

Abstract
Ghrelin (Gh), a small peptide, which was originally discovered as a gastrointestinal (GI) tropic hormone, has shown to have a presence and function within multiple tissue systems. Recently, Gh has shown to exhibit anti-inflammatory and regenerative abilities in response to both chemical and mechanical stressors within neural tissues. By continuing to elucidate the potential applications of Gh on pathological neural states, the viability of this peptide hormone for therapeutic uses can be explored for future clinical application.
Keywords: Gastrointestinal peptide hormone, Anti-inflammatory, Neuropathic pain, Neural apoptosis
Neuropathy: A general overview
Neuropathy (NP) is a unique process that involves the death or damage of nerve cells in the peripheral or central nervous systems as result of a physical, chemical or electrical stimulus.1 The resultant effects of neuropathy vary greatly and encompass a wide spectrum of symptoms ranging from the complete loss of function of downstream muscles to the hypersensitivity of tissues in response to marginal levels of stimuli.1,2,3 Although this process has been extensively studied for many decades, the mechanism in which cells undergo natural and stress response neuropathies has yet to be determined definitively. The complexity behind this process can be accredited to the wide range of methods in which neuropathy can be induced, as well as, the many systems that are affected through its initiation. To further compound the ambiguous nature of the issue, there is an eclectic mix of symptoms that an individual suffering from neuropathy may display4: tiredness, mechanical hypo and hypersensitivity, unregulated muscle stimulation, thermo hypersensitivity and most notably a lowered threshold for pain induction through nociception stimulation.1,3,4 This lowered threshold can result in a lowered tolerance to marginal introduction of mechanical stressors or in the worst of cases, a continuous state of NP prompted pain followed by the eventual loss of both sensory and motor function.
Several trials have proven successful in discovering different therapeutic methods which are effective in targeting the underlying mechanisms of neuropathic pain.6,7,8 These studies have shown promise, both within in-vitro studies as well as several rat model in-vivo studies, but have not been shown to be effective for long-term large sample size human trials. The inconsistency in translating these model systems to human trials has proven to be a barrier in treating NP. By continuing to isolate specific components of NP for targeted treatment, there is hope that a better understanding of the mechanisms that lead to NP can lead to a more promising treatment.
To further categorize NP, many clinical researchers address this disease as either acute or chronic in nature.6 Acute NP involves a short-term sensation, which traditionally leads to a moderate level of neural regeneration that is accompanied by a nearly complete return of normal neural function. Conversely, chronic pain involves prolonged damage that ends with minimal or no regeneration of symptomatic tissues. As a result, chronic NP pain has a higher likelihood of life altering and long term complications.6,9 Although there are many causes of NP, two common causes of chronic neuropathy, as well as the focus of this review, are diabetic neuropathy (DN) and chronic contusion injury (CCI) NP.1,10 The potential application of the peptide hormone Gh will also be discussed in relation to these to NP states.
Diabetic Neuropathy: mechanism of action and related symptoms
DN is one of the most widespread diseases affecting several regions worldwide. Many countries, including India, have already begun to experience the detrimental effects of DN while other western society countries are projected to have a dramatic increase in patient diagnosis due to the high prevalence of related risk factors.11 Historically, the indefinite mode of induction of DN has limited the research potential in finding a treatment. Now that the mechanism of action is better understood, studies are establishing the sequence of events that ultimately lead to the clinical presentation of DN.12,13 These studies are laying the groundwork for the potential reduction and inhibition of DN related symptoms through metabolic and tissue specific therapeutic agents.
