

Abstract
Available research data in Autism suggests the role of a network of brain areas, often known as the ‘social brain’. Recent studies highlight the role of genetic mutations as underlying patho-mechanism in Autism. This mini review, discusses the basic concepts behind social brain networks, theory of mind and genetic factors associated with Autism. It critically evaluates and explores the relationship between the behavioral outcomes and genetic factors providing a conceptual framework for understanding of autism.
Keywords: Autism, Behavioral Genetics, Brain Networks
Introduction
It is hypothesized that the deficits in social cognition and related cognitive functions in Autism results from reduced synchronization between these key brain regions during different social and emotional tasks: recent research suggests autism to be a ‘neural connectivity disorder’.
These interconnected neural systems can be understood through the relationship between functionally relevant anatomic areas and neurochemical pathways, the programming of which is genetically modulated during neurodevelopment and mediated through a range of neuropeptides and interacting neurotransmitter systems. It has been suggested that autism emerges from a developmental cascade in which a fundamental deficit in attention to social stimuli beginning as early as infancy leads to impaired interactions with primary caregivers. This results in abnormal development of social cognition, which in turn adversely affects later behavioral and functional domains such as language development which are dependent on these early processes.
A common neuroanatomical theme in autism is over-connectivity in closely related areas and decreased connectivity in longer circuitry needing large scale integration. Disordered development of grey and white matter in autistic individuals has been demonstrated in the frontal and temporal cortices, where selective increase in late developing white matter and narrow mini columns in frontal and temporal cortex has been associated with early accelerated postnatal head growth.1Studies have found fewer, abnormally small and densely packed neurons especially in lateral nucleus of the thalamus and the Purkinje cells of the cerebellum.2 The corpus callosum and major inter-hemispheric connection tracts are smaller than non-autistic, age- and gender-matched individuals.
Social brain network in autism
Neuropsychiatric and neuropsychological evaluations in Autism have revealed selective dysfunction of ‘social cognition’, with sparing of motor, perceptual and basic cognitive skills. Social cognition includes a range of skills and functions required for successful interpersonal interaction, mediated by a ‘Social Brain Network’, consisting brain regions that are dysfunctional in autism: Fusiform face area (perception of personal identity), inferior frontal gyrus (facial expression imitation), posterior superior temporal sulcus (perception of facial expressions and eye gaze tasks), superior frontal gyrus (theory of mind, i.e., taking another person’s perspective) and the amygdala (emotion processing).3-5
Theory of mind in autism
One theory of autism proposes that the core deficit is an inability to metalize and infer the state of mind of another person, or “Theory of Mind” (TOM).6 Autistic individuals perform poorly on typical ToM tasks, which involve guessing what a character is thinking based on a vignette presented in words or pictorially. Difficulty in metalizing leads to being unable to share or express emotions as they cannot anticipate thoughts and actions of others or even understand that others have their own intentions, feelings and points of view is been inferred from the study. Communication is a way of influencing others to construct a picture of the world similar to ones own, but in autism, individuals cannot conceive that others have inner worlds. They can master complex technical operations but cannot learn from verbal instructions and environmental clues, act on hints or understand humor or irony.
ToM deficits in autism have been linked to abnormal patterns of hypo activation in superior temporal gyrus, superior temporal sulcus, and basal temporal areas and hyper activation in Brodmann’s area 9/10, compared to healthy subjects who performed well on ToM tasks. Furthermore, it has been demonstrated that the amygdala and left medial prefrontal cortex, which are core regions in healthy subjects were not involved at all in autistic subjects.7 While reduced amygdala and medial PFC function has also been associated with difficulty in attributing emotional states to others.8
Mirror neuron system in autism
The Mirror Neuron System, which is postulated to underlie the ability to mimic, learn and understand the actions of others 9 has also been implicated in autism.10 Mirror neurons are those in the ventral motor regions that fire when subjects observe actions performed by con-specifics, particularly when the subject has to mimic or learn that action. Although mirror neuron dysfunction has been proposed in autism behavioral paradigms, but has not revealed differences between autistic and non-autistic children in imitating and understanding hand gestures.11 It has been proposed that lack of empathy, or the difficulty to ‘feel what you feel’ is linked to mirror neuron system dysfunction, but the evidence is sparse.
