

Abstract
Objective:
To assess the utility and safety of rituximab in pediatric autoimmune and inflammatory disorders of the CNS.
Methods:
Multicenter retrospective study.
Results:
A total of 144 children and adolescents (median age 8 years, range 0.7–17; 103 female) with NMDA receptor (NMDAR) encephalitis (n = 39), opsoclonus myoclonus ataxia syndrome (n = 32), neuromyelitis optica spectrum disorders (n = 20), neuropsychiatric systemic lupus erythematosus (n = 18), and other neuroinflammatory disorders (n = 35) were studied. Rituximab was given after a median duration of disease of 0.5 years (range 0.05–9.5 years). Infusion adverse events were recorded in 18/144 (12.5%), including grade 4 (anaphylaxis) in 3. Eleven patients (7.6%) had an infectious adverse event (AE), including 2 with grade 5 (death) and 2 with grade 4 (disabling) infectious AE (median follow-up of 1.65 years [range 0.1–8.5]). No patients developed progressive multifocal leukoencephalopathy. A definite, probable, or possible benefit was reported in 125 of 144 (87%) patients. A total of 17.4% of patients had a modified Rankin Scale (mRS) score of 0–2 at rituximab initiation, compared to 73.9% at outcome. The change in mRS 0–2 was greater in patients given rituximab early in their disease course compared to those treated later.
Conclusion:
While limited by the retrospective nature of this analysis, our data support an off-label use of rituximab, although the significant risk of infectious complications suggests rituximab should be restricted to disorders with significant morbidity and mortality.
Classification of evidence:
This study provides Class IV evidence that in pediatric autoimmune and inflammatory CNS disorders, rituximab improves neurologic outcomes with a 7.6% risk of adverse infections.
Rituximab is an anti-CD20 chimeric monoclonal antibody that results in B-cell depletion. Initially used to treat B-cell neoplasms such as non-Hodgkin lymphoma,1 rituximab has more recently been licensed for use in autoimmune disorders such as rheumatoid arthritis and anti-neutrophil cytoplasmic antibody–associated vasculitides.2,3 In a double-blind placebo-controlled trial, rituximab reduced disease activity in relapsing-remitting multiple sclerosis,4 and open-label studies have described benefit in NMDA receptor (NMDAR) encephalitis, neuromyelitis optica (NMO) spectrum disorders (NMOSD), and opsoclonus myoclonus ataxia syndrome (OMAS).5–7 New biologic therapies such as rituximab have provided clinical benefits, but concerns about safety remain, particularly regarding serious infection and viral reactivation.8,9 Rituximab usage in children has increased over the last 10 years for a broad spectrum of indications including post-transplant complications, malignancy, immunodeficiency, and autoimmune disease.8 Serious infection in children following rituximab is more common for transplant and malignancy indications than autoimmune indications, although long-term safety data and formal clinical trials are lacking.8 In order to improve knowledge regarding the risk/benefit ratio of rituximab usage, we conducted a retrospective multicenter review of the utility and safety of rituximab in children with autoimmune and inflammatory CNS conditions.
METHODS
Standard protocol approval and registration.
The study gained new ethical approval or used preexisting ethically approved studies to collect de-identified clinical data from children who received rituximab using a designed pro forma (QIE-2013-02-17).
Cohort data collection.
The primary research aim was to define the utility and safety of rituximab in a retrospective cohort study (Class IV evidence). Using a network of 15 pediatric international centers with an interest in neuroimmunology, our primary aims were to survey the use, safety, and efficacy of rituximab. We collected data from patients treated for autoimmune or inflammatory CNS indications prior to their 18th birthday. Patients were captured using combinations of electronic medical records, clinical databases, or pharmacy records. The data were collected and rated by one primary investigator per site, with cross-checking with attending clinicians and coinvestigators.
The designed pro forma (appendix e-1 on the Neurology® Web site at Neurology.org) collected demographic data, age at presentation, age at rituximab administration, the dosage regimen used, and medication to reduce allergic phenomena. The clinical syndrome, diagnosis, and diagnostic investigations were recorded, as were immunotherapies that were used before rituximab or at any time during the patient’s illness. Hematologic and immunologic analyses were based upon available laboratory tests that were ordered for clinical purposes. The presence of hematologic effects was recorded as previously described.10 B-cell depletion was assessed using CD19 count, and we also recorded whether the B-cell depletion was present >12 months after rituximab. Infusion adverse events (AEs) were symptoms or signs that occurred during the infusion that were considered allergic, hypersensitive, or other unwanted effects of the infusion. The management of the side effects was not recorded. Infusion AEs were recorded as described on product data sheets and classified using Common Terminology Criteria for Adverse Events (CTCAE v4.0), as has been previously used in this context.10–12 We recorded any side effect that may have been attributed to rituximab usage, with particular focus on infectious complications, which were classified using CTCAE v4.0.11 The development of progressive multifocal leukoencephalopathy (PML) was specifically queried.
