

Abstract
Background
Amyotrophic Lateral Sclerosis, in which motor neurons degenerate, leading to paralysis, not only the affected motor neurons, but the surrounding non-neuronal cells also contribute significantly to the disease. However, the disease mechanism is not known.
Purpose
In this study we have addressed the disease mechanism by expressing the ALS associated mutant SOD1G37R in the motor neurons (mMN) and astrocytes (mA) cell lines.
Methods
A series of cell culture assays, immunostaining, RT-PCR and Western blot analysis were performed.
Results
We noticed impairments in both these cell types. The mMN motor neurons were insensitive to forskolin, a known activator of adenylate cyclase, which leads to motor neuron death. In addition, less number of mMN were positive for phosphorylated neurofilament-H (pNFH) unlike the normal motor neurons. Similarly, the mutant SOD1 expressing astrocytes (mA) had two impairments: The inability to activate the oxidative stress protection and the absence of secretory factor(s). Normal astrocytes and their secreted factors could restore the pNFH in the mMN but not the mA. In addition, we show that pNFH restoration is a specific function since the insensitivity of mMN to forskolin could be rescued by neither normal astrocytes nor their secreted factors.
Conclusion
Thus we demonstrate some of the abnormalities caused by the ALS associated mutant SOD1G37R and a potential way, to reverse an abnormality through cell replacement.
Keywords: cAMP, Oxidative stress, Reserpine, Motor neurons, Forskolin, ALS
Introduction
Amyotrophic Lateral Sclerosis (ALS) is an adult onset neurodegenerative disease, caused by degeneration of motor neurons leading to paralysis and eventually death. The disease occurs in sporadic and familial forms. Several gene mutations are linked to ALS. One of them is in the enzyme Cu-Zn superoxide dismutase I (SOD1).1 Around 90 individual SOD1 mutations2 have been identified. Expression of the mutant human SOD1 (G93C, G37R, G85R and others) in mice lead to motor neuron disease development.3-6 Mutant SOD1 (mSOD1) causes ALS through a gain of toxic function in a autosomal dominant fashion.1
ALS development is a complex phenomenon due to abnormalities in motor neurons and non-neuronal cells.7,8 Though the disease initiation affects the motor neurons, the trigger or the defining event is not yet known. The major non-neuronal cells surrounding, supporting and dynamically interacting with the motor neurons are the astrocytes. Though the identity of the non-neuronal cells is not known, lack of mSOD1 in them is speculated to delay the disease development in ALS mice despite mSOD1 in the motor neurons.
Alteration in the signal cascade is manifested as phosphorylation changes in both ALS patients9 and mSOD1 mice.10,11 The differentiation of the triggering event versus secondary consequence is however not known. Here, we set out to address the role of cyclic AMP (cAMP) a second messenger signal cascade essential for a cell’s normal functions. Forskolin, which activates adenylate cyclase increases intracellular cAMP leading to motor neuron death.12 another signal cascade event we addressed was neurofilament-H (NFH) phosphorylation. NFs are intermediary filament proteins expressed exclusively in the neurons. These include NFH, NFM and NFL of 200kD, 160kD and 68kD, respectively.13 These NFs are dynamically phosphorylated by second messenger dependent and independent kinases14 in the carboxyl terminal KSP repeats. Because of the contribution of mSOD1 non-neuronal cells to the disease we addressed the impairments in the mA. mSOD1 mediated oxidative stress in the motor neurons15 but not the astrocytes is well known. Similarly, there are reports of toxic factors secretion by the mA.16,17 but the role of trophic factors is not adequately investigated.
In this report, we address and identify gross level impairments in the mSOD1G37R motor neurons and astrocytes and the reversal of one of them with normal astrocytes / their secreted factors. In addition, we show that in the milieu of mutant SOD1G37R expressing astrocytes, normal astrocytes could retain their function and contribute to the reversal of the impairment in the motor neurons. But to get complete protection, additional impairments need to be addressed.
Methods
Mammalian cell culture
The adherent mouse spinal cord motor neuron cell line, NSC3418 (a kind gift from Neil Cashmann) and rat C6 glioma cell line was grown in DMEM medium with high glucose (Hyclone), 5% fetal bovine serum, penicillin G (100 units/ml), streptomycin (100 µg/ml), and amphotericin B (1 µg/ml) at 37°C in 5% CO2 and 95% air in a humidified incubator.