One hypothesis for the clinical presentation of DN involves the accumulation of carbohydrate derivatives in response to hyperglycemia.14,15 To summarize, while in the hyperglycemic state the body begins storing the accumulated glucose as sorbitol through the actions of the enzyme aldose reductase. This glucose to sorbitol reduction is important in DN because it requires the utilization of coenzyme NADPH, an essential substrate for several other biological processes. When utilized for minimal sorbitol synthesis this reaction causes no adverse side effects; however, when relied upon as a major control mechanism in reducing chronically elevated blood glucose levels this conversion quickly exhausts the supply of NADPH thereby inhibiting other necessary reactions (Fig. 1). One important reaction that demands the presence of NADPH is the use of this coenzyme as the regenerating factor for glutathione. Glutathione is an important element in regulating reactive oxidative species (ROS) of aerobic tissues. If not restrained to low levels, these ROS have detrimental effects on cell stability and can lead to apoptosis of neural cells and demyelination of neural tissue via the death of supportive glial cells. Once damage to the neural or glial cells occurs, microglia cells begin the induction of inflammation, which further compounds the damage by inducing apoptosis of various cells. This immune induced apoptosis disrupts signaling processes, which develops into the improper coordination of both sensory and motor signaling leading to the classical symptoms of NP that were discussed above.
Fig 1:
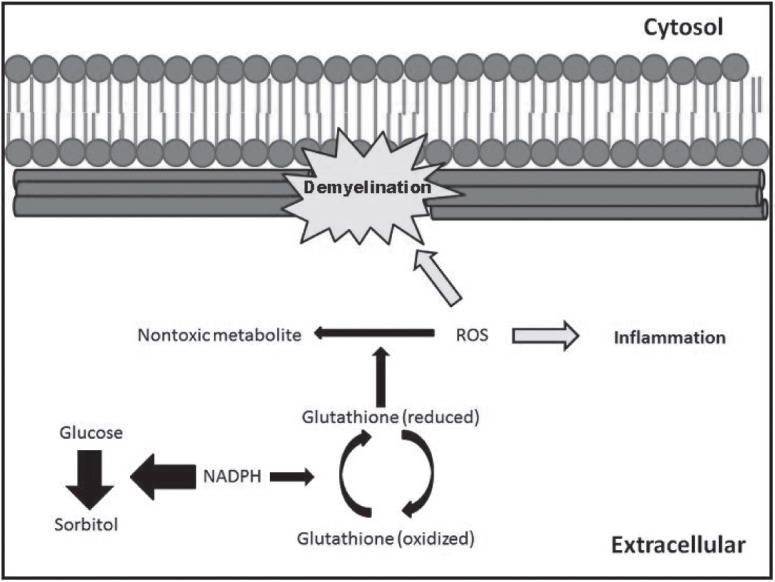
Proposed mechanism for neural demyelination in Diabetic Neuropathy. The shunting of NADPH from the glutathione recycling process limits the reduction of ROS within the aerobic neural tissue. A build-up of ROS causes demyelination of neurons as well as the induction of inflammation from the resultant cell damage.
The inflammation response that is believed to mediate the apoptosis of neural tissues is thought to depend on the presence of several essential pro-inflammatory cytokines IL-1, IL-6 and TNF-α.16,17 Several studies have shown the increased presence of these specific cytokines in relation to chemical induced apoptosis of neuronal tissues. A treatment that targets these specific cytokines may have the potential to inhibit the microglia induced apoptosis and in coordination of neuronal processes that is believed to be at the center of DN.
Spinal Cord Neuropathy: mechanism of action and related symptoms
Although there are an extensive variety of spinal cord neuropathy systems, which operate through several mechanisms, this review will focus on the CCImodel and its effects on chronic pain stimulation. The CCImodel involves the introduction of a contusion to the dorsal horn in order to simulate blunt force spinal cord lesion. On the cellular level, the similar microglia mediated apoptosis begins the disruption of neuronal signaling and leads to the immediate incoordination of both sensory and motor functioning. This disturbance clinically presents itself as hypersensitivity to external stimuli as well paralysis of caudal muscle tissues. When correlated with the DN system, the cellular apoptosis may also play a key role in the induction of peripheral pain as the loss of myelin directly effects the functioning of the sensory neurons. At the molecular level, shortly following the contusion, spinal cord neuropathy begins to take place as gamma-aminobutyric acid (GABA) inhibitory effects begin to decrease as shown through the reduction of decarboxylase 65 and 67 mRNA expression.18,19 The regression of GABA signaling causes a lack of nociceptor inhibition which is essential when down regulating peripheral pain within normally functioning tissues. Although this has yet to be completely resolved, studies have shown through the use of Gabapentin, a GABA analogue, that the introduction of hypersensitivity to mechanical and thermo stimulus in a variety of neuropathic syndromes is strongly associated to the deterioration of GABAsecretion/receptor presence.20,21 The loss of this essential inhibition is believed to enable once insignificant membrane depolarizations to go unregulated and through signal summation cause hypersensitivity to both mechanical and temperature stimulants. To summarize, the loss of GABA inhibition following CCI allows for both the elevation of action potential intensity as well as an increase in neural stimulation. Both of these mechanisms of unregulated neural signaling leads to the introduction of nociceptor induced pain activity.