While therapeutic efficacy could not be formally evaluated in a retrospective analysis, we evaluated the clinical disease state at initiation and following rituximab using the modified Rankin Scale (mRS) for children.6,13 The mRS was recorded for the “worst at any stage,” at initiation of rituximab, and at outcome. An mRS of 0–2 was used as a marker of a better outcome, as previously described.6 We also subjectively classified response to rituximab, as recorded by the attending clinician as “definite improvement,” “probable improvement,” “possible improvement,” “no benefit,” or “disease worsening during therapy.” The duration of disease (DoD) at the time of rituximab administration was calculated, and the median DoD for each of the main subgroups was calculated (NMDAR encephalitis, OMAS, NMOSD, and neuropsychiatric systemic lupus erythematosus). In order to determine the change in mRS related to the timing of rituximab therapy, we divided patients according to the median DoD at the time of rituximab initiation into early (≤ median) and late (> median) in the 4 main subgroups. The duration of follow-up and ongoing disability at the last documented visit was recorded. Therapies being received at last clinic follow-up were also recorded. Figures were generated using Prism software version 4.0b (GraphPad Software, Inc., San Diego, CA).
RESULTS
Clinical cohort and subgroup demographics.
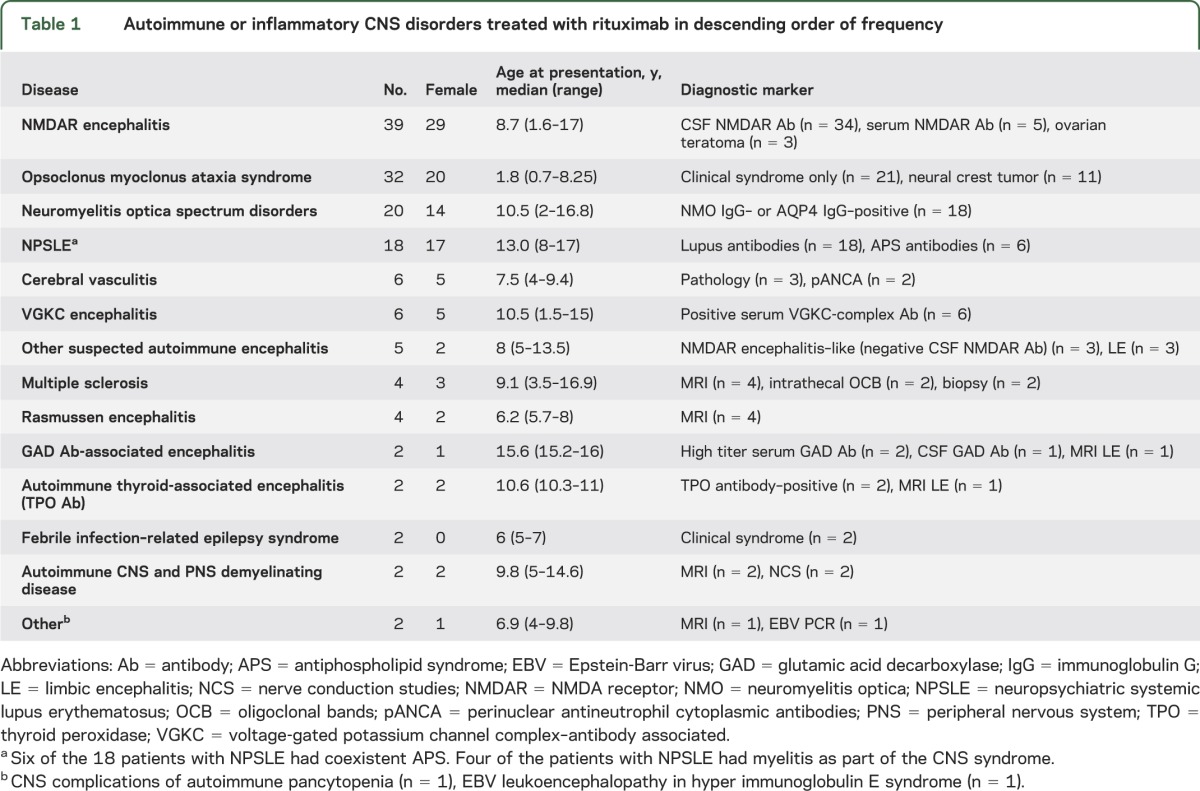
Data from 144 patients (103 female) were available. The age at first neurologic presentation was a median of 8 years (range 0.7–17 years). The disease subgroups are presented in table 1. There was a total duration of follow-up of 307 patient-years after rituximab administration (median 1.65 years, range 0.1–8.5 years). A total of 134 of 144 patients were followed for longer than 6 months.