Cloning and generation of SOD1 expression cell lines
The human genomic wildtype and mutant SOD1G37R fragments of 12 kb3 were obtained by EcoR1 digestion and cloned in pcDNA3.1. These recombinant plasmids were transfected with Lipofectamine 2000 into the NSC34 and C6 cell lines and the stable lines were obtained by geneticin (G418) treatment. Individual clones were obtained by limited dilution clonal selection.
Forskolin treatment
Astrocytes and neuronal cultures were plated onto 12-mm diameter cover slips in 12 well plates with cell density of 3x103/ml and 4x103/ml, respectively. After 24h, cells were treated with 100 µM forskolin (Sigma, USA). After 24h, cells were mounted with Hoechst and were counted in three sets.
Co-culture and conditioned medium assays
The normal and mutant SOD1 expressing motor neurons and astrocytes were plated at the cell density of 6 X 103 and 2X103 respectively. The immunostaining was carried out after 2 days of co-culture grown in the standard medium described above. The specific cell density combination was obtained after optimization.
Conditioned medium treatment
Astrocytes were plated at cell density of 3x103/ml in 12 well plates. Neuronal cultures were plated onto 12-mm diameter cover slips with cell density of 4x103/ml in 12 well plates. After 24h, medium of neuronal cultures was replaced with cell free conditioned medium of astrocytes. Proper controls had also been taken. After 48h, they were immunostained for pNFH. The numbers of pNFH positive and negative cells were counted from 10 fields in duplicates, in three sets.
Immunocytochemistry
The neuronal/astrocytic cells were fixed with 4% paraformaldehyde (pH 7.8) for 15 min at room temperature. Cells were then blocked with 3% normal goat serum, 0.1% Triton X-100 in PBS (pH 7.4) followed by primary antibody staining ( anti-rat-phosphorylated NFH, MAB5448, Chemicon;1:50 /anti-rabbit- S-100, Sigma;1:200) for 2h and secondary antibody staining (anti-rat FITC1:100, anti-rabbit FITC1:1000) for 1h at room temperature.
Reverse Transcriptase -PCR
RNA was extracted from neuronal cultures using TRI reagent (Sigma, USA). RNA was then reverse transcribed (RT) with MMLV reverse transcriptase using random hexamers in the presence of RNAse inhibitor. Primer sequence used is given in Table 1. The cycling parameters used were, 94ºC for 30 min, 30 cycles of 94ºC for 30 s, 54ºC for 30 s, 72ºC for 45 s and finally, 72ºC for 10 min. PCR was carried out using Veriti thermal cycler (Applied Biosystems, USA). The amplicons were then separated by agarose gel electrophoresis (1.8%).
Table 1: PCR primers used for RT-PCR analysis of neurofilament expression.
| Forward primer (5’ to 3’) | Reverse primer (5’ to 3’) | |
|---|---|---|
| NFL | TAGCGCCATGCAGGACACAATC | TCTTCCTGGACGTGGCTGGTAT |
| NFM | CGCCACAACCACGACCTCA | CGGCCTCTTCCTTCTCCTCTTT |
| NFH | TCGGCCCAAGAGGAGATAAC | ATGGAGGGAATTTTTGGGAGTC |
| β–Actin | T C TA CGAGGGC TAT GCTCTCC | ACGGATGTCAACGTCACACTT |
Oxidative stress assay and reserpine treatment
For the oxidative stress induction and cell death assays we followed the published protocol19 with minor modifications. Approximately 3X102 cells/well of the mammalian rat glioma cell line, C6 were plated in 24 well plates on 11mm glass coverslips. On day 3 the cells were treated with DMSO or reserpine and on day 4 with 0.2 mM tBH (Tert-butyl hydroperoxide). On day 5, cells were tested for viability. The medium was removed and the cells on the coverslips were fixed with 4% paraformaldehyde in PBS, followed by Hoechst 33342 staining and mounting.20 The slides were viewed with 10X, 40X objectives in Axioskop fluorescence microscope (Zeiss, Germany). Normal/ live cells showed moderate blue fluorescence of the nucleus with the nucleus appearing bigger whereas dead/sick cells showed highly dark/dense staining indicative of condensed chromatin which is indicative of apoptotic cell death.21 The dose response was determined for both tBH and reserpine. The lowest concentration of tBH required to kill the C6 cells was 0.2 mM. On day 3, the cells were treated with different concentrations like 165 nM, 410 nM and 820 nM of reserpine (in DMSO) in duplicates. On day 4, cells were induced for oxidative stress by 0.2 mM tBH. The minimal concentration of reserpine (165nM) that could provide protection against tBH was selected and further assayed for the extent of protection against oxidative stress.