Genetic factors in autism
The programming of various brain networks is genetically modulated during neurodevelopment and mediated through a range of neuropeptides and interacting neurotransmitter systems. Studies have reported that there are approximately 103 disease genes, 44 genomic loci are associated with autism.12 A recent review of genetic studies of autism identified three basic phenotype/genotype combinations:13
Autism plus phenotype consisting of Autism Spectrum Disorders (ASD) caused by rare, single-gene mutations; for e.g., fragile X in 5-10% in Autism Plus.
Broad autism phenotype caused by genetic variations in single or multiple genes. These variations are common and are present in the general population, but result in varying clinical phenotypes when they cross a certain threshold through complex gene-gene and gene-environment interactions.
A severe and specific phenotype caused by ‘de-novo’ mutations in the patient or transmitted through asymptomatic carriers of such mutation.
Table I. Disease genes and genetic disorders reported in individual with ASD.
| Gene | Locus | Mutations/CNVs | Encoded protein/gene function | Clinical features | References |
|---|---|---|---|---|---|
| NTNG1 | 1p13.3 | mutations | Protein acting as axon guidance cues during nervous system development | Schizophrenia, ASDs | (14) |
| CLCN6 | 1p36.22 | mutations | Member of voltage-dependent chloride channel in the nervous system | ASDs | (15) |
| NRXN1 | 2p16.3 | mutations, CNVs | Cell adhesion molecule and a receptor in the nervous system, formation and maintenance of synaptic junctions | ASDs, schizophrenia, epilepsy, ADHD, ID, speech delay, hyperactivity, depression, learning difficulties | (16-20) |
| TBR1 | 2q24.2 | mutations | Transcription factor required for normal brain development | Schizophrenia, ASDs | (21) |
| SCN2A | 2q24.3 | mutations | Sodium channel, voltage-gated, type II, alpha subunit | ASDs epilepsy | (22, 23) |
| SCN1A | 2q24.3 | mutations | Sodium channel, voltage-gated, type I, alpha subunit | ASDs epilepsy | (12, 24) |
| CNTN4 | 3p32.2 | mutations | Axonal-associated cell adhesion molecule | ASDs | (25) |
| FOXP1 | 3p13 | mutations | Transcription factor | ID, ASDs | (12, 24) |
| TBL1XR1 | 3q26.32 | mutations | Transcription activation | ASDs | (21) |
| CDH10 | 5p14.2 | mutations | Neuronal cell-adhesion molecule | ASDs | (26) |
| CDH9 | 5p14.1 | mutations | Neuronal cell-adhesion molecule | ASDs | (26) |
| SLIT3 | 5q34q35.1 | mutations | Axonal guidance regulator | Depression, schizophrenia, ASDs | (21) |
| SYNGAP1 | 6p21.32 | mutations, CNVs | Development of cognition and proper synapse function | ID, ASDs | (27) |
| AHI1 | 6q23.3 | mutations | Cerebellar and cortical development in humans | Joubert syndrome | (12, 25) |
| HOXA1 | 7p15.3 | mutations | Transcription factor | ASDs | (12) |
| RELN | 7q22.1 | deletions | Cell positioning and neuronal migration during brain development | ASDs | (28) |
| CNTNAP2 | 7q36.1 | mutations, CNVs | Cell adhesion molecule and receptor in the nervous system | Focal cortical dysplasia, ASDs, ID, epilepsy, schizophrenia, bipolar disorder | (12) |
| DLGAP2 | 8p23.3 | CNVs | Molecular organization of synapses and neuronal cell signaling | ASDs | (27) |
| CHD7 | 8q12.2 | mutations, deletions | Chromatin remodeling | CHARGE syndrome, ASDs | (12) |
| RIPK2 | 8q21.3 | mutations | Interacts with p38 kinase | ASDs | (15) |
