guinea pig cardiac neurons express TRPC3, TRPC4, TRPC5 and TRPC6 (Merriam et al., 2012).
The presence of multiple FFA-sensitive channels must be considered when analyzing the effect of the drug at the whole-cell or systems levels. Because FFA has different affinities for different ion channels, interpreting and analyzing its effects depend on the sensitivity of each possibly affected channel type and the drug concentration used. For example, FFA modifies fictive swim patterns of the lamprey spinal cord, which is attributable to modulation of both Ca2+ channels and NMDA receptors (Wang et al., 2006). Similarly, FFA targets different channels in Aplysia bag cell neurons, modulating K+ channels, voltage-gated Ca2+ channels and Ca2+-dependent cation conductances (Gardam et al., 2008).
In addition to ion channels, FFA also affects other targets that indirectly impact ion channels and excitable cell behavior. For example, FFA activates the cAMP-activated protein kinase, (Chi et al., 2011), and yet inhibits the mouse GABA transporter GAT4 (Liantonio et al., 2007) and glycine transporters (Steinmeyer et al., 1991). Finally, FFA can also alter mitochondrial Ca2+homeostasis, impacting Ca2+-dependent channels (Macdonald et al., 2010). Since our review focuses on the direct effects of FFA on ion channels, we will not describe the effects above in detail, but we emphasize that there are other biochemical and integral membrane proteins that may be affected by FFA. Therefore, these other targets must be taken into account when analyzing the effects of FFA in the context of physiological experiments.
A recent publication reevaluating the chemical structure of FFA demonstrated that this molecule possesses at least nine polymorphs (Lopez-Mejias et al., 2012), which may influence the bioavailability of the drug and thus provide new opportunities for the investigating the channels types targeted by FFA, depending on these polymorphs.
5.2. Opposite effects on the same channel
Another FFA-related caveat comes from its ability to exert opposite effects on the same channel, depending on concentration. FFA inhibits TRPC6 with an IC50 of 17.1 μM (Klose et al., 2011) but 100 μM FFA activates the same channel (Inoue et al., 2001). TRPM8 is inhibited at 100 μM FFA but slightly activated at higher concentrations (Hu et al., 2010). A worse situation was reported for BKCa modulation since FFA activates the channel below 10 μM, inhibits the channel between 10 to 50 μM, and then activates the channel above 50 μM (Kochetkov et al., 2000).
5.3. Flufenamic acid or other fenamates
Other NSAIDs, including fenamates, are also known to modulate a variety of ion channels (Gwanyanya et al., 2012). Most ion channels modulated by FFA are also affected by other fenamates. A few studies provide a comparative analysis of the effects of several fenamates on the same ion channel, ranking fenamates according to their potencies to block or activate channels. Because the rank order of efficacy among fenamates differs from one channel to the other, we will not review all of them. However, in the majority of reports, FFA appears to be more effective than niflumic acid (NA) and mefenamic acid (MFA), two of the most commonly tested fenamates. This sequence was observed for TRPM2, TRPV4 and TRPC6 inhibition (Klose et al., 2011; Chen et al., 2012), TRPC4 and TRPC5 inhibition (Jiang et al., 2012), TRPA1 activation (Hu et al., 2010), BKCa activation (Ottolia & Toro, 1994), Cx43 inhibition (Harks et al., 2001), as well as K2P 2.1 and K2P 10.1 channel activation (Takahira et al., 2005). The sequence of fenamate sensitivity might be somewhat different for chloride channels, since MFA is more effective than FFA in ClC-K and GABAA receptor modulation (Woodward et al., 1994; Liantonio et al., 2006), whereas NA is more effective than FFA on IClCa (Greenwood & Large, 1995; Oh et al., 2008). For the Slo2.1 potassium channel, the sequence is MFA > FFA > NA (Garg & Sanguinetti, 2012).
Most of the FFA-targeted ion channels are sensitive to other fenamates, but this does not necessitate non-specificity of the FFA binding site within channel proteins. Indeed, FFA and NA do not use same binding site on the ClC-Ka channel (Zifarelli et al., 2010).
5.4. Assets of flufenamic acid
Despite its promiscuity, FFA remains a convenient toll for physiological studies. FFA can be used in a wide variety of experimental models ranging from molecular preparations such as inside-out single-channel recordings, to cellular preparations such as whole-cell recordings on isolated cells as well as isolated tissue slices in vitro and in situ. Instead of reviewing all these preparations, which have been already presented in the above sections and table 1, we illustrate several examples of FFA applications using different experimental models (Fig. 3).
FFA is lipophilic and thus membrane permeable (McCarty et al., 1993; Hill et al., 2004). Accordingly, FFA can access intracellular or extracellular targets whatever is its side of application, as illustrated for TRPM4 inhibition (Fig. 3). FFA access can be achieved by drug application in the bath during inside-out patch recordings, when the inside of the channel faces the bath (Guinamard et al., 2006b) or in the whole cell-configuration when external side is exposed (Pena et al., 2004; Pace et al., 2007).
The effects of FFA develop and reverse rapidly. Examples in Fig. 3 show that, even when applied on a multicellular isolated tissue preparation (mouse right ventricle, Fig. 3B; (Simard et al., 2012)) or a rhythmically active respiratory rhythmogenic network (Fig. 3C; (Picardo et al., 2012)), the effect of FFA develops within a few minutes and washes out with a commensurate time course. When applied to isolated cells, the effects of FFA occur (and reverse) in the range of few seconds.