Western Blotting
Proteins were extracted from neuronal/astrocytic cultures by sonication in hypotonic extraction buffer (50mM Tris, 1mM EDTA pH 8.0) and centrifugation. The supernatant was collected and protein concentration was determined using Bio-Rad protein assay dye reagent. 20 μg of protein extract was separated by poly acrylamide gel electrophoresis and blotted to nitrocellulose membrane (Pall Corporation, USA). Membranes were subjected to amido-black staining for loading control followed by destaining. Membranes were blocked in 5% w/v dried skimmed milk in TBS-T Buffer (20mM Tris-HCl, 137mM NaCl, 0.1% Tween) for 1h at room temperature. The membranes were incubated overnight with primary antibody (rat phosphorylated NFH, Chemicon International, USA, rabbit anti-GDNF, Santa Cruz Biotechnology, USA) at 4ºC (1:200 dilution in 1% blocking buffer). After washing with TBS-T buffer three times, membranes were incubated with secondary antibody (anti rat-HRP/Protein A-peroxidase) for 1h at room temperature at 1:1000 dilutions in TBS-T buffer. After washing with TBS-T buffer for three times, membranes were subjected to detection using ChemiLucent detection system kit (Chemicon International, USA). The Western blots were carried out three times with three different cell preparations and quantitated by densitometry.
Immunoprecipitation
Astrocytes were seeded at 3x103/ml in 60mm plates. After 48h, 2ml media were collected and subjected to immunoprecipitation22 with rabbit anti-GDNF antibody (Santa Cruz Biotechnology, USA) for 16 h at 4ºC followed by addition of Protein A-Agarose beads (Sigma, USA) for 16 h at 4ºC. In addition, neat medium was taken as control. Immunoprecipitated beads were then collected and washed three times with 1X PBS buffer, boiled 8-10 minutes in sample loading buffer and then subjected to western blotting analysis.
Statistical Analysis
Statistical analysis was carried out using SigmaPlot. The student t-test was done to determine the statistical significance.
Results
Though mutant SOD1 has been implicated to cause the motor neuron disease through gain of a toxic function, the exact nature of the toxic function is not known. Reports and evidence suggest that ALS progresses by a complex pathway wherein several cell types contribute to the disease. Our results addressed the abnormalities in motor neurons and astrocytes.
Generation and characterization of mutant SOD1 expressing motor neurons and astrocytic cell lines
In order to get the in vitro cell culture conditions closer to the disease system, we chose the mouse spinal cord motor neuron cell line, NSC34 and the rat glioma astrocytic cell line, C6 to represent the motor neurons and astrocytes respectively. We stably transfected these cell lines with the wildtype or mutant human genomic SOD1.G37R For motor neurons we used two individual clones and the mixed culture and for astrocytes we used only the mixed culture stably expressing mutant SOD1. The motor neurons and astrocyte cell lines were confirmed by choline acetyl transferase and S-100 immunostaining respectively (Fig. 1A). Human SOD1 expression was confirmed by Western blots analysis against human/ mouse SOD1 (Fig. 1B).
Fig. 1:

Generation of SOD1 expressing motor neuron and astrocyte cell lines. A. a. The motor neuron cell line, NSC34, immunostained for choline acetyltransferase. Scale bar 100 µg/m. b. The astrocytic cell line, C6, immunostained for S-100. Scale bar µg/m B. human SOD1 expressing: a) 1- non transgenic 2) human WT SOD1 expressing NSC34 cell line. b. Individual clones of mutant SOD1G37R motor neuron cell line. b) mutant SOD1 expressing (1) and not expressing Astrocytic cell line, C6.
I. Impairments in the motor neurons
Mutant SOD1 expressing motor neurons manifested two impairments.
i) cAMP signaling
Cyclic AMP is a ubiquitous second messenger system used by various signaling cascades like GPCR signaling, insulin signaling and growth hormone signaling. cAMP is an essential second messenger for normal functioning of a cell. Strangely, mutant SOD1 motor neurons (mMN) are unresponsive to forskolin, an adenylate cyclase activator leading to increase in cAMP levels, unlike the wildtype NSC34 motor neurons, which succumb to forskolin leading to cell death (Fig. 2). We evaluated whether this is a generic response to mutant SOD1 phenotype. When mutant SOD1 astrocytes were treated with forskolin, similar result resembling the non SOD1 expressing astrocytes were seen (Fig. 2).