| UNC13B | 9p13.3 | mutations | Synaptic vesicle maturation in a subset of excitatory/glutamatergic synapses | ASDs | (15) |
| ABCA1 | 9q31.1 | mutations | Neuronal structure and function | Bipolar disorder, schizophrenia, ASDs | (15) |
| LAMC3 | 9q34.12 | mutations | Laminin, plays a role in forming the con-volution of the cerebral cortex | ASDs, ID | (24) |
| TSC1 | 9q34.13 | mutations | Regulation of protein synthesis in a wide range of cell types including neurons | Tuberous sclerosis, ASDs | (29) |
| ANK3 | 10q21.2 | mutations | Protein that link the integral membrane proteins to the underlying spectrin-actin cytoskeleton | Bipolar disorder, ASDs | (15) |
| PTEN | 10q23.3 | mutations | Modulating cell cycle, inhibition of the AKT signaling pathway | Cowden syndrome, ASDs, macro-cephaly | (25, 30-32) |
| DHCR7 | 11q13.2 | mutations | 7-Dehydrocholesterol Reductase | Smith-Lemli-Opitz syndrome, ASDs | (12, 33) |
| q13.5 | |||||
| SHANK2 | 11q13.3 | mutations, deletions | Structural and functional organization of the dendritic spine and synaptic junction | Schizophrenia, ASDs, ID | (34) |
| HTR3A | 11q23.2 | mutations | 5-hydroxytryptamine (serotonin) receptor 3A | ASDs | (24) |
| GRIN2B | 12p13.1 | mutations | Glutamate receptor ionotropic, NMDA 2B | ASDs, ADHD, schizophrenia | (21, 24) |
| CACNA1C | 12p13.3 | mutations | Calcium channel, voltage-dependent, L type, alpha 1C subunit | Timothy syndrome, ASDs | (12, 25) |
| CHD8 | 14q11.2 | mutations | Chromatin remodeling | ASDs, macrocephaly | (21, 22) |
| TSC2 | 16p13.3 | mutations | Regulation of protein synthesis in a wide range of cell types including neurons | Tuberous sclerosis | (25, 29) |
| NF1 | 17q11.2 | mutations | Stimulates the GTPase activity of Ras signaling pathway | Neurofibromatosis, ASDs | (12) |
| KATNAL2 | 18q21.1 | mutations | Microtubule-severing ATPase activity | ASDs | (22, 23) |
| DYRK1A | 21q22.13 | mutations, CNVs | Plays a role in a signaling pathway regulating cell proliferation | Majority of phenotypic features in down syndrome, ASDs, ID, microcephaly | (21, 35) |
| SHANK3 | 22q13.33 | mutations, deletions | Structural and functional organization of the dendritic spine and synaptic junction | Phelan-McDermid syndrome, ASDs, schizophrenia | (29, 36-38) |
| PTCHD1 | Xp22.11 | mutations, CNVs | Synaptic functioning | ASDs, ID | (27, 39) |
| NLGN4 | Xp22.31 p22.32 | mutations, CNVs | Neuronal cell surface protein involved in the formation and remodeling of central nervous system synapses | ASDs, ID | (28, 40-42) |
| PHF8 | Xp11.22 | mutations | Cell cycle progression, rDNA transcription and brain development | ASDs, ID | (43) |
| HUWE1 | Xp11.22 | mutations | Neural differentiation and proliferation | ASDs, ID | (43) |
| NLGN3 | Xq13.1 | mutations, CNVs | Neuronal cell surface protein, involved in the formation and function of synapses | ASDs, ID | (28, 29) |
| FMR1 | Xq27.3 | mutations | Translation repressor | Fragile X syndrome, ID, ASDs | (25, 32) |
| MECP2 | Xq28 | mutations, CNVs | Chromosomal protein that binds to methylated DNA, neuron maturation, negative regulation of neuron apoptotic process, cerebellum development, regulation of postsynaptic membrane potential, regulation of transcription | Re& syndrome, ASDs, ID | (25, 32) |
| SLC6A8 | Xq28 | mutations | Creatine transporter | Creatine deficiency syndrome, ID, ASDs | (12) |
| TMLHE | Xq28 | mutations | Enzyme in the carnitine biosynthesis pathway | ASDs | (43, 44) |
When is a gene mutation pathogenic