6. Conclusion
FFA appears to be a broad spectrum ion channel modulator, with preference for non-selective cation channels and chloride channels. However, it remains a convenient tool if used with precaution, keeping in mind the caveats recapped above. That is particularly true for studies investigating the role of channels with higher sensitivity for FFA such as TRPM4. In combination with other more specific tools, FFA can provide a useful tool to identify ion channels and probe their physiological role(s) in a range of reduced preparations in vitro or in situ.
Extensive knowledge of ion channels targeted by FFA may revive interest in the use of this molecule for therapeutic purposes, as was suggested for NSAIDs, especially fenamates, in the treatment of neurological disorders (Khansari & Coyne, 2012). The recently developed FFA hydrophobic derivative nanoprodrugs show an increase in the drug efficiency (Lee et al., 2011a). Accordingly, lower doses might be efficient in medical use and, thus, a better targeting of different physiological actors might be achieved.
Acknowledgments
Christophe Simard is a recipient of a fellowship from the French Ministère de l’Enseignement et de la Recherche.
Christopher A. Del Negro is supported by US National Institutes of Health grants 1R21NS070056-01 and 5R01HL104127-03.
ABBREVIATIONS
- BKCa
big K+ channel
- BLINaC
brain liver intestine Na+ channel
- CaCC
Ca2+-activated chloride current
- CFTR
cystic fibrosis transmembrane conductance regulator
- ClC
chloride channel
- ClC-K
chloride channel kidney
- Cx
connexin
- EC50
concentration for half maximal effect
- FFA
flufenamic acid
- GABA
γ-aminobutyric acid
- HEK-293
human embryonic kidney cell line 293
- HERG
human ether-a-gogo-related gene
- ICl,swell
swelling-activated chloride current
- IC50
concentration for half maximal inhibition
- KCa
Ca2+-activated K+ channel
- Kv
voltage-gated K+ channel
- K2P
two pores K+ channel
- MFA
mefenamic acid
- nAchR
nicotinic acetylcholine receptor
- NA
niflumic acid
- NMDA
N-methyl-d-aspartate
- NSC
non-selective cation channels
- NSCCa
Ca2+-activated non-selective cation channels
- PanX
pannexin
- TMEM16A
transmembrane protein 16A
- TRP
transient receptor potential channels
- TRPA
transient receptor potential ankyrin
- TRPC
transient receptor potential canonical
- TRPM
transient receptor potential melastatin
- TRPV
transient receptor potential vanilloid
Footnotes
Conflict of Interest Statement:
The authors declare that there are no conflicts if interest.
Written assurance:
This manuscript has not been published and is not under consideration for publication elsewhere.
References
- Albert AP, Pucovsky V, Prestwich SA, Large WA. TRPC3 properties of a native constitutively active Ca2+-permeable cation channel in rabbit ear artery myocytes. J Physiol. 2006;571:361–369. doi: 10.1113/jphysiol.2005.102780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly FA, Al-Tamimi SA, Alwarthan AA. Determination of flufenamic acid and mefenamic acid in pharmaceutical preparations and biological fluids using flow injection analysis with tris(2,2’-bipyridyl)ruthenium(II) chemiluminescence detection. Analitica Chmica Acta. 2000;416:87–96. [Google Scholar]
- Barish ME. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J Physiol. 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nature genetics. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Progress in biophysics and molecular biology. 2003;82:25–42. doi: 10.1016/s0079-6107(03)00003-8. [DOI] [PubMed] [Google Scholar]
- Becq F, Mettey Y. Pharmacological interventions for the correction of ion transport defect in cystic fibrosis. Expert Opin Ther Patents. 2004;14:1465–1483. [Google Scholar]
- Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NV, Antignac C, Sudbrak R, Kispert A, Hildebrandt F. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nature genetics. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- Bodendiek SB, Raman G. Connexin modulators and their potential targets under the magnifying glass. Current medicinal chemistry. 2010;17:4191–4230. doi: 10.2174/092986710793348563. [DOI] [PubMed] [Google Scholar]
- Bondarava M, Li T, Endl E, Wehner F. alpha-ENaC is a functional element of the hypertonicity-induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Archiv : European journal of physiology. 2009;458:675–687. doi: 10.1007/s00424-009-0649-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Herzer T, Wagner CA, Schmidt F, Raber G, Waldegger S, Lang F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol Pharmacol. 1994;46:750–753. [PubMed] [Google Scholar]
- Catterall WA. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett. 2010;486:107–116. doi: 10.1016/j.neulet.2010.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GL, Zeng B, Eastmond S, Elsenussi SE, Boa AN, Xu SZ. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. British journal of pharmacology. 2012 doi: 10.1111/j.1476-5381.2012.02058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Li K, Yan Q, Koizumi S, Shi L, Takahashi S, Zhu Y, Matsue H, Takeda M, Kitamura M, Yao J. Nonsteroidal anti-inflammatory drug flufenamic acid is a potent activator of AMP-activated protein kinase. J Pharmacol Exp Ther. 2011;339:257–266. doi: 10.1124/jpet.111.183020. [DOI] [PubMed] [Google Scholar]