Another perspective involving the GABA induced inhibition focuses on a study by Wallace et al. in which a contusion onto the spinal column causes a reduction in signal inhibition without a corresponding reduction in GABA receptors.19 The findings from this study suggest that the decrease in inhibitory post synaptic currents (ISPCs) following a contusion to the spinal cord is not due to the regression of GABA receptors, but rather a transformation of its activity and resultant function. Pitcher et al. focused on this observation as it linked the activities of Na+-K+-Cl- co-transporter isoform 1 (NKCC1) and antagonist K+-Cl- co-transporter 2 (KCC2) expression to the change in the GABAergic response.21,22 Pitcher hypothesizes that the overexpression of the ion pump NKCC1 in transporting chloride ions into the neuron and the regression of the ion pump KCC2 in the removal of chloride ions out of the neuron following neural tissue inflammation causes the once inhibitory activity of GABA to become excitatory upon binding with its GABAA receptor (Fig. 2). This change in flux has been shown to be characteristic of CCI cases and is postulated to be a key cause of both primary and secondary hyperalgesia.22,23,24 Although the location of these ion channels has yet to be definitively determined, they are believed to be located within the interneurons, C/D pain fibers or the sensory tract leading to the thalamus, which would allow for the change in activity to directly affect key pain signaling processes.21 As of right now the channels have been determined to be in the CNS spinal tissue with indefinite cell discrimination.
Fig 2:

NKCC1 and KCC2 activity in ion flux changes of damaged neural tissue. The increase in NKCC1 activity with the combined decrease in KCC2 activity allow for the accumulation of chloride ions within the damaged cell. Upon GABA binding with its receptor, the resultant opening of the GABA chloride channel allows for a chloride ion flux out of the cell leading to depolarization. This is counteractive to GABA’s purpose in a non-pathological state as an inhibitory or repolarizing effector.
One may note that the findings of Pitcher appear to contradict the study in which the therapeutic use of Gabapentin had shown to have potential in the treatment of hyperalgesia. Pitcher’s study indicates that GABA action is creating a depolarization effect due to the presence of several key membrane pumps while the Gabapentin study had shown the effectiveness of Gabapentin in reducing hyperalgesia following CCI. One possible cause for this discrepancy is the delay time allowed for each individual study. The Gabapentin study focused on individuals with prolonged pain (3 months) while Pitcher began his study to assess NKCC1’s implications shortly after the introduction of injury. When taken together the two studies suggest a dimorphic origin of pain, which is dependent on a similar loss of signal inhibition that is caused by two distinctly separate mechanisms. One being caused by reversal of a previously inhibitory signal while the other being caused by regression of signal activation. Although there is no definite explanation as to the change in causes for hypersensitivity within these two studies, factors such as this add to the complications that delay the discovery of a long-term treatment of CCI induced NP. These studies suggest that there is both a short term and long-term molecular change, which individually cause hyperalgesia. Although not yet resolved, it suggests the need of a multiple phase treatment method, which accommodates to the changing cause of hyperalgesia or prevents late onset while treating the early molecular changes.