Fig. 2:

Mutant SOD1 expression impairs cAMP signaling in motor neurons and makes these motor neurons resistant to cell death induced by forskolin. A. a), c) and e) – without forskolin treatment; b), d) and f) – forskolin treatment; a) and b) are normal NSC 34 cells; c) and d) are mutant SOD1 expressing NSC34 cells; e) and f) are C6 astrocytes. Green – phosphorylated NFH: blue – Hoechst staining of the nuclei. Scale bar -5μm. B. The quantitative estimation of the motor neuron cell death upon forskolin treatment. The NSC34 cells and the wildtype SOD1 expressing cells are sensitive to forskolin treatment and show severe cell death whereas mutant SOD1 expressing cells were insensitive to forskolin treatment. ** P< 0.005
ii) Mutant SOD1 expression in the motor neuron reduces the pNFH positive motor neurons
The NSC34 cells showed robust expression of phosphorylated NFH. The phosphorylated NFH was identified using the pNFH antibody (Chemicon, USA). This antibody is exclusive for the phosphorylated KSP repeats present in the carboxy terminal of the NFH. While both the NSC34 and the wildtype SOD1 expressing motor neuron lines showed extensive pNFH expression, the mutant SOD1 expressing motor neurons showed reduced staining for the pNFH, especially, in terms of the number of motor neurons. While 70-80% of NSC34 and SOD1WT motor neurons showed pNFH (Fig. 3a, b), only 30-40% of the mMN showed pNFH (Fig. 3c). In order to identify the specific type of downregulation of pNFH, we evaluated the expression of all the three NFs expression by RT-PCR (data not shown). We did not see any change in the expression of NFs. Similarly, when the pNFH protein was detected by Western blots analysis with the same pNFH antibody we could not find a significant difference in the pNFH level between NSC, NSCWT and NSCG37R (data not shown). Therefore, the observed down regulation could be ascribed to reduction in pNFH which was detected at the cellular level (Fig. 3a–c).
Fig. 3:
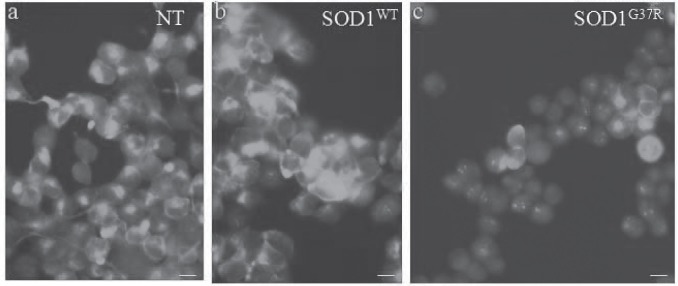
Mutant SOD1 downregulates phosphorylated NFH (pNFH) levels in the motor neuron cell line. In comparison with a) normal and b) wildtype SOD1 expressing motor neuron cell lines, the number of pNFH positive cells were drastically less in mMN (c). Scale bar -5μm.
II. Impairments in the astrocytes
1. Mutant SOD1 expressing astrocytes could not restore the pNFH
As constant crosstalk occurs between the astrocytes and motor neurons, we asked whether the astrocytes could restore the pNFH in the mutant SOD1 motor neurons. For this we co-cultured the motor neurons and astrocytes. While normal astroyctes- motor neurons did not alter the pNFH in the mutant SOD1 expressing coculture, the mMN showed reduction in pNFH (Fig. 5).