For a mutation to be pathogenic in autism, it should involve neurodevelopmental genes that regulate neuronal development, migration, circuitry formation and synapse function. Some candidate molecules are NGLs (neuronal cell adhesion molecules) NRX/CBLN/GIuD2 complex (synapse organizer), LRRs (transmembrane proteins), SHANK3 (multiple ankyrin repeat domains), which are all involved in synaptogenesis. This is mediated via signaling molecular pathways through ubiquitin, mammalian target of rapamycin(mTOR), kinase and adenosine phosphorylation pathway. Mutation of genes leads to cascade of events linking transcription (e.g. MECP2 transcriptional regulator), translation (fragile X mental retardation related protein FMRP; translational regulator) and specific synaptic proteins important for maintenance of excitation/inhibition (E/I) ratio during synapse formation. The disruption of E/l ratio results in alternated in a) structure of synaptic connections, b) molecular assembly of synapses c) functional synaptogenesis.45 E/I ratio imbalance also leads to high glutamatergic& low GABAergic activity and shift to excitatory hyper transmission states, leading to the development of a circuit which is hyperexcitable; i.e., a non-tunable circuit with poor differentiation and stability. (Fig 1.)46
Fig. 1:
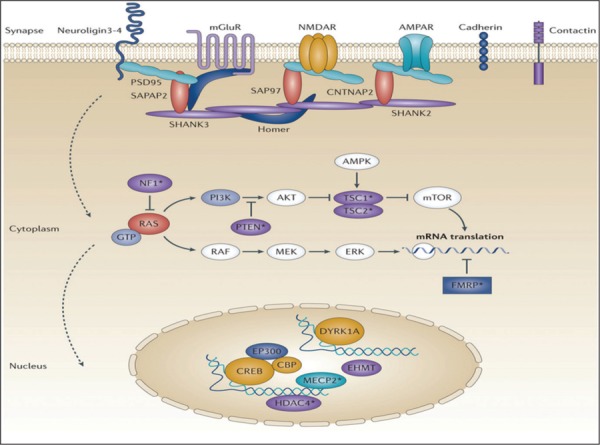
Proteins with genetic variants associated with autism spectrum disorder (ASD) (excluding those in white ovals) are clustered in specific intracellular processes. In colour, proteins with genetic variants associated with ASD; in white, proteins not directly associated with ASD. From Ghosh et al., Nat Rev Drug Discov. 2013;12(10):777-90. Reprinted with permission.
Mutations of genes regulating neuronal migration may result in abnormal organization of cortical mini-columns and poor synchronization between neural regions, such as the hippocampus and prefrontal cortex, which is fundamental for learning and memory.47
Conceptual framework for autism: from behavior to genes
The putative underlying mechanism of local over-connectivity and long-range over connectivity is supported by the following cognitive deficits:
Repetition of domain-specific routines in the absence of domain general executive integration; for e.g., echolalia but no functional spontaneous speech.
Deficit in long-range communication between parallel specialized sub-circuits, such as the amygdalae and fusiform face area, contributing to impaired emotion perception.
No cortical global workspace for integration of past and present experience.
Lack of learning by trial and error through social experience indicates domain general executive integration and generalizability.
Therefore, in autism, an initial domain specific deficit results in secondary lack of normal social experience. Dependence on local domain specific networks leads to cognitive rigidity. The link of modular deficit to mirror phenomena leads to repetitive behaviors in the absence of functional imitation.
Disturbed patterns of neuronal activity underlying specific types of behavior could be correlated with specific genetic alleles thus linking gene to brain development to behavior.
Conclusion
Learning is genetically programmed but environmental activity dependent. This bi-directional interface offers an opportunity for intervention. Through modeling, observational and imitation learning in the preschool years that enhance social -emotional and social-cognitive development can build stronger circuitry.