Table 1.
Autoimmune or inflammatory CNS disorders treated with rituximab in descending order of frequency
Preceding or concurrent therapies.
Most patients had a prolonged or relapsing course requiring multiple immunosuppressive or immune-modifying therapies. At some point prior to the first rituximab infusion, 138 patients were treated with one or more doses of corticosteroids, 104 received one or more courses of IV immunoglobulin, 43 were treated with a single or multiple dose regimen of cyclophosphamide, and 21 underwent plasma exchange (as summarized in table e-1).
Rituximab administration.
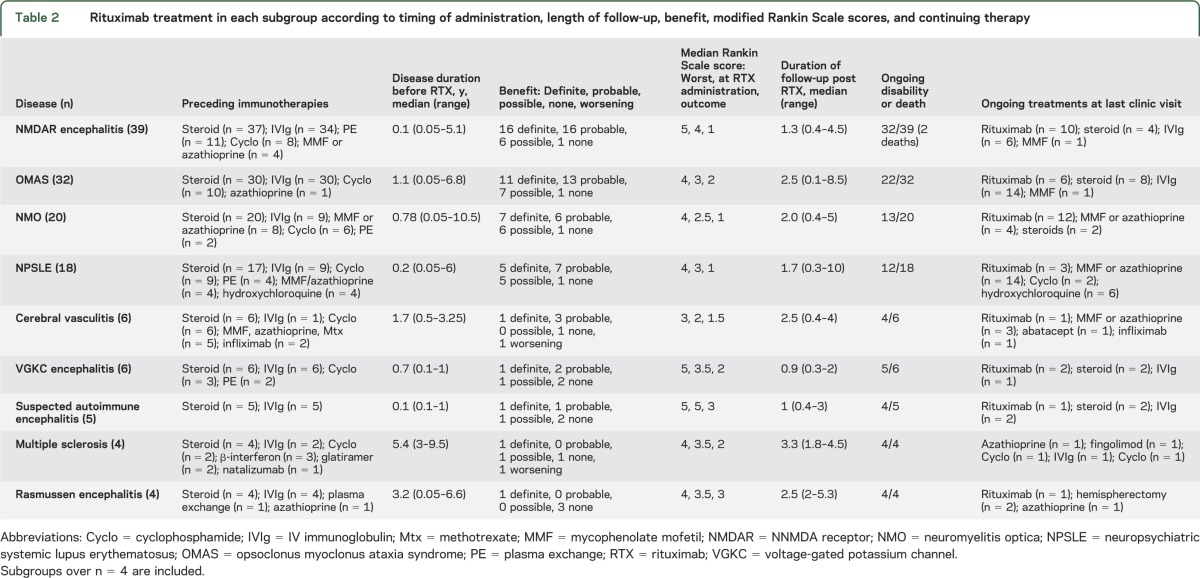
The age at rituximab administration was a median of 9.9 years (range 1.6–17.9 years). The duration of disease before rituximab usage was a median of 0.5 years (range 0.05–9.5 years), and varied by disease (table 2). At the time of rituximab initiation, the median mRS was 3 (range 0–5). A variety of dosage regimens were employed as outlined in table e-2, with 375 mg/m2 weekly for 4 weeks being the most commonly used regimen (n = 57). Antihistamine was administered with the infusion in 106 patients. A total of 112 patients received corticosteroid with the infusion or were receiving ongoing corticosteroid at the time of rituximab. Twenty-six patients were commenced on ongoing prophylactic antibiotic (usually cotrimoxazole); 15 of these patients also received cyclophosphamide during their disease. Monitoring for potential viral reactivation syndromes described after rituximab10 was inconsistent: viral serology was performed for JC virus (n = 14), varicella-zoster virus (n = 14), hepatitis B and C (n = 13), and BK virus (n = 3). Thirty-eight patients had repeat rituximab courses, with patients with NMO most likely to receive repeat courses (table 2). The timing of the repeat courses was not reported.
Table 2.
Rituximab treatment in each subgroup according to timing of administration, length of follow-up, benefit, modified Rankin Scale scores, and continuing therapy
Hematologic and immunologic effects.