Fig. 5:
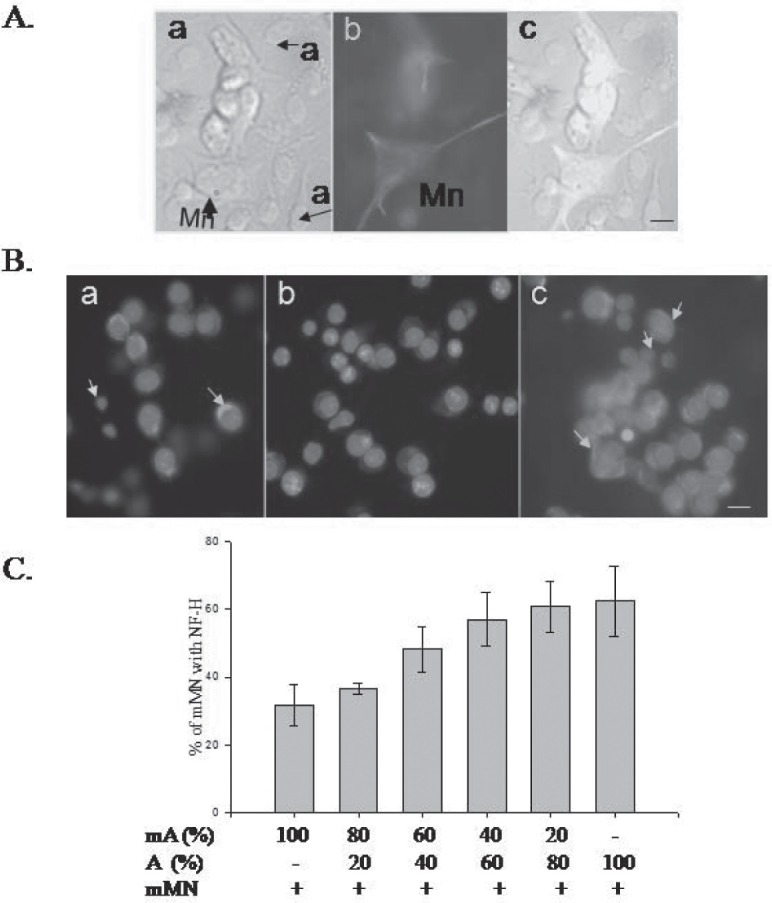
Crosstalk between the astrocytes and motor neurons. Restoration of phosphorylated NFH levels by normal astrocytes. A. Coculture of astrocytes and motor neurons are shown. a) bright field C6 glioma cells and NSC34 motor neurons. MN- motor neurons; a- astrocytes; b) motor neurons immunostained with pNFH antibody. c) overlay of motor neurons and astrocytes. Scale bar-5μm. The yellow arrow indicates motor neurons and the white arrow indicates astrocytes nucleus. Green –pNFH; blue – Hoechst33342. B a. C6 glioma cells and NSC 34 coculture – pNFH staining in the NSC34 cells b) Reduced pNFH immunostaining in a coculture of mutant SOD1 expressing NSC34 motor neurons and C6 astrocytes. c). Coculture with normal C6 astrocytes restores pNFH expression in the mMN. C. Determination of minimal number of normal astrocytes required to restore pNFH in mMN in the milieu of mA.
2. Mutant SOD1 expressing astrocytes could not be protected against oxidative stress
The susceptibility of mMN to oxidative stress is well documented. But, so far we do not know how mutant SOD1 expressing astrocytes respond to stress and whether they could be protected against oxidative stress. We tested oxidative stress protection by treating the astrocytes with reserpine, an antihypertensive drug which instills stress tolerance23 in C. elegans (unpublished results). Hence, we assayed the normal and mutant SOD1 astrocytes, with and without reserpine treatment for their protection against tert-butyl hydroperoxide (tBH) mediated oxidative stress. First, we evaluated whether reserpine will be able to provide protection against oxidative stress in mammalian cell culture (Fig. 4) as in C.elegans (unpublished results). For this, we treated normal C6 astrocytic cell line with the ROS generating tBH at 0.2 mM concentration. This caused cell death which is within the reported range in cell culture systems.19 The C6 cells showed extensive cell death (Fig. 4A-a) upon tBH treatment. First we identified the concentration of reserpine required to provide protection against oxidative stress (Fig. 4B) and arrived at 165 nM. When the C6 cells were first treated with 165 nM reserpine for 24h and then subjected to oxidative stress by tBH, reserpine provided protection against cell death [Fig. 4A-c and 4C]. The normal astrocytes but not the mutant SOD1 astrocytes could be significantly protected against oxidative stress (Fig. 4C). Thus, mutant SOD1 astrocytes have severe impairment in their oxidative stress protection machinery.