Genetically mediated deficits and consequent functional impairments involve activity-dependent synapse development, which depend on postnatal learning and experience. Understanding these neurobiological underpinnings can lead to the design of interventions that accommodate the way the brains of children with autism function and may lead to the promotion of more flexible thinking and learning. Furthermore, since genetically mediated deficits and consequent functional impairments involve activity-dependent synapse development that depends on postnatal learning and experience, early intervention can prevent or reduce the risk of these deficits cascading into a trajectory toward full expression of the disorder. Such a model implies the importance of intervening early to prevent downstream effects, and is supported by studies showing greater efficacy with early intervention programs which seek to counteract this early deficit and normalize the development of social and communicative capacities through provision of heavily enriched social stimuli by therapists and caregivers. This offers an opportunity to interrupt the sequence of events that would otherwise have resulted in an abnormal developmental trajectory, but instead promote interactions that normalizes basic brain responses to social stimuli and alter the course of development by exploiting the neuronal maturation and brain plasticity in the early years of life.
Footnotes
Article complies with International Committee of Medical Journal editor’s uniform requirements for manuscript.
Competing Interests: None, Source of Funding: None
REFERENCES
- 1.Hutsler J, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain research. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- 2.Casanova M, Buxhoeveden D, Switala A et al. Neuronal density and architecture (Gray Level Index) in the brains of autistic patients. Journal of child neurology. 2002;17(7):515–21. doi: 10.1177/088307380201700708. [DOI] [PubMed] [Google Scholar]
- 3.Schultz R. Developmental deficits in social perception in autism: the role of the amygdala and fusiform face area. International journal of developmental neuroscience: the official journal of the International Society for Developmental Neuroscience. 2005;23(2–3):125–41. doi: 10.1016/j.ijdevneu.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Grelotti D, Klin A, Gauthier I et al. fMRI activation of the fusiform gyrus and amygdala to cartoon characters but not to faces in a boy with autism. Neuropsychologia. 2005;43(3):373–85. doi: 10.1016/j.neuropsychologia.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 5.Pelphrey K, Morris J, McCarthy G. Neural basis of eye gaze processing deficits in autism. Brain: a journal of neurology. 2005;128(Pt 5):1038–48. doi: 10.1093/brain/awh404. [DOI] [PubMed] [Google Scholar]
- 6.Frith U, Morton J, Leslie A. The cognitive basis of a biological disorder: autism. Trends in neurosciences. 1991;14(10):433–8. doi: 10.1016/0166-2236(91)90041-r. [DOI] [PubMed] [Google Scholar]
- 7.Brambilla P, Hardan A, di Nemi S et al. The functional neuroanatomy of autism. Functional neurology. 2004;19(1):9–17. [PubMed] [Google Scholar]
- 8.Castelli F, Frith C, Happé F et al. Autism, Asperger syndrome and brain mechanisms for the attribution of mental states to animated shapes. Brain: a journal of neurology. 2002;125(Pt 8):1839–49. doi: 10.1093/brain/awf189. [DOI] [PubMed] [Google Scholar]
- 9.Cattaneo L, Rizzolatti G. The mirror neuron system. Archives of neurology. 2009;66(5):557–60. doi: 10.1001/archneurol.2009.41. [DOI] [PubMed] [Google Scholar]
- 10.Iacoboni M, Dapretto M. The mirror neuron system and the consequences of its dysfunction. Nature reviews Neuroscience. 2006;7(12):942–51. doi: 10.1038/nrn2024. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton A, Brindley R, Frith U. Imitation and action understanding in autistic spectrum disorders: how valid is the hypothesis of a deficit in the mirror neuron system? Neuropsychologia. 2007;45(8):1859–68. doi: 10.1016/j.neuropsychologia.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 12.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 13.Eapen V. Genetic basis of autism: is there a way forward? Current opinion in psychiatry. 2011;24(3):226–36. doi: 10.1097/YCO.0b013e328345927e. [DOI] [PubMed] [Google Scholar]
- 14.O’Roak BJ, Vives L, Girirajan S et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–50. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi C, Wu J, Jiang T et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Human mutation. 2012;33(12):1635–8. doi: 10.1002/humu.22174. [DOI] [PubMed] [Google Scholar]
- 16.Bill BR, Geschwind DH. Genetic advances in autism: heterogeneity and convergence on shared pathways. Current opinion in genetics & development. 2009;19(3):271–8. doi: 10.1016/j.gde.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glessner JT, Wang K, Cai G et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–73. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, Schroer R, Yan J et al. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neuroscience letters. 2006;409(1):10–3. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Shen Y, Zhang F et al. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. American journal of human genetics. 2013;92(3):375–86. doi: 10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wis´niowiecka-Kowalnik B, Nesteruk M, Peters SU et al. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2010;153B(5):983–93. doi: 10.1002/ajmg.b.31064. [DOI] [PubMed] [Google Scholar]