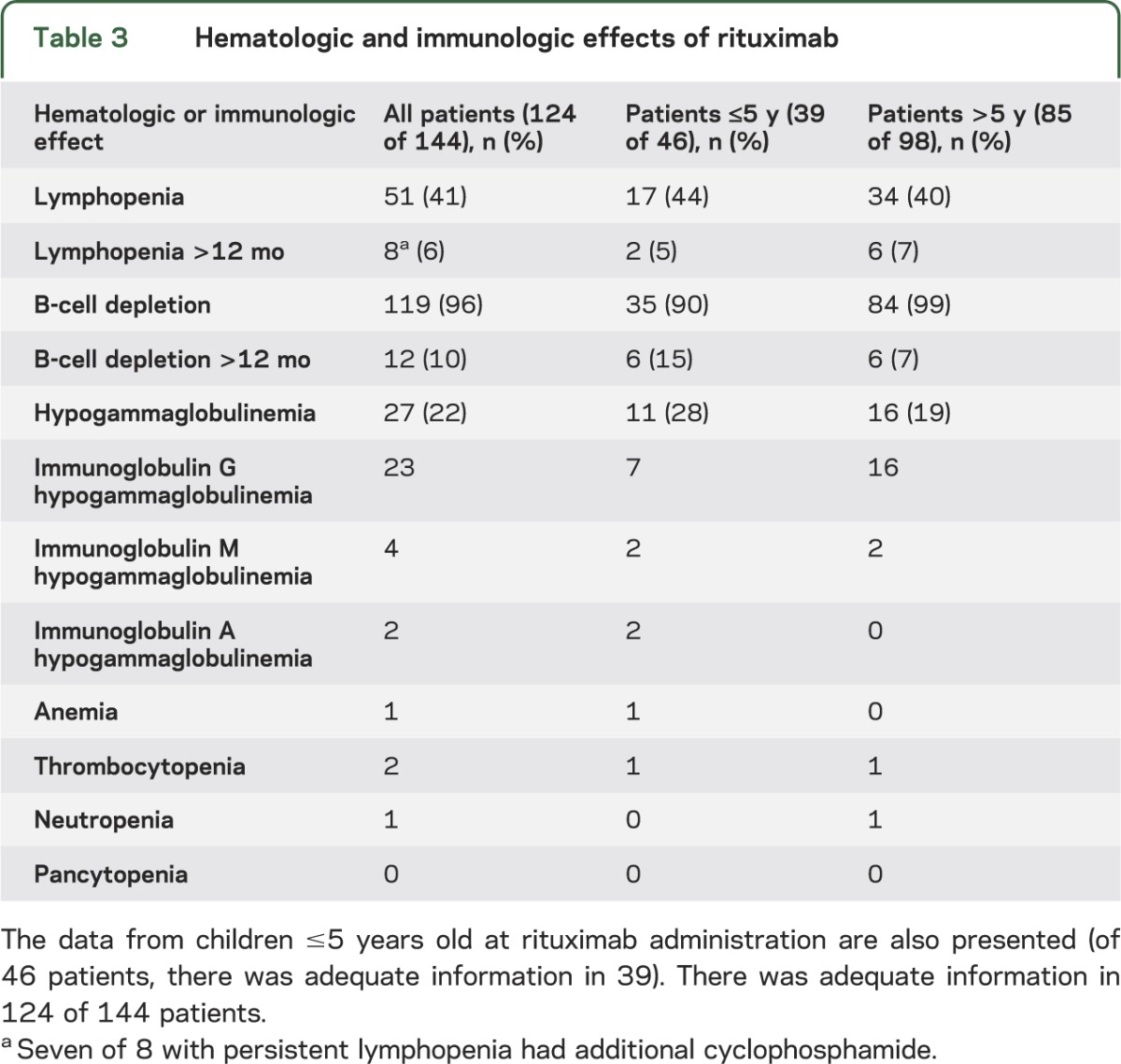
Measurement of hematologic and immunologic effects was recorded in 124 of 144 patients after rituximab (there was inadequate data in 20 patients). Complete blood count and differential, lymphocyte subsets (B-cell count), and immunoglobulin were performed, and results are presented in table 3. A total of 119 of 124 (96%) had B-cell depletion (actual values not recorded), which was present >12 months in 12 children, and 27 children had documented hypogammaglobulinemia (table 3). Other hematologic effects were uncommon, including neutropenia.
Table 3.
Hematologic and immunologic effects of rituximab
Infusion AEs.
Of 144 patients, 18 patients (12.5%) had one or more infusion-related symptoms (table 4). The timing of the infusion side effects (e.g., first infusion, second infusion) was not recorded. Three patients (2%) had a grade 4 reaction (anaphylaxis) that resolved without complication with standard therapy including antihistamine and corticosteroid doses. One of these children did not receive antihistamine prophylaxis. In one patient, infusion-related fever created a transient exacerbation of seizures. Only one patient was unable to tolerate rituximab due to progressively worse hypersensitivity reactions despite premedication and hence was suspended after the fourth incomplete dose. This patient, with NMOSD, subsequently tolerated and completed a course of ofatumumab. There was no difference in infusion AEs between those who received antihistamine prophylaxis (13/106, 12%), and those who did not (5/38, 13%).