- Cliff WH, Frizzell RA. Separate Cl- conductances activated by cAMP and Ca2+ in Cl(-)-secreting epithelial cells. Proc Natl Acad Sci U S A. 1990;87:4956–4960. doi: 10.1073/pnas.87.13.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortright DN, Crandall M, Sanchez JF, Zou T, Krause JE, White G. The tissue distribution and functional characterization of human VR1. Biochemical and biophysical research communications. 2001;281:1183–1189. doi: 10.1006/bbrc.2001.4482. [DOI] [PubMed] [Google Scholar]
- Crowder EA, Saha MS, Pace RW, Zhang H, Prestwich GD, Del Negro CA. Phosphatidylinositol 4,5-bisphosphate regulates inspiratory burst activity in the neonatal mouse preBotzinger complex. J Physiol. 2007;582:1047–1058. doi: 10.1113/jphysiol.2007.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Garg V, Sanguinetti MC. Activation of Slo2.1 channels by niflumic acid. J Gen Physiol. 2010;135:275–295. doi: 10.1085/jgp.200910316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Negro CA, Hayes JA, Pace RW, Brush BR, Teruyama R, Feldman JL. Synaptically activated burst-generating conductances may underlie a group-pacemaker mechanism for respiratory rhythm generation in mammals. Progress in brain research. 2010;187:111–136. doi: 10.1016/B978-0-444-53613-6.00008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Hayes JA, Mackay DD, Pace RW, Crowder EA, Feldman JL. Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:446–453. doi: 10.1523/JNEUROSCI.2237-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demion M, Guinamard R, El Chemaly A, Rahmati M, Bois P. An outwardly rectifying chloride channel in human atrial cardiomyocytes. Journal of cardiovascular electrophysiology. 2006;17:60–68. doi: 10.1111/j.1540-8167.2005.00255.x. [DOI] [PubMed] [Google Scholar]
- Do CW, Peterson-Yantorno K, Civan MM. Swelling-activated Cl- channels support Cl- secretion by bovine ciliary epithelium. Investigative ophthalmology & visual science. 2006;47:2576–2582. doi: 10.1167/iovs.05-0851. [DOI] [PubMed] [Google Scholar]
- Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J Physiol. 2009;587:2163–2177. doi: 10.1113/jphysiol.2008.165860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Hume JR, Nattel S. Evidence that outwardly rectifying Cl- channels underlie volume-regulated Cl- currents in heart. Circulation research. 1997a;80:103–113. doi: 10.1161/01.res.80.1.103. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997b;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, Yamboliev I, Horowitz B, Hume JR. Functional inhibition of native volume-sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti-ClC-3 antibody. J Physiol. 2001;531:437–444. doi: 10.1111/j.1469-7793.2001.0437i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annual review of physiology. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari S, Zampighi GA, Leung DW, Wright EM, Loo DD. Inhibition of gap junction hemichannels by chloride channel blockers. The Journal of membrane biology. 2002;185:93–102. doi: 10.1007/s00232-001-0115-0. [DOI] [PubMed] [Google Scholar]
- Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Fischer M. Physiology and pathophysiology of ClC-K/barttin channels. Frontiers in physiology. 2010;1:155. doi: 10.3389/fphys.2010.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famaey JP, Whitehouse MW. Effects of nonsteroidal anti-inflammatory drugs on the uptake of various cations by lymphoid cells. Archives internationales de physiologie et de biochimie. 1976;84:719–734. doi: 10.3109/13813457609067047. [DOI] [PubMed] [Google Scholar]
- Fernandez M, Lao-Peregrin C, Martin ED. Flufenamic acid suppresses epileptiform activity in hippocampus by reducing excitatory synaptic transmission and neuronal excitability. Epilepsia. 2010;51:384–390. doi: 10.1111/j.1528-1167.2009.02279.x. [DOI] [PubMed] [Google Scholar]
- Fischer H, Illek B, Sachs L, Finkbeiner WE, Widdicombe JH. CFTR and calcium-activated chloride channels in primary cultures of human airway gland cells of serous or mucous phenotype. American journal of physiology Lung cellular and molecular physiology. 2010;299:L585–594. doi: 10.1152/ajplung.00421.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower R, Gryglewski R, Herbaczynska-Cedro K, Vane JR. Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nature: New biology. 1972;238:104–106. doi: 10.1038/newbio238104a0. [DOI] [PubMed] [Google Scholar]
- Flower RJ. Drugs which inhibit prostaglandin biosynthesis. Pharmacological reviews. 1974;26:33–67. [PubMed] [Google Scholar]
- Foster RR, Zadeh MA, Welsh GI, Satchell SC, Ye Y, Mathieson PW, Bates DO, Saleem MA. Flufenamic acid is a tool for investigating TRPC6-mediated calcium signalling in human conditionally immortalised podocytes and HEK293 cells. Cell calcium. 2009;45:384–390. doi: 10.1016/j.ceca.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Gardam KE, Geiger JE, Hickey CM, Hung AY, Magoski NS. Flufenamic acid affects multiple currents and causes intracellular Ca2+ release in Aplysia bag cell neurons. Journal of neurophysiology. 2008;100:38–49. doi: 10.1152/jn.90265.2008. [DOI] [PubMed] [Google Scholar]
- Garg P, Sanguinetti MC. Structure-activity Relationship of Fenamates as Slo2.1 Channel Activators. Mol Pharmacol. 2012 doi: 10.1124/mol.112.079194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harbor perspectives in biology. 2010;2:a003962. doi: 10.1101/cshperspect.a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogelein H, Dahlem D, Englert HC, Lang HJ. Flufenamic acid, mefenamic acid and niflumic acid inhibit single nonselective cation channels in the rat exocrine pancreas. FEBS letters. 1990;268:79–82. doi: 10.1016/0014-5793(90)80977-q. [DOI] [PubMed] [Google Scholar]
