

Abstract
Genomes assembled de novo from short reads are highly fragmented relative to the finished chromosomes of H. sapiens and key model organisms generated by the Human Genome Project. To address this, we need scalable, cost-effective methods enabling chromosome-scale contiguity. Here we show that genome-wide chromatin interaction datasets, such as those generated by Hi-C, are a rich source of long-range information for assigning, ordering and orienting genomic sequences to chromosomes, including across centromeres. To exploit this, we developed an algorithm that uses Hi-C data for ultra-long-range scaffolding of de novo genome assemblies. We demonstrate the approach by combining shotgun fragment and short jump mate-pair sequences with Hi-C data to generate chromosome-scale de novo assemblies of the human, mouse and Drosophila genomes, achieving – for human – 98% accuracy in assigning scaffolds to chromosome groups and 99% accuracy in ordering and orienting scaffolds within chromosome groups. Hi-C data can also be used to validate chromosomal translocations in cancer genomes.
The Human Genome Project (HGP) defined and achieved high standards for the de novo assembly of reference genomes for H. sapiens and key model organisms. For example, the public draft human genome, reported in 2001, contained 90% of the euchromatic sequence with an N50 (defined as the length L at which 50% of sequence is in contigs of length ≥L) of 82 kilobases (Kb)1,2. The finished human genome, reported in 2004, contained 99% of the euchromatic sequence with an N50 of 38.5 megabases (Mb) and an error rate of 1 event per 100,000 bases2. At both stages, nearly all sequences were assigned, ordered and oriented to chromosomes, although many errors were corrected during finishing2.
Massively parallel DNA sequencing technologies produce billions of short reads per instrument run at a very low cost per sequenced base, empowering a wide range of experiments3,4. However, although extensive progress has been made in developing algorithms for de novo genome assembly from short reads5, we remain remarkably distant from routinely assembling genomes to the standards set by the HGP. For example, the human genome was assembled with less than 40 gigabases (Gb) of Sanger sequencing, but de novo assemblies of short reads relying on 5- to 10-fold more sequence are highly fragmented relative to the finished chromosomes of the H. sapiens reference build6,7.
It is important to recognize that the high quality of the HGP’s genome assemblies is not solely attributable to the length and accuracy of Sanger sequencing reads. Rather, a diversity of approaches was brought to bear to achieve long-range contiguity. For the human genome, this included dense genetic maps, dense physical maps and hierarchical shotgun sequencing of a tiling path of long insert clones1,2. Whole-genome shotgun assemblies – typically based on end sequencing of both short and long insert clones – also relied on dense genetic and physical maps to assign, order and orient sequence contigs or scaffolds to chromosomes8.