Ghrelin: newly discovered applications
Gh, a gastrointestinal derived protein hormone, was originally discovered as a ligand that interacts with its analogous receptor, growth hormone secretagogue receptor (GHS-R), to regulate the fluidity between the fed and fasting states.25 Although Gh is secreted predominantly from the stomach and the receptors are mostly concentrated within the hypothalamus, both the receptor and peptide hormone have recently been found to exist ubiquitously within various mammalian organ systems. Although predominantly present in the deaceylated form in circulation, only the acetylated form has shown to interact with the GHS-R while the deacylated form is believed to be either an inactive pro-hormone or deactivated hormone state.26
Several recent studies, which have looked into trace expression of acetylated Gh within tissues unrelated to digestion and homeostasis regulation have prompted investigations into other potential sites and purposes of Gh activity (Fig. 3). One function in particular is centered on work which has shown Gh to have the ability to counteract inflammatory signals that are known to interrupt proper neural signaling through neural tissue apoptosis.27,28 The application of Gh, in-vitro and in-vivo, has shown to specifically inhibit apoptotic processes induced by cytokines IL-1 and TNF-α (as well as several other cytokines) which are normally active within damaged neural tissues.27,28 Studies such as the one by Lee showed that Gh can work with its receptor in order to prevent oligodendrocyte damage from reactive oxygen species, similar to what is believed to cause DN.29 Although interaction with GHS-R has shown to be beneficial when utilizing Gh’s effects, another study by Bulgarelli shows an implication for ghrelin to work independently of its GHS-R receptor upon CD36 receptors of microglia cells to prevent their activation and pro-inflammatory response.30 The lack of need for GHS-R has allowed for Gh to be applied to neural, glia and immune cells that may not express RNA for GHS-R but rather any cell that merely interacts with microglia, a common neural immune cell.
Fig 3:
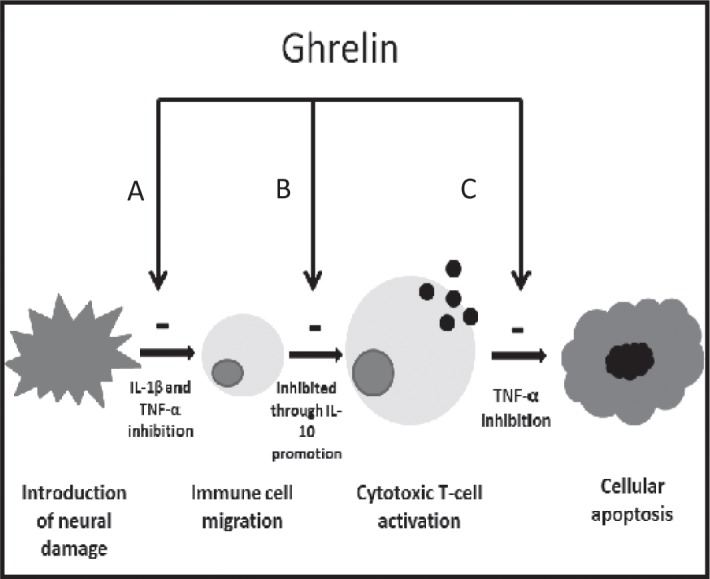
Effects of Ghrelin on Inflammation Induced Neuropathic Pain. Gh has shown to be influential at inhibiting cytokines that are effective at several necessary stages of the inflammatory response which create a multistep effect towards the inhibition of neural tissue apoptosis. A) inhibition of both IL-1β and TNF-α result in the reduction of cell migration to the site of injury B) promotion of T cell regulating cytokine IL-10 inhibit the differentiation of cytotoxic T cells C) inhibition of TNF-α also inhabits cytotoxic T cell induced apoptosis.
In addition to the inhibition of pro-inflammatory signals, Gh’s also functions as a stimulant to T-regulator cells through the promotion of the cytokine IL-10. This is beneficial when regulating inflammation because T-cells are instrumental in the inactivation of already present inflammatory responses and thus act indirectly as an anti-inflammatory cytokine as they inhibit further microglia acitivation.31 T regulatory cells specifically reduce the effects of Th-1 mediated cytotoxic cells which when contacted by damaged tissue, induce apoptosis of both neurons and glia cells. The elimination of the TH-1 pathway during neural cell damage helps IL-10 and subsequently Gh function as a neural protective agent for tissues that are exposed to a stressor, which has a tendency to promote apoptosis, or controlled tissue salvaging, such as the case in both DN and CCI.