- Gradogna A, Pusch M. Molecular Pharmacology of Kidney and Inner Ear CLC-K Chloride Channels. Frontiers in pharmacology. 2010;1:130. doi: 10.3389/fphar.2010.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. British journal of pharmacology. 1995;116:2939–2948. doi: 10.1111/j.1476-5381.1995.tb15948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff VK, Lum-Ragan JT, Boissard CG, Post-Munson DJ, Meanwell NA, Starrett JE, Jr, Kozlowski ES, Romine JL, Trojnacki JT, McKay MC, Zhong J, Dworetzky SI. Effects of channel modulators on cloned large-conductance calcium-activated potassium channels. Mol Pharmacol. 1996;50:206–217. [PubMed] [Google Scholar]
- Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. American journal of physiology Cell physiology. 2009;297:C493–502. doi: 10.1152/ajpcell.00624.2008. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Chatelier A, Bois P. Calcium-activated nonselective cation channels in mammalian cardiomyocytes. Trends in cardiovascular medicine. 2006a;16:245–250. doi: 10.1016/j.tcm.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Magaud C, Potreau D, Bois P. Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension. 2006b;48:587–594. doi: 10.1161/01.HYP.0000237864.65019.a5. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Paulais M, Lourdel S, Teulon J. A calcium-permeable non-selective cation channel in the thick ascending limb apical membrane of the mouse kidney. Biochimica et biophysica acta. 2012;1818:1135–1141. doi: 10.1016/j.bbamem.2011.12.024. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Rahmati M, Lenfant J, Bois P. Characterization of a Ca2+-activated nonselective cation channel during dedifferentiation of cultured rat ventricular cardiomyocytes. The Journal of membrane biology. 2002;188:127–135. doi: 10.1007/s00232-001-0180-4. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Salle L, Simard C. The non-selective monovalent cationic channels TRPM4 and TRPM5. Advances in experimental medicine and biology. 2011;704:147–171. doi: 10.1007/978-94-007-0265-3_8. [DOI] [PubMed] [Google Scholar]
- Gwanyanya A, Macianskiene R, Bito V, Sipido KR, Vereecke J, Mubagwa K. Inhibition of the calcium-activated chloride current in cardiac ventricular myocytes by N-(p-amylcinnamoyl)anthranilic acid (ACA) Biochemical and biophysical research communications. 2010;402:531–536. doi: 10.1016/j.bbrc.2010.10.069. [DOI] [PubMed] [Google Scholar]
- Gwanyanya A, Macianskiene R, Mubagwa K. Insights into the effects of diclofenac and other non-steroidal anti-inflammatory agents on ion channels. The Journal of pharmacy and pharmacology. 2012;64:1359–1375. doi: 10.1111/j.2042-7158.2012.01479.x. [DOI] [PubMed] [Google Scholar]
- Han S, Kim JS, Jung BK, Han SE, Nam JH, Kwon YK, Nah SY, Kim BJ. Effects of ginsenoside on pacemaker potentials of cultured interstitial cells of Cajal clusters from the small intestine of mice. Molecules and cells. 2012;33:243–249. doi: 10.1007/s10059-012-2204-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harks EG, de Roos AD, Peters PH, de Haan LH, Brouwer A, Ypey DL, van Zoelen EJ, Theuvenet AP. Fenamates: a novel class of reversible gap junction blockers. J Pharmacol Exp Ther. 2001;298:1033–1041. [PubMed] [Google Scholar]
- Hill K, Benham CD, McNulty S, Randall AD. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47:450–460. doi: 10.1016/j.neuropharm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Hu H, Tian J, Zhu Y, Wang C, Xiao R, Herz JM, Wood JD, Zhu MX. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Archiv : European journal of physiology. 2010;459:579–592. doi: 10.1007/s00424-009-0749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Wong X, Jan LY. International Union of Basic and Clinical Pharmacology. LXXXV: calcium-activated chloride channels. Pharmacological reviews. 2012a;64:1–15. doi: 10.1124/pr.111.005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY. Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron. 2012b;74:179–192. doi: 10.1016/j.neuron.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circulation research. 2006;99:119–131. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circulation research. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zeng B, Chen GL, Bot D, Eastmond S, Elsenussi SE, Atkin SL, Boa AN, Xu SZ. Effect of non-steroidal anti-inflammatory drugs and new fenamate analogues on TRPC4 and TRPC5 channels. Biochemical pharmacology. 2012;83:923–931. doi: 10.1016/j.bcp.2012.01.014. [DOI] [PubMed] [Google Scholar]
- Jiang X, Newell EW, Schlichter LC. Regulation of a TRPM7-like current in rat brain microglia. J Biol Chem. 2003;278:42867–42876. doi: 10.1074/jbc.M304487200. [DOI] [PubMed] [Google Scholar]
- Jin NG, Kim JK, Yang DK, Cho SJ, Kim JM, Koh EJ, Jung HC, So I, Kim KW. Fundamental role of ClC-3 in volume-sensitive Cl- channel function and cell volume regulation in AGS cells. American journal of physiology Gastrointestinal and liver physiology. 2003;285:G938–948. doi: 10.1152/ajpgi.00470.2002. [DOI] [PubMed] [Google Scholar]