Table 4.
Infusion and infectious adverse events in 144 patients

Infectious side effects.
Eleven patients (7.6%) had a recorded infectious complication in the follow-up period (table 4). Two children had a grade 5 (death) AE, and 2 children a grade 4 (life-threatening or disabling) AE (table 4, and described in appendix e-2).14 The grade 4 and 5 infectious AEs occurred a median of 30 days (range 3–38 days) after initiation of rituximab. A further 7 patients had a grade 3 infectious AE (hospitalization or IV antibiotics) in the follow-up period that may have been partly related to rituximab immunosuppression (table 4). An infectious AE occurred in 2 of the 26 patients on antibiotic prophylaxis (7.7%), compared to 9 of 118 patients not on prophylaxis (7.6%). An infectious AE occurred in 2 of 58 patients (3.4%) who received cyclophosphamide at any stage during their disease, compared to 9 of 86 (7.6%) who did not receive cyclophosphamide. There were no cases of PML, nor any secondary malignancy identified during the reporting period (total 307 patient-years of follow-up).
Young patients.
We compared the safety data in preschool children ≤5 years old (n = 46) with the children >5 years of age. There was no difference in hematologic or immunologic effects, except an increased rate of hypogammaglobulinemia (table 3). In the children ≤5 years, there was no increased risk of infusion AEs (7/46, 15% vs 11/98, 11%) and no increased risk of infectious AE (3/46, 6.5% vs 8/98, 8%) compared to the children >5 years. Two of the 3 children ≤5 years with infectious AE had persistent hypogammaglobulinemia.
Outcome of cohort.
A total of 35 of 144 (24%) patients required intensive care management. After a median follow-up of 1.65 years after rituximab (range 0.1–8.5), 3 patients (2%) died. Two patients who died had NMDAR encephalitis, as described in the supplementary material. The third patient who died had GAD antibody–associated encephalitis and died 3 months into the illness due to refractory status epilepticus. A total of 101 patients (70%) had residual problems including cognitive or developmental impairment (n = 66), motor impairment (n = 46), psychiatric disease (n = 34), or visual impairment (n = 13). Sixteen children continue to experience seizures. Forty children (28%) experienced a full clinical recovery at the end of the observation period.
According to the impression of the treating clinician, rituximab had a definite benefit in 45 patients, probable benefit in 49, possible benefit in 31, no benefit or unclear in 17, and the disease worsened in 2 patients. The median mRS in the total cohort was 4 (worst score), 3 (at rituximab administration), and 2 (at outcome determination). The mRS at initiation of rituximab and outcome separated by etiology is presented in table 2. The percentage of patients with mRS of 0–2 was variable between subgroups, but showed a general improvement in the main indications (figure, A). The change in mRS 0–2 was compared according to the timing of rituximab administration (early or late, separated according to the median duration of disease at the time of rituximab initiation). For the 4 main indications, the change in mRS 0–2 and the change in median mRS was greater in the “early” groups compared to the “late” groups (figure, B, and table e-3).
Figure. Modified Rankin Scale score at initiation of rituximab and at outcome.

Modified Rankin Scale (mRS) score compared in all patients (total) and subgroups. (A) Groups are as follows (all patients n = 144 [total]): NMDAR encephalitis (NMDAR), opsoclonus myoclonus ataxia syndrome (OMAS), neuromyelitis optica spectrum disorders (NMOSD), neuropsychiatric systemic lupus erythematosus (NPSLE), and other groups (others). The follow-up was too short to create a mRS in 2 patients (one with OMAS, one other). mRS 0–2 infers a better outcome and is presented for each group as a percentage. The dotted line represents the change in mRS 0–2 between rituximab initiation and outcome. (B) mRS at rituximab initiation and outcome is compared according to the timing of rituximab administration. For each group, we separated an early and late group using the median duration of disease at rituximab initiation. Details of the timing, the median mRS, and the difference (Δ) in median mRS between initiation and outcome are also presented in table e-3.
DISCUSSION
We describe the clinical features, treatment protocols, laboratory features, and clinical outcome of 144 children and adolescents treated with the CD20 monoclonal antibody rituximab. To date, unlike in adults,4,15 there are no randomized clinical trials of any immunotherapy in pediatric CNS inflammatory disease, leading to off-label use in severely ill children. While retrospective analysis of treatment efficacy and safety has methodologic limitations, it is a critical first step toward enhanced appreciation of the role of such therapies in child and adolescent patient populations.