- 21.O’Roak BJ, Vives L, Fu W et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (New York, NY) 2012;338(6114):1619–22. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders SJ, Murtha MT, Gupta AR et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neale BM, Kou Y, Liu L et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Roak BJ, Deriziotis P, Lee C et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43(6):585–9. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miles JH. Autism spectrum disorders-a genetics review. Genetics in medicine: official journal of the American College of Medical Genetics. 2011;13(4):278–94. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 26.Wang K, Zhang H, Ma D et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–33. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinto D, Pagnamenta AT, Klei L et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaefer GB, Mendelsohn NJ, Professional P et al. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15(5):399–407. doi: 10.1038/gim.2013.32. [DOI] [PubMed] [Google Scholar]
- 29.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493(7432):327–37. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varga EA, Pastore M, Prior T et al. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genetics in medicine: official journal of the American College of Medical Genetics. 2009;11(2):111–7. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- 31.Santini E, Huynh TN, MacAskill AF et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493(7432):411–5. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter MT, Scherer SW. Autism spectrum disorder in the genetics clinic: a review. Clinical genetics. 2013;83(5):399–407. doi: 10.1111/cge.12101. [DOI] [PubMed] [Google Scholar]
- 33.Scherer SW, Dawson G. Risk factors for autism: translating genomic discoveries into diagnostics. Human genetics. 2011;130(1):123–48. doi: 10.1007/s00439-011-1037-2. [DOI] [PubMed] [Google Scholar]
- 34.Berkel S, Marshall CR, Weiss B et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature genetics. 2010;42(6):489–91. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 35.van Bon BW, Hoischen A, Hehir-Kwa J et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clinical genetics. 2011;79(3):296–9. doi: 10.1111/j.1399-0004.2010.01544.x. [DOI] [PubMed] [Google Scholar]
- 36.Durand CM, Betancur C, Boeckers TM et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature genetics. 2007;39(1):25–7. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moessner R, Marshall CR, Sutcliffe JS et al. Contribution of SHANK3 mutations to autism spectrum disorder. American journal of human genetics. 2007;81(6):1289–97. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boeckers TM, Bockmann J, Kreutz MR et al. ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. Journal of neurochemistry. 2002;81(5):903–10. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- 39.Noor A, Whibley A, Marshall CR et al. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Science translational medicine. 2010;2(49) doi: 10.1126/scitranslmed.3001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jamain S, Quach H, Betancur C et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature genetics. 2003;34(1):27–9. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laumonnier F, Bonnet-Brilhault F, Gomot M et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. American journal of human genetics. 2004;74(3):552–7. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daoud H, Bonnet-Brilhault F, Védrine S et al. Autism and nonsyndromic mental retardation associated with a de novo mutation in the NLGN4X gene promoter causing an increased expression level. Biological psychiatry. 2009;66(10):906–10. doi: 10.1016/j.biopsych.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 43.Nava C, Lamari F, Héron D et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Translational psychiatry. 2012:2. doi: 10.1038/tp.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Celestino-Soper PB, Violante S, Crawford EL et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(21):7974–81. doi: 10.1073/pnas.1120210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doll C, Broadie K. Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Frontiers in cellular neuroscience. 2014;8:30. doi: 10.3389/fncel.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh A, Michalon A, Lindemann L, Fontoura P, Santarelli L. Drug discovery for autism spectrum disorder: challenges and opportunities. Nature reviews Drug discovery. 2013;12(10):777–90. doi: 10.1038/nrd4102. [DOI] [PubMed] [Google Scholar]
- 47.Sigurdsson T, Stark K, Karayiorgou M et al. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464(7289):763–7. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]