- Jones SM, Palmer MJ. Pharmacological analysis of the activation and receptor properties of the tonic GABA(C)R current in retinal bipolar cell terminals. PloS one. 2011;6:e24892. doi: 10.1371/journal.pone.0024892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiri LR, Kwan AC, Webb WW, Harris-Warrick RM. Dopamine-induced oscillations of the pyloric pacemaker neuron rely on release of calcium from intracellular stores. Journal of neurophysiology. 2011;106:1288–1298. doi: 10.1152/jn.00456.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar R, Batra N, Riquelme MA, Jiang JX. Biological role of connexin intercellular channels and hemichannels. Archives of biochemistry and biophysics. 2012;524:2–15. doi: 10.1016/j.abb.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khansari PS, Coyne L. NSAIDs in the treatment and/or prevention of neurological disorders. Inflammopharmacology. 2012;20:159–167. doi: 10.1007/s10787-011-0116-2. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Park KJ, Kim HW, Choi S, Jun JY, Chang IY, Jeon JH, So I, Kim SJ. Identification of TRPM7 channels in human intestinal interstitial cells of Cajal. World journal of gastroenterology : WJG. 2009;15:5799–5804. doi: 10.3748/wjg.15.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose C, Straub I, Riehle M, Ranta F, Krautwurst D, Ullrich S, Meyerhof W, Harteneck C. Fenamates as TRP channel blockers: mefenamic acid selectively blocks TRPM3. British journal of pharmacology. 2011;162:1757–1769. doi: 10.1111/j.1476-5381.2010.01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochetkov KV, Kazachenko VN, Marinov BS. Dose-dependent potentiation and inhibition of single Ca2+-activated K+ channels by flufenamic acid. Membrane & cell biology. 2000;14:285–298. [PubMed] [Google Scholar]
- Krey RA, Goodreau AM, Arnold TB, Del Negro CA. Outward Currents Contributing to Inspiratory Burst Termination in preBotzinger Complex Neurons of Neonatal Mice Studied in Vitro. Front Neural Circuits. 2010;4:124. doi: 10.3389/fncir.2010.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucka M, Kretschmannova K, Stojilkovic SS, Zemkova H, Tomic M. Dependence of spontaneous electrical activity and basal prolactin release on nonselective cation channels in pituitary lactotrophs. Physiological research / Academia Scientiarum Bohemoslovaca. 2012 doi: 10.33549/physiolres.932301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Yoon CW, Osipov A, Moghavem N, Nwachokor D, Amatya R, Na R, Pantoja JL, Pham MD, Black KL, Yu JS. Nanoprodrugs of NSAIDs: Preparation and Characterization of Flufenamic Acid Nanoprodrugs. Journal of drug delivery. 2011a;2011:980720. doi: 10.1155/2011/980720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Witkovsky P, Rice ME. Regulation of Substantia Nigra Pars Reticulata GABAergic Neuron Activity by H(2)O(2) via Flufenamic Acid-Sensitive Channels and K(ATP) Channels. Frontiers in systems neuroscience. 2011b;5:14. doi: 10.3389/fnsys.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim YD, Park CG, Kim MY, Chang IY, Zuo DC, Shahi PK, Choi S, Yeum CH, Jun JY. Neurotensin modulates pacemaker activity in interstitial cells of Cajal from the mouse small intestine. Molecules and cells. 2012;33:509–516. doi: 10.1007/s10059-012-2290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Kim BJ, Kim HJ, Yang DK, Zhu MH, Lee KP, So I, Kim KW. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. American journal of physiology Gastrointestinal and liver physiology. 2003;284:G604–616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- Lentjes EG, van Ginneken CA. Pharmacokinetics of flufenamic acid in man. International journal of clinical pharmacology, therapy, and toxicology. 1987;25:185–187. [PubMed] [Google Scholar]
- Lerma J, Martin del Rio R. Chloride transport blockers prevent N-methyl-D-aspartate receptor-channel complex activation. Mol Pharmacol. 1992;41:217–222. [PubMed] [Google Scholar]
- Li C, Meng Q, Yu X, Jing X, Xu P, Luo D. Regulatory effect of connexin 43 on basal Ca2+ signaling in rat ventricular myocytes. PloS one. 2012;7:e36165. doi: 10.1371/journal.pone.0036165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liantonio A, Giannuzzi V, Picollo A, Babini E, Pusch M, Conte Camerino D. Niflumic acid inhibits chloride conductance of rat skeletal muscle by directly inhibiting the CLC-1 channel and by increasing intracellular calcium. British journal of pharmacology. 2007;150:235–247. doi: 10.1038/sj.bjp.0706954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liantonio A, Gramegna G, Camerino GM, Dinardo MM, Scaramuzzi A, Potenza MA, Montagnani M, Procino G, Lasorsa DR, Mastrofrancesco L, Laghezza A, Fracchiolla G, Loiodice F, Perrone MG, Lopedota A, Conte S, Penza R, Valenti G, Svelto M, Camerino DC. In-vivo administration of CLC-K kidney chloride channels inhibitors increases water diuresis in rats: a new drug target for hypertension? Journal of hypertension. 2012;30:153–167. doi: 10.1097/HJH.0b013e32834d9eb9. [DOI] [PubMed] [Google Scholar]
- Liantonio A, Picollo A, Babini E, Carbonara G, Fracchiolla G, Loiodice F, Tortorella V, Pusch M, Camerino DC. Activation and inhibition of kidney CLC-K chloride channels by fenamates. Mol Pharmacol. 2006;69:165–173. doi: 10.1124/mol.105.017384. [DOI] [PubMed] [Google Scholar]
- Liantonio A, Picollo A, Carbonara G, Fracchiolla G, Tortorella P, Loiodice F, Laghezza A, Babini E, Zifarelli G, Pusch M, Camerino DC. Molecular switch for CLC-K Cl- channel block/activation: optimal pharmacophoric requirements towards high-affinity ligands. Proc Natl Acad Sci U S A. 2008;105:1369–1373. doi: 10.1073/pnas.0708977105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER. The Ca2+-Activated TRP Channels: TRPM4 and TRPM5. In: Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): 2007a. [PubMed] [Google Scholar]
- Liman ER. TRPM5 and taste transduction. Handbook of experimental pharmacology. 2007b:287–298. doi: 10.1007/978-3-540-34891-7_17. [DOI] [PubMed] [Google Scholar]