Rituximab was administered after a median of 0.5 years of disease, indicating that rituximab is generally given as a second-line immunosuppressant, typically after corticosteroids. Rituximab was also given for relapse prevention and to allow corticosteroid reduction. Despite an apparent benefit in many patients, after a median follow-up of 1.65 years after rituximab treatment, there was mortality of 2% and ongoing morbidity of 70%, indicating that this cohort of patients had severe and often refractory disease.
The variable dosing regimen utilized across the different pediatric centers reflects the different dosage regimens for differing indications in adult medicine.10 A recent report has shown that significantly lower doses of rituximab (100 mg weekly for 3 weeks) can produce effective B-cell depletion in Chinese adults with NMO,16 although lower doses may not result in improved safety profiles. A standardized infusion process was not consistent between sites. Antihistamines and concomitant corticosteroids are recommended to reduce the incidence of infusion-related side effects.10 Given the infusion reactions experienced by the children reported (18/144), treating clinicians need to be alert for allergic complications. We could not document a difference in infusion reactions between those children provided antihistamine treatment concomitantly with rituximab, but this does not negate the potential value of such preventative strategies.
B-cell depletion was induced in 119/124 (96% of patients who had lymphocyte subset measurement). Rituximab is often detectable in blood for up to 4 months after administration, and B cells typically start to return in the peripheral circulation after 6 months, and by 12 months in the majority of patients.9,17 B-cell depletion beyond 12 months was only observed in 12 patients, although this may be an underestimate as serial measurement was not routinely performed.
The rationale for repeat rituximab dosing is based on clinical diagnosis and the perceived risk of new attacks. For example, patients with NMO who are NMO immunoglobulin G (IgG)–positive are prone to a relapsing course and accrue disability with relapses.18 Understanding the timing of B-cell repopulation is one way of determining the timing of subsequent doses. An alternative approach increasingly employed in autoimmune disease is to administer regular rituximab every 6 months irrespective of B-cell counts to maintain remission of autoimmune disease. Reductions in immunoglobulin M (IgM), immunoglobulin A, or IgG were reported in 27/124 patients, although immunoglobulin measurements were not routinely performed prior to the initial rituximab dose and could have been preexisting in some patients. IgG or IgM hypogammaglobulinemia is more common when repeated courses of rituximab are given,19 or when given with cyclophosphamide.20 Other hematologic effects were uncommon (table 3).
The primary aim of the study was to define the potential infectious side effects of rituximab in this pediatric population. There were multiple confounders with this analysis. The patients were often sick, with 24% requiring intensive care admission, and the majority of patients received corticosteroids and other immunotherapies. In total, 11/144 (7.6%) of the cohort had an infectious (or suspected infectious) AE, although it is probable there were other more minor infectious illness not reported. Although some of the infectious complications could have been multifactorial in etiology, 4 patients had grade 4 or 5 AEs. Two of these patients had symptomatic cytomegalovirus (CMV) infection while immunosuppressed from rituximab, one of whom had also received cyclophosphamide. Although B-cell depletion would not typically be considered a risk factor for viral infection, reactivation of CMV, hepatitis B, varicella-zoster, herpes simplex, and CNS infection with enterovirus are recognized AEs with rituximab,10,21 particularly in patients treated for organ transplantation and malignant indications.8 These viral reactivation syndromes possibly represent some of the secondary effects of B-cell depletion on T-cell function.22,23 Rituximab should therefore be considered a broad-acting immunosuppressive agent. No cases of PML were reported in a total of 307 patient-years of follow-up, although the numbers are too small and follow-up too short to determine PML risk in children with CNS disease. PML was reported in only 1 of 2,875 children treated with rituximab for a broad range of indications in an International Classification of Diseases coding survey.8,24 There were a further 7 patients who had grade 3 AEs (respiratory and gastrointestinal infections) that were treated without complication. It is conceivable that some of these infections were related to the acuity and severity of the neurologic disease, rather than immunosuppression alone.
There was no apparent reduction in serious infection in the small proportion of patients who received antibiotic prophylaxis. Antibiotic prophylaxis could be considered in “higher-risk” patients with preexisting hypogammaglobulinemia, receiving cyclophosphamide, with known respiratory disease, or with documented significant hypogammaglobulinemia after rituximab, although the complications of chronic antibiotic usage should also be considered.
In this cohort, there were 46 children under 5 years of age at the time of rituximab administration, and there was no apparent increase in side effects in these young patients apart from a possible increase in hypogammaglobulinemia.