Fig. 4:

Mutant SOD1 expressing astrocytes could not be rescued from oxidative stress. Reserpine provides resistance against oxidative stress in the rat glioma cell line, C6 but the mutant SOD1G37R astrocytes does not respond to reserpine treatment. A.Hoechst 33342- stained a) control b) TBH treated c) reserpine followed by TBH treated cells. The sick cells show condensed nuclei which are indicated by white arrows. The healthy cell nucleus is indicated by white arrowhead. The control panel (a) shows more number of cells compared to the tBH treatment (b). Scale bar – 5μm. B. Reserpine dose response curve to determine the minimal concentration of reserpine required for protection against oxidative stress. The condensed nucleus/chromatin containing cells were considered as dead cells and the moderate staining with bigger clear nuclear appearance were counted as normal live cells. C. Reserpine protects against oxidative stress in the normal C6 astrocytes but not the mutant SOD1 expressing astrocytes. The reserpine concentration is 165 nM. ** P<0.005
III. Crosstalk between astrocytes and motor neurons could reverse the impaired mutant SOD1 motor neurons
1. Cell non-autonomous regulation of pNFH in the mutant SOD1 motor neurons
As mutant SOD1 astrocytes could not restore the pNFH in mMN, we asked whether this effect is due to mutant SOD1 or pNFH is non cell autonomously regulated in the motor neurons. When the mutant SOD1 motor neurons were co-cultured with normal astrocytes, the pNFH- expressing motor neurons could be restored close to normal (Fig. 5B-c, C).
2. The crosstalk occurs through the astrocytes secreted factors
In order to identify whether the communication is driven by neurotrophic factors, we cultured the mutant SOD1 motor neurons with the conditioned medium of normal or mutant SOD1 astrocytes. The normal astrocytes conditioned medium (Fig. 6A-b, c) but not that of mutant SOD1 astrocytes (Fig. 6A-a) was able to restore the pNFH expression (Fig. 6B). Thus, astrocytes derived secreted factors regulate the expression of pNFH and this is lacking in the mutant SOD1 astrocytes.
Fig. 6:

Secretory factors of normal astrocytes restore the pNFH expression. A. Supplementation of secretory factors of C6 astrocytes can restore the pNFH in mMN. a). mMN- mA b). mMN- mA treated with conditioned medium from MN-A. c) Conditioned medium of A in mMN- mA. Scale bar-5 μm. B. Increase in the fraction of pNFH positive motor neurons upon C6 (A) conditioned medium treatment. ** - P = 0.008.
3. The pNFH level restoration is independent of GDNF
As several trophic factors are shown to provide protection in the ALS mice, we next addressed the role of the most prominent trophic factor secreted by glia, the glial derived neurotrophic factor, GDNF, in the pNFH upregulation. For this, we determined the level of the secreted GDNF by immunoprecipitation and Western blot analysis (Fig. 7). The GDNF level was not significantly altered when normal and mutant SOD1 astrocytes were analyzed suggesting GDNF is not a trophic factor. It is possible the other secreted factor(s) could be playing a role in the pNFH upregulation.
Fig. 7:
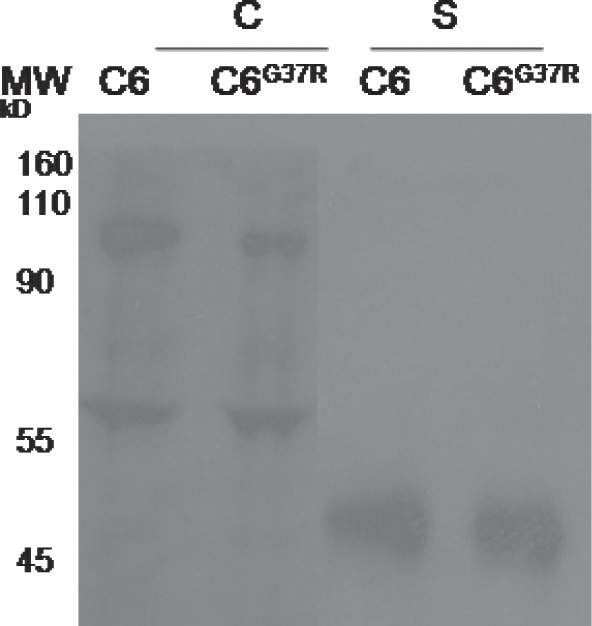
GDNF secretion is unaltered upon expression of mutant SOD1 in the C6 astrocytes. C- Cell lysate; S- secreted. The secreted GDNF was detected after immunoprecipation with GDNF antibody.