- Liu GJ, Kalous A, Werry EL, Bennett MR. Purine release from spinal cord microglia after elevation of calcium by glutamate. Mol Pharmacol. 2006;70:851–859. doi: 10.1124/mol.105.021436. [DOI] [PubMed] [Google Scholar]
- Lopez-Mejias V, Kampf JW, Matzger AJ. Nonamorphism in flufenamic acid and a new record for a polymorphic compound with solved structures. Journal of the American Chemical Society. 2012;134:9872–9875. doi: 10.1021/ja302601f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Compan V, Zheng W, Martin E, North RA, Verkhratsky A, Surprenant A. Pannexin 1 forms an anion-selective channel. Pflugers Archiv : European journal of physiology. 2012;463:585–592. doi: 10.1007/s00424-012-1077-z. [DOI] [PubMed] [Google Scholar]
- Ma W, Hui H, Pelegrin P, Surprenant A. Pharmacological characterization of pannexin-1 currents expressed in mammalian cells. J Pharmacol Exp Ther. 2009;328:409–418. doi: 10.1124/jpet.108.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Rogers CJ, Twyman RE. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol. 1989;410:479–499. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macianskiene R, Gwanyanya A, Sipido KR, Vereecke J, Mubagwa K. Induction of a novel cation current in cardiac ventricular myocytes by flufenamic acid and related drugs. British journal of pharmacology. 2010;161:416–429. doi: 10.1111/j.1476-5381.2010.00901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Tsukihara T. Structure of the gap junction channel and its implications for its biological functions. Cell Mol Life Sci. 2011;68:1115–1129. doi: 10.1007/s00018-010-0551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malykhina AP, Shoeb F, Akbarali HI. Fenamate-induced enhancement of heterologously expressed HERG currents in Xenopus oocytes. Eur J Pharmacol. 2002;452:269–277. doi: 10.1016/s0014-2999(02)02330-0. [DOI] [PubMed] [Google Scholar]
- Marimuthu M, Park C, Kim S, Choi CS. Real-time electrical measurement of L929 cellular spontaneous and synchronous oscillation. International journal of nanomedicine. 2012;7:83–92. doi: 10.2147/IJN.S28465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty NA, McDonough S, Cohen BN, Riordan JR, Davidson N, Lester HA. Voltage-dependent block of the cystic fibrosis transmembrane conductance regulator Cl- channel by two closely related arylaminobenzoates. J Gen Physiol. 1993;102:1–23. doi: 10.1085/jgp.102.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam LA, Roman CW, Baran CN, Girard BM, May V, Parsons RL. Pretreatment with Nonselective Cationic Channel Inhibitors Blunts the PACAP-Induced Increase in Guinea Pig Cardiac Neuron Excitability. Journal of molecular neuroscience : MN. 2012 doi: 10.1007/s12031-012-9763-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B. Drosophila mutant with a transducer defect. Biophysics of structure and mechanism. 1977;3:59–64. doi: 10.1007/BF00536455. [DOI] [PubMed] [Google Scholar]
- Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- Moore RA, Tramer MR, Carroll D, Wiffen PJ, McQuay HJ. Quantitative systematic review of topically applied non-steroidal anti-inflammatory drugs. Bmj. 1998;316:333–338. doi: 10.1136/bmj.316.7128.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naziroglu M, Luckhoff A, Jungling E. Antagonist effect of flufenamic acid on TRPM2 cation channels activated by hydrogen peroxide. Cell biochemistry and function. 2007;25:383–387. doi: 10.1002/cbf.1310. [DOI] [PubMed] [Google Scholar]
- Naziroglu M, Ozgul C, Celik O, Cig B, Sozbir E. Aminoethoxydiphenyl borate and flufenamic acid inhibit Ca2+ influx through TRPM2 channels in rat dorsal root ganglion neurons activated by ADP-ribose and rotenone. The Journal of membrane biology. 2011;241:69–75. doi: 10.1007/s00232-011-9363-9. [DOI] [PubMed] [Google Scholar]
- Numata T, Sato K, Christmann J, Marx R, Mori Y, Okada Y, Wehner F. The DeltaC splice-variant of TRPM2 is the hypertonicity-induced cation channel in HeLa cells, and the ecto-enzyme CD38 mediates its activation. J Physiol. 2012;590:1121–1138. doi: 10.1113/jphysiol.2011.220947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SJ, Park JH, Han S, Lee JK, Roh EJ, Lee CJ. Development of selective blockers for Ca(2)(+)-activated Cl channel using Xenopus laevis oocytes with an improved drug screening strategy. Molecular brain. 2008;1:14. doi: 10.1186/1756-6606-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol. 2009;587:965–979. doi: 10.1113/jphysiol.2008.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolia M, Toro L. Potentiation of large conductance KCa channels by niflumic, flufenamic, and mefenamic acids. Biophys J. 1994;67:2272–2279. doi: 10.1016/S0006-3495(94)80712-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace RW, Mackay DD, Feldman JL, Del Negro CA. Inspiratory bursts in the preBotzinger complex depend on a calcium-activated non-specific cation current linked to glutamate receptors in neonatal mice. J Physiol. 2007;582:113–125. doi: 10.1113/jphysiol.2007.133660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Oh SJ, Han KS, Woo DH, Park H, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge LD, Valenzuela CF. Block of hippocampal CAN channels by flufenamate. Brain research. 2000;867:143–148. doi: 10.1016/s0006-8993(00)02275-7. [DOI] [PubMed] [Google Scholar]