Finnaly, the presence of GHS-R within deep dorsal horn regions of the spinal cord have been shown to have an inhibitory effect on nociceptor stimulation. The exact mechanism has yet to be determined but the interaction of Gh with GHS-R has shown to decrease the expression of c-Fos, a secondary messenger which is often utilized as a marker to indicate recent neuronal stimulation,32 within stimulated pain transmitting neurons. With this newly observed inhibition of pain signaling through a system, which bypasses the dysfunctional GABA mediated pathways, Gh may have the ability to reduce and treat hyperalgesia based symptoms of NP by compensating for the loss of IPSCs that results from the regression of proper GABA function.
Future Outlook
Gh’s function as a general anti-inflammatory agent through the inhibition of common inflammation signaling pathways has allowed for it to be applied to a variety of NP states that often function through common means, such as DN’s and CCI’s shared utilization of cytokine induced apoptosis to compensate for chemical and mechanical stressors respectively. By recognizing and exploring the similarities between these two neuropathy states as well as applying the newly evolved discoveries of Gh to other neural disease systems with similar mediators, the use of Gh can be foreseen to transition between various NP states interchangeably as a general therapeutic agent to combat inflammation induced apoptosis.
Footnotes
The article complies with International Committee of Medical Journal Editor’s uniform requirements for the manuscripts.
Competing interests – None
Source of Funding – None
References
- 1.Fear C. Neuropathic pain: Clinical features, assessment and treatment. Nurs Stand. 2010;25:35–40. doi: 10.7748/ns2010.10.25.6.35.c8037. [DOI] [PubMed] [Google Scholar]
- 2.Serra J, Sola R, Aleu J et al. Double and triple spikes in in c-nociceptors in neuropathic pain states: An additional peripheral mechanism of hyperalgesia. Pain. 2011;152(2):343–353. doi: 10.1016/j.pain.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzman R, Maleki J. Post Injury neuropathic pain syndromes. Med Clin North Am. 1999;83:597–626. doi: 10.1016/s0025-7125(05)70126-7. [DOI] [PubMed] [Google Scholar]
- 4.Taylor B. The pathophysiology of neuropathic pain. Neurosurgical Pain Management. 2004:29–37. [Google Scholar]
- 5.Vadakkan K, Xu Hu. Common characteristics and pathways in neuropathic pain of etiologies. Drug Discov Today Dis Models. 2006;3:413–417. [Google Scholar]
- 6.Zimmerman M. Pathobiology of neuropathic pain. Euro J Pharmacol. 2001;429:23–27. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]
- 7.Holtman JR, Jr, Crooks PA, Johnson-Hardy J et al. Antinociceptive effects and toxicity of morphine-6-O-sulfate sodium salt in rat models of pain. Euro J Pharmacol. 2010;648:87–94. doi: 10.1016/j.ejphar.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 8.Melli G, Hoke A. Dorsal root ganglion sensory neuronal cultures: a tool or drug discovery for peripheral neuropathies. Expert Opin Drug Discov. 2009;4:1035–1045. doi: 10.1517/17460440903266829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osspov M, Porreca F. Challenges in the development of novel treatment strategies for neuropathic pain. NeuroRX. 2005;2:650–661. doi: 10.1602/neurorx.2.4.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yagihashi S, Yamagishi S, Wada R. Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes Res Clin Pract. 2007;77:S184–189. doi: 10.1016/j.diabres.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 11.Tahrani AA, Askwith T, Stevens MJ. Emerging drugs for diabetic neuropathy. Exprt Opin Emerg Drugs. 2010;15:661–683. doi: 10.1517/14728214.2010.512610. [DOI] [PubMed] [Google Scholar]
- 12.Petit W, Upender R. Medical evaluation and treatment of diabetic peripheral neuropathy. Clin Podiatr Med Surg. 2003;20:671–688. doi: 10.1016/S0891-8422(03)00068-5. [DOI] [PubMed] [Google Scholar]