4. Astrocytes can increase the pNFH, despite the presence of mutant SOD1 astrocytes
We also studied the effect of normal astrocytes in the milieu of mutant SOD1 astrocytes. For this, we co-cultured varying numbers of normal astrocytes with the mutant SOD1 astrocytes and motor neurons. If we could provide 50% normal astrocytes, then the pNFH level could be restored to normalcy. Thus, in essence we have shown that reduction in pNFH could be rescued by normal astrocytes (Fig. 5) or their secreted factors (Fig. 6) even in the presence of mutant SOD1 expressing astrocytes.
Discussion
In ALS, though the motor neurons degenerate, the disease seems to be progressing due to involvement of various cells of the central nervous system.8 Here, we show evidence for impaired motor neurons and astrocytes in an in vitro system elicited due to the expression of the ALS associated mutant SOD1.G37R The mutant SOD1 expression-mediated impairment could be ascribed to the lack of response to cAMP activation through the adenylate cyclase activator, forskolin, which is crucial to respond to extracellular signals (Fig. 2). Next, we noticed the reduction in the number of pNFH positive cells in the mMNs. NFH is highly phosphorylated by second messenger dependent and independent kinases like glycogen synthase kinase-3 and cdk5,24 Erk2, Erk525 and protein kinase A,14 protein kinase C26 in the carboxyl terminal KSP repeats. Hence, the two impairments could be interrelated or independent. These two impairments together suggest impairments in the signaling cascade.
In this regard, so far the cAMP cascade had not been evaluated in ALS. But a recent report suggests that in a cell culture model similar to ours, oxidative stress induces histone deacetylase downregulation.27 The major donwregulated histone deacetylase is the cAMP response element binding protein (CBP), which is a transcriptional coactivator. This CBP is also known to be the target of caspase-6.28 The histone deacetylase inhibitors could protect the motor neurons but not against the disease in the ALS model mice. Putting together, we suggest that the overall cAMP cascade needs to be addressed to secure protection against the disease. Moreover, though forskolin mainly acts through activation of adenylate cyclase, it is known to act through other mechanisms like ion channel inactivation,29 hence, the delineation of the cAMP cascade would help to address the mechanism of forskolin action as well. This in turn could identify new targets impaired in ALS motor neurons.
As non- neuronal cells are becoming imminent players in ALS, we asked about the potential abnormalities in the mSOD1 astrocytes. We noticed two impairments namely, lack of certain secretory factors and the inability to activate the oxidative stress mechanism. Oxidative stress, as a cause for ALS, has long been suggested. So far, the reports have focused on the oxidative stress in the motor neurons. Here, what we find that there is the impairment of oxidative stress protection machinery in the astrocytes.
The major impairments in astrocytes so far reported are the reduction in the glutamate transporter; GLT-1(EAAT2), in the ALS patients30 and ALS model mice/rats.31 The only drug, riluzole, which provides modest protection in ALS serves to decrease glutamate. In addition, some toxic factors which are detrimental to the motor neurons have been reported to be secreted by the mutant SOD1 astrocytes.16,17 Also, astrocytes are known to influence the type of AMPA receptors expressed by the motor neurons and hence their vulnerability to excitotoxicity.32 Here, we report a new impairment, namely, the inability to restore the pNFH expression in the mSOD1 expressing motor neurons. It is possible that this is the result of absence of a factor rather than secretion of toxic factor. Interestingly, GDNF, long thought to be a potential protective factor, is not impaired in the mSOD1 astrocytes and GDNF secretion is maintained (Fig. 7). Hence, it is possible that pNFH is regulated independent of GDNF. This provides a potential way to restore the pNFH related pathway to normalcy in ALS.
Upon treatment of mMN with normal astrocytes or their conditioned medium we could not reverse the insensitivity of mMN to forskolin (data not shown). Future studies to understand the impairments in the cAMP system and ways to reverse them will greatly help to provide protection against ALS.
Acknowledgements
We thank Dr. Neil Cashman for the NSC-34 cell line, Prof. Joseph Neale for the C6 glioma cell line and Prof. Philip Wong for the wildtype and mutant human SOD1 containing plasmids. We thank Department of Biotechnology for the financial support.
Footnotes
The article complies with International Committee of Medical Journal Editor’s uniform requirements for the manuscripts.