- Pena F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. 2004;43:105–117. doi: 10.1016/j.neuron.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Pezier A, Grauso M, Acquistapace A, Monsempes C, Rospars JP, Lucas P. Calcium activates a chloride conductance likely involved in olfactory receptor neuron repolarization in the moth Spodoptera littoralis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:6323–6333. doi: 10.1523/JNEUROSCI.0261-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardo MC, Weragalaarachchi K, Akins VT, Del Negro CA. Physiological and Morphological Properties of Dbx1-derived Respiratory Neurons in the preBötzinger Complex of Neonatal Mice. J Physiol [Lond] 2012 doi: 10.1113/jphysiol.2012.250118. submitted, 2nd round evaluation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocrnich CE, Shao Q, Liu H, Feng MM, Harasym S, Savage M, Khimdas S, Laird DW, Hutnik CM. The effect of connexin43 on the level of vascular endothelial growth factor in human retinal pigment epithelial cells. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2012;250:515–522. doi: 10.1007/s00417-011-1871-x. [DOI] [PubMed] [Google Scholar]
- Rae MG, Hilton J, Sharkey J. Putative TRP channel antagonists, SKF 96365, flufenamic acid and 2-APB, are non-competitive antagonists at recombinant human alpha1beta2gamma2 GABA(A) receptors. Neurochemistry international. 2012;60:543–554. doi: 10.1016/j.neuint.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Ravi S, Keat AC, Keat EC. Colitis caused by non-steroidal anti-inflammatory drugs. Postgraduate medical journal. 1986;62:773–776. doi: 10.1136/pgmj.62.730.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MS, Walters KA. Dermal absorption and toxicity assessment. Informa Healthcare; New York, N.Y: 2008. [Google Scholar]
- Sakai H, Lingueglia E, Champigny G, Mattei MG, Lazdunski M. Cloning and functional expression of a novel degenerin-like Na+ channel gene in mammals. J Physiol. 1999;519(Pt 2):323–333. doi: 10.1111/j.1469-7793.1999.0323m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller Y. Activation of a calcium-activated cation current during epileptiform discharges and its possible role in sustaining seizure-like events in neocortical slices. Journal of neurophysiology. 2004;92:862–872. doi: 10.1152/jn.00972.2003. [DOI] [PubMed] [Google Scholar]
- Schlichter LC, Mertens T, Liu B. Swelling activated Cl- channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels (Austin) 2011;5:128–137. doi: 10.4161/chan.5.2.14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatric nephrology. 2011;26:1789–1802. doi: 10.1007/s00467-011-1871-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura K, Zhou M, Ito Y, Kimura S, Zou LB, Sekiguchi F, Kitramura K, Sunano S. Effects of flufenamic acid on smooth muscle of the carotid artery isolated from spontaneously hypertensive rats. Journal of smooth muscle research = Nihon Heikatsukin Gakkai kikanshi. 2002;38:39–50. doi: 10.1540/jsmr.38.39. [DOI] [PubMed] [Google Scholar]
- Simard C, Salle L, Rouet R, Guinamard R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. British journal of pharmacology. 2012;165:2354–2364. doi: 10.1111/j.1476-5381.2011.01715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nature genetics. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- Simon F, Varela D, Riveros A, Eguiguren AL, Stutzin A. Non-selective cation channels and oxidative stress-induced cell swelling. Biological research. 2002;35:215–222. doi: 10.4067/s0716-97602002000200013. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Oxley B, Malpas S, Pillai GV, Simpson PB. Compounds exhibiting selective efficacy for different beta subunits of human recombinant gamma-aminobutyric acid A receptors. J Pharmacol Exp Ther. 2004;311:601–609. doi: 10.1124/jpet.104.070342. [DOI] [PubMed] [Google Scholar]
- Srinivas M, Spray DC. Closure of gap junction channels by arylaminobenzoates. Mol Pharmacol. 2003;63:1389–1397. doi: 10.1124/mol.63.6.1389. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991;354:301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- Stumpff F, Boxberger M, Thieme H, Strauss O, Wiederholt M. Flufenamic acid enhances current through maxi-K channels in the trabecular meshwork of the eye. Curr Eye Res. 2001;22:427–437. doi: 10.1076/ceyr.22.6.427.5485. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Morita T, Iwamoto T. Diversity of Cl(-) channels. Cell Mol Life Sci. 2006;63:12–24. doi: 10.1007/s00018-005-5336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahira M, Sakurai M, Sakurada N, Sugiyama K. Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflugers Archiv : European journal of physiology. 2005;451:474–478. doi: 10.1007/s00424-005-1492-5. [DOI] [PubMed] [Google Scholar]
- Tang CY, Chen TY. Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia. Journal of biomedicine & biotechnology. 2011;2011:685328. doi: 10.1155/2011/685328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB) British journal of pharmacology. 2008;153:1324–1330. doi: 10.1038/sj.bjp.0707675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, Nilius B. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell calcium. 2005;37:267–278. doi: 10.1016/j.ceca.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Waldegger S, Jeck N, Barth P, Peters M, Vitzthum H, Wolf K, Kurtz A, Konrad M, Seyberth HW. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Archiv : European journal of physiology. 2002;444:411–418. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nature genetics. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Wallen P, Robertson B, Cangiano L, Low P, Bhattacharjee A, Kaczmarek LK, Grillner S. Sodium-dependent potassium channels of a Slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J Physiol. 2007;585:75–90. doi: 10.1113/jphysiol.2007.138156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Grillner S, Wallen P. Effects of flufenamic acid on fictive locomotion, plateau potentials, calcium channels and NMDA receptors in the lamprey spinal cord. Neuropharmacology. 2006;51:1038–1046. doi: 10.1016/j.neuropharm.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Wang S, Lee J, Ro JY, Chung MK. Warmth suppresses and desensitizes damage-sensing ion channel TRPA1. Molecular pain. 2012;8:22. doi: 10.1186/1744-8069-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kuehl-Kovarik MC. Flufenamic acid modulates multiple currents in gonadotropin-releasing hormone neurons. Brain research. 2010;1353:94–105. doi: 10.1016/j.brainres.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner F, Shimizu T, Sabirov R, Okada Y. Hypertonic activation of a non-selective cation conductance in HeLa cells and its contribution to cell volume regulation. FEBS letters. 2003;551:20–24. doi: 10.1016/s0014-5793(03)00868-8. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Anderson MP, Rich DP, Berger HA, Denning GM, Ostedgaard LS, Sheppard DN, Cheng SH, Gregory RJ, Smith AE. Cystic fibrosis transmembrane conductance regulator: a chloride channel with novel regulation. Neuron. 1992;8:821–829. doi: 10.1016/0896-6273(92)90196-k. [DOI] [PubMed] [Google Scholar]
- Weylandt KH, Valverde MA, Nobles M, Raguz S, Amey JS, Diaz M, Nastrucci C, Higgins CF, Sardini A. Human ClC-3 is not the swelling-activated chloride channel involved in cell volume regulation. J Biol Chem. 2001;276:17461–17467. doi: 10.1074/jbc.M011667200. [DOI] [PubMed] [Google Scholar]
- White MM, Aylwin M. Niflumic and flufenamic acids are potent reversible blockers of Ca2(+)-activated Cl- channels in Xenopus oocytes. Mol Pharmacol. 1990;37:720–724. [PubMed] [Google Scholar]
- Wiemuth D, Grunder S. The pharmacological profile of brain liver intestine Na+ channel: inhibition by diarylamidines and activation by fenamates. Mol Pharmacol. 2011;80:911–919. doi: 10.1124/mol.111.073726. [DOI] [PubMed] [Google Scholar]
- Wiemuth D, Sahin H, Falkenburger BH, Lefevre CM, Wasmuth HE, Grunder S. BASIC--a bile acid-sensitive ion channel highly expressed in bile ducts. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:4122–4130. doi: 10.1096/fj.12-207043. [DOI] [PubMed] [Google Scholar]
- Winder CV, Wax J, Serrano B, Jones EM, Mc PM. Anti-inflammatory and antipyretic properties of N-(alpha,alpha,alpha-trifluoro-m-tolyl) anthranilic acid (CI-440; flufenamic acid) Arthritis and rheumatism. 1963;6:36–47. doi: 10.1002/art.1780060105. [DOI] [PubMed] [Google Scholar]
- Winpenny JP, Marsey LL, Sexton DW. The CLCA gene family: putative therapeutic target for respiratory diseases. Inflammation & allergy drug targets. 2009;8:146–160. doi: 10.2174/187152809788462590. [DOI] [PubMed] [Google Scholar]
- Wisnoskey BJ, Sinkins WG, Schilling WP. Activation of vanilloid receptor type I in the endoplasmic reticulum fails to activate store-operated Ca2+ entry. The Biochemical journal. 2003;372:517–528. doi: 10.1042/BJ20021574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R. Effects of fenamates and other nonsteroidal anti-inflammatory drugs on rat brain GABAA receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1994;268:806–817. [PubMed] [Google Scholar]
- Xia R, Dekermendjian K, Lullau E, Dekker N. TRPV1: a therapy target that attracts the pharmaceutical interests. Advances in experimental medicine and biology. 2011;704:637–665. doi: 10.1007/978-94-007-0265-3_34. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Hartzell HC, Yu K. Bestrophins and retinopathies. Pflugers Archiv : European journal of physiology. 2010;460:559–569. doi: 10.1007/s00424-010-0821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Waniishi Y, Inoue R, Ito Y. Fenamates potentiate the alpha 1-adrenoceptor-activated nonselective cation channels in rabbit portal vein smooth muscle. Japanese journal of pharmacology. 1996;70:81–84. doi: 10.1254/jjp.70.81. [DOI] [PubMed] [Google Scholar]
- Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca -activated Cl channels contributes to regulating receptor- and store-operated Ca entry in human pulmonary artery smooth muscle cells. Pulmonary circulation. 2011;1:269–279. doi: 10.4103/2045-8932.83447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau HJ, Baranauskas G, Martina M. Flufenamic acid decreases neuronal excitability through modulation of voltage-gated sodium channel gating. J Physiol. 2010;588:3869–3882. doi: 10.1113/jphysiol.2010.193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G, Liantonio A, Gradogna A, Picollo A, Gramegna G, De Bellis M, Murgia AR, Babini E, Camerino DC, Pusch M. Identification of sites responsible for the potentiating effect of niflumic acid on ClC-Ka kidney chloride channels. British journal of pharmacology. 2010;160:1652–1661. doi: 10.1111/j.1476-5381.2010.00822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Oortgiesen M, Vijverberg HP. Differential modulation of alpha 3 beta 2 and alpha 3 beta 4 neuronal nicotinic receptors expressed in Xenopus oocytes by flufenamic acid and niflumic acid. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995;15:2168–2178. doi: 10.1523/JNEUROSCI.15-03-02168.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]