The apparent benefit of therapy was recorded in this cohort, although we acknowledge the following limitations. First, assessment of benefit was unblinded, retrospective, and subjective. Secondly, the follow-up after rituximab was relatively short (median 1.65 years) and variable between subgroups, and thus long-term benefits, or risks, remain to be defined. Third, most patients received other immunotherapies before and after rituximab; thus ascribing therapeutic benefit and risk to a single agent is inaccurate. Fourth, assessing efficacy is difficult in heterogeneous disorders using crude markers of disability such as the mRS. The outcome data in this cohort must also be considered in the context of the natural history of these serious disorders. For example, NMDAR encephalitis in adults and children has a mortality of 5%,6 and more than 50% of adult patients with relapsing NMO are blind in one or both eyes or require ambulatory help within 5 years of onset.6,18 A further confounder is the fact that some patients can improve independent of immunosuppression. The timing of rituximab varied according to etiology as described in table 2, with relatively earlier usage in autoimmune encephalitis and during the relapsing or chronic phases in other indications. With improved understanding of the risk/benefit ratio, it is expected that rituximab will be administered earlier in the disease course when its benefits could be maximized, as has been recently proposed in opsoclonus myoclonus ataxia syndrome,25 and as suggested in this cohort (figure, B). Although subjective, treating clinicians often considered rituximab to be beneficial, and only 2 patients had a disease “worsening,” which is occasionally observed after rituximab treatment, and may be secondary to a loss of T-cell regulation.23
Further work to optimize dosing regimens, standardize laboratory and safety monitoring, and consider patient registries would greatly enhance the ability to counsel patients and families on the risks and benefits of rituximab treatment. Formal clinical trials will be limited by the rarity of each individual CNS inflammatory disease, and by the need to consider other therapeutic options in these severely ill patients.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Eileen Baildam, Susana Camposano, Julie Curtin, David D'Cruz, Jonathan Gadian, Deepak Gill, Yvonne Koh, Bahadir Konuskan, Ram Kumar, Sunny Philip, Mike Pike, Peter Procopis, Davinder Singh-Grewal, Manish Sinha, Stefan Spinty, Kathryn Torok, Christopher Troedson, and Rachel Worthington for providing clinical information.
GLOSSARY
- AE
adverse events
- CMV
cytomegalovirus
- CTCAE
Common Terminology Criteria for Adverse Events
- DoD
duration of disease
- mRS
modified Rankin Scale
- NMDAR
NMDA receptor
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorders
- OMAS
opsoclonus myoclonus ataxia syndrome
- PML
progressive multifocal leukoencephalopathy
Footnotes
Editorial, page 111
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Project conception, design, and modification: R.C.D., F.B., S.B., M.L. Data acquisition: all authors. Data analysis and first drafts: R.C.D., F.B., M.L., M.P.G., B.B., M.T. Editing and approval of final draft: all authors.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
R. Dale has received research funding from the National Health and Medical Research Council, MS Research Australia, Star Scientific Foundation, Pfizer Neuroscience, Tourette Syndrome Association, University of Sydney, and the Petre Foundation. Russell Dale has received honoraria from Biogen-Idec as an invited speaker. F. Brilot has received research funding from the National Health and Medical Research Council, MS Research Australia, Star Scientific Foundation, Pfizer Neuroscience, Tourette Syndrome Association, and University of Sydney. L. Duffy and M. Twilt report no disclosures relevant to the manuscript. A. Waldman has received research funding from the NIH, National MS Society, and American Brain Foundation. Amy Waldman has received an honorarium from Teva as an invited speaker. S. Narula reports no disclosures relevant to the manuscript. E. Muscal has received research funding from the Lupus Foundation of America (LFA) and Texas Children's Hospital. K. Deiva received funds from Merck Serono and travel subsidies and speaker fees from Biogen-Idec. E. Andersen reports no disclosures relevant to the manuscript. M. Eyre is supported by a National Institute for Health Research (UK) Academic Clinical Fellowship. D. Eleftheriou has received funding from Arthritis Research UK. P. Brogan has received consultancy fees from Roche and Novartis. R. Kneen, G. Alper, and B. Anlar report no disclosures relevant to the manuscript. E. Wassmer has received research grants from Action Medical Research and MS Society, receives research support grants from BCH Research Charity, has received travel grants from UCB, Shire, and Biogen Idec, has received educational grants to organize meetings from Merck Serono, Novartis, Bayer, and Biogen Idec, received speaker's fees from Merck Serono, Biogen Idec, Shire, and Teva, and has received consultancy fees from Genzyme. K. Heineman reports no disclosures