- 13.Greene D, Stevens M, Obrosova I et al. Glucose induced oxidative stress and programmed cell death in diabetic neuropathy. Euro J Pharmacol. 1999;375:217–233. doi: 10.1016/s0014-2999(99)00356-8. [DOI] [PubMed] [Google Scholar]
- 14.Anderson K, Seed T, Harris J. Free radicals and reactive oxygen species in programmed cell death. Med Hypotheses. 1999;52:451–463. doi: 10.1054/mehy.1997.0521. [DOI] [PubMed] [Google Scholar]
- 15.Talbot S, Chahmi E, Dias J et al. Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J Neuroinflammation. 2010;7:36. doi: 10.1186/1742-2094-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527–1534. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 17.Miyazato M, Sasatomi K, Hiragata S et al. GABA receptor activation in the lumbosacral spinal cord decreases detrusor over activity in spinal cord injured rats. J Urol. 2008;179:1178–1183. doi: 10.1016/j.juro.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore K, Kohno T, Karchewsky LA et al. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace MS, Irving G, Cowles VE. Gabapentin extended release tablets for the treatment of patients with postherpetic neuralgia: a randomized, double blind, placebo controlled, multicenter study. Clin Drug Investig. 2010;30:765–776. doi: 10.2165/11539520-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Bar Ad V. Gabapentin for treatment of cancer related pain syndromes. Rev Recent Clin Trials. 2010;5(3):174–178. doi: 10.2174/157488710792007310. [DOI] [PubMed] [Google Scholar]
- 21.Hasbargen T, Ahmed M, Miranpuri G et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci. 2010;1198:168–172. doi: 10.1111/j.1749-6632.2010.05462.x. [DOI] [PubMed] [Google Scholar]
- 22.Pitcher MH, Cervevo F. Role of NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151:756–762. doi: 10.1016/j.pain.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Liedtke CM, Cole TS. Activation of NKCC1 by hyperosmotic stress in human tracheal epithelial cells involves PKC-delta and ERK. Biochim Biophys Acta. 2002;1589(1):77–88. doi: 10.1016/s0167-4889(01)00189-6. [DOI] [PubMed] [Google Scholar]
- 24.Cramer SW, Baggott C, Cain J et al. The role of cation dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;17(4):36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojima M, Hosoda H. Ghrelin is a growth hormone releasing acetylated peptide from the stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 26.Guneli E, Kazikdas K, Kolatan E. Ghrelin may attenuate pro-inflammatory cytokine-mediated neuropathic pain. Med Hypothesis. 2007;69(2):356–360. doi: 10.1016/j.mehy.2006.12.042. [DOI] [PubMed] [Google Scholar]
- 27.Guneli E, Onal A, Ates M et al. Effects of repeated administered ghrelin on chronic constriction injury of sciatic rats. Neurosci Lett. 2010;479(3):226–230. doi: 10.1016/j.neulet.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 28.Lee JY, Chung H, Yoo YS et al. Inhibition of apoptotic cell death by ghrelin improves functional recovery after spinal cord injury. Endocrinology. 2010;151(8):3815–3826. doi: 10.1210/en.2009-1416. [DOI] [PubMed] [Google Scholar]
- 29.Lee JY, Oh TH, Yune TY. Ghrelin inhibited hydrogen peroxide-induced apoptotic cell death of oligodendrocytes via ERK and p38MAPK signaling. Endocrinology. 2011;152(6):2377–2386. doi: 10.1210/en.2011-0090. [DOI] [PubMed] [Google Scholar]
- 30.Bulgarelli I, Tamiazzo L, Bresciani E et al. Desacyl-ghrelin and synthetic GH-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J Neurosci Res. 2009;87(12):2718–2727. doi: 10.1002/jnr.22088. [DOI] [PubMed] [Google Scholar]
- 31.Vergnano A, Ferrini F, Salio C et al. The gastrointestinal hormone ghrelin modulates inhibitory neurotransmission in deep laminea in mouse spinal cord dorsal horn. Endocrinology. 2008;149(5):2306–2312. doi: 10.1210/en.2007-1164. [DOI] [PubMed] [Google Scholar]
- 32.Inoue Y, Nakahara K, Kangawa K et al. Transitional change in rat fetal cell proliferation in response to ghrelin and de-sacyl ghrelin during the late stage of pregnancy. Biochem Biophys Res Commun. 2010;393(3):455–460. doi: 10.1016/j.bbrc.2010.02.022. [DOI] [PubMed] [Google Scholar]