Competing interests – None
Source of Funding – Department of Biotechnology
References
- 1.Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(11):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 3.Wong PC, Pardo CA, Borchelt DR et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 4.Gurney ME, Pu H, Chiu A et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 5.Bruijn LI, Becher MW, Lee MK et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 6.Subramaniam JR, Lyons WE, Liu J et al. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–330. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 7.Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85(1):94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Clement AM, Nguyen MD, Roberts EA et al. Wild-type non-neuronal cells extend surviv.al of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 9.Hu JH, Zhang H, Wagey R et al. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2003;85(2):432–442. doi: 10.1046/j.1471-4159.2003.01670.x. [DOI] [PubMed] [Google Scholar]
- 10.Hu JH, Chernoff K, Pelech S et al. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem. 2003b;85(2):422–431. doi: 10.1046/j.1471-4159.2003.01669.x. [DOI] [PubMed] [Google Scholar]
- 11.Dewil M, dela Cruz VF, Van Den Bosch L et al. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1 (G93A)-induced motor neuron death. Neurobiol Dis. 2007;26(2):332–341. doi: 10.1016/j.nbd.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 12.Aglah C, Gordon T, Posse de Chaves EI. cAMP promotes neurite outgrowth and extension through protein kinase A but independently of Erk activation in cultured rat motoneurons. Neuropharmacology. 2008;55(1):8–17. doi: 10.1016/j.neuropharm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Liem RKH Molecular biology of neuronal intermediate filaments. Curr Opin Cell Biol. 1993;5:12–16. doi: 10.1016/s0955-0674(05)80003-1. [DOI] [PubMed] [Google Scholar]
- 14.Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy. J Neuropathol Exp Neurol. 2005;64(8):649–664. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- 15.Harraz MM, Marden JJ, Zhou W et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai M, Re DB, Nagata T et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Giorgio FP, Carrasco MA, Siao MC et al. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cashman NR, Durham HD, Blusztajn JK et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194(3):209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 19.Pias E, Aw TY. Early redox imbalance mediates hydroperoxide-induced apoptosis in mitotic competent undifferentiated PC12 cells. Cell Death and Differentiation. 2002;9:1007–1016. doi: 10.1038/sj.cdd.4401064. [DOI] [PubMed] [Google Scholar]
- 20.Kondo Y, Rusnak JM, Hoyt DG et al. Enhanced apoptosis in metallothionein null Cell. Molecular Pharmacol. 1997;52:195–201. doi: 10.1124/mol.52.2.195. [DOI] [PubMed] [Google Scholar]
- 21.Joselin AP, Schulze-Osthoff K, Schwerk C. Loss of acinus inhibits oligonucleosomal DNA Fragmentation but not chromatin condensation during apoptosis. J Biol Chem. 2006;281(18):12475–12484. doi: 10.1074/jbc.M509859200. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima K, Hida H, Shimano Y et al. GDNF is a major component of trophic activity in DA-depleted striatum for survival and neurite extension of DAergic neurons. Brain Res. 2001;916:76–84. doi: 10.1016/s0006-8993(01)02866-9. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava D, Arya U, Soundara Rajan T et al. Reserpine can confer stress tolerance and lifespan extension in the nematode C. elegans. Biogerontology. 2008;9(5):309–316. doi: 10.1007/s10522-008-9139-5. [DOI] [PubMed] [Google Scholar]
- 24.Bajaj NP, Miller CC. Phosphorylation of neurofilament heavy-chain side-arm fragments by cyclin-dependent kinase-5 and glycogen synthase kinase-3alpha in transfected cells. J Neurochem. 1997;69(2):737–743. doi: 10.1046/j.1471-4159.1997.69020737.x. [DOI] [PubMed] [Google Scholar]
- 25.Veeranna , Amin ND, Ahn NG et al. Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci. 1998;18(11):4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julien J-P, Mushynski WE. The distribution of phosphorylation sites among identified proteolytic fragments of mammalian neurofilaments. J Biol Chem. 1983;258:4019–4025. [PubMed] [Google Scholar]
- 27.Rouaux C, Panteleeva I, René F et al. Loeffler JPSodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27(21):5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouaux C, Jokic N, Mbebi C et al. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laurenza A, Sutkowski EM, Seamon KB. A specific stimulator of adenylate cyclase or a diterpine with multiple sites of action. Trds in Pharmacol Sci. 1989;10:442–447. doi: 10.1016/S0165-6147(89)80008-2. [DOI] [PubMed] [Google Scholar]
- 30.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 31.Howland DS, Liu J, She Y et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Damme P, Bogaert E, Dewil M et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci USA. 2007;104(37):14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]