relevant to the manuscript. C. Hemingway has received educational grants from Bayer Schering, Biogen, Terumo, and Merck Serono. K. Riney has received support from Novartis to attend a meeting and an honorarium for participation in the Asia Pacific and South African Tuberous Sclerosis Advisory Board. A. Kornberg has received research funds from the National Health and Medical Research Council, Muscular Dystrophy Association (USA), and the MS Research Foundation. Dr. Kornberg has received honoraria from Biogen-Idec and Genzyme as an invited speaker. M. Tardieu and A. Stocco report no disclosures relevant to the manuscript. B. Banwell serves as a consultant to Biogen-Idec, Novartis, Teva Neuroscience, and Merck/Serono. Dr. Banwell is chief editor for Multiple Sclerosis and Related Disorders. Dr. Banwell has been funded by the Canadian MS Research Foundation, the Canadian MS Society, and CIHR. M. Gorman has received research funding from the National MS Society, NIH, and Department of Defense. S. Benseler reports no disclosures relevant to the manuscript. M. Lim receives research grants from Action Medical Research and MS Society, receives research support grants from the London Clinical Research Network and Evelina Appeal, has received consultation fees from CSL Behring; received travel grants from Merck Serono, and has been awarded educational grants to organize meetings by Novartis, Biogen Idec, Merck Serono, and Bayer. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Maloney DG, Grillo-Lopez AJ, White CA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood 1997;90:2188–2195 [PubMed] [Google Scholar]
- 2.Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363:221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006;54:2793–2806 [DOI] [PubMed] [Google Scholar]
- 4.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688 [DOI] [PubMed] [Google Scholar]
- 5.Pranzatelli MR, Tate ED, Travelstead AL, et al. Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 2006;28:585–593 [DOI] [PubMed] [Google Scholar]
- 6.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008;65:1443–1448 [DOI] [PubMed] [Google Scholar]
- 8.Kavcic M, Fisher BT, Seif AE, et al. Leveraging administrative data to monitor rituximab use in 2875 patients at 42 freestanding children's hospitals across the United States. J Pediatr 2013;162:1252–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimby E. Tolerability and safety of rituximab (MabThera). Cancer Treat Rev 2005;31:456–473 [DOI] [PubMed] [Google Scholar]
- 10.Highlights of prescribing information. Available at: http://www.gene.com/download/pdf/rituxan_prescribing.pdf. Accessed February 1, 2014
- 11.Common Terminology Criteria for Adverse Events v3.0 (CTCAE). Available at: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 1, 2014 [Google Scholar]
- 12.McLaughlin P, Grillo-Lopez AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998;16:2825–2833 [DOI] [PubMed] [Google Scholar]
- 13.Bigi S, Fischer U, Wehrli E, et al. Acute ischemic stroke in children versus young adults. Ann Neurol 2011;70:245–254 [DOI] [PubMed] [Google Scholar]
- 14.Le Sache N, Afanetti M, Deiva K, Chevret L, Tissieres P. Fulminant toxic shock syndrome following rituximab therapy in an 11-year-old boy. J Neurol 2013;260:2892–2893 [DOI] [PubMed] [Google Scholar]
- 15.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910 [DOI] [PubMed] [Google Scholar]
- 16.Yang CS, Yang L, Li T, et al. Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology 2013;81:710–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pranzatelli MR, Tate ED, Verhulst SJ, et al. Pediatric dosing of rituximab revisited: serum concentrations in opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 2010;32:e167–e172 [DOI] [PubMed] [Google Scholar]
- 18.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 19.De La Torre I, Leandro MJ, Valor L, Becerra E, Edwards JC, Cambridge G. Total serum immunoglobulin levels in patients with RA after multiple B-cell depletion cycles based on rituximab: relationship with B-cell kinetics. Rheumatology 2012;51:833–840 [DOI] [PubMed] [Google Scholar]
- 20.Venhoff N, Effelsberg NM, Salzer U, et al. Impact of rituximab on immunoglobulin concentrations and B cell numbers after cyclophosphamide treatment in patients with ANCA-associated vasculitides. PLoS One 2012;7:e37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quartier P, Tournilhac O, Archimbaud C, et al. Enteroviral meningoencephalitis after anti-CD20 (rituximab) treatment. Clin Infect Dis 2003;36:e47–e49 [DOI] [PubMed] [Google Scholar]
- 22.Stasi R, Del Poeta G, Stipa E, et al. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 2007;110:2924–2930 [DOI] [PubMed] [Google Scholar]
- 23.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol 2010;68:369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger JR, Houff SA, Major EO. Monoclonal antibodies and progressive multifocal leukoencephalopathy. MAbs 2009;1:583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pranzatelli MR, Tate ED, Swan JA, et al. B cell depletion therapy for new-onset opsoclonus-myoclonus. Mov Disord 2010;25:238–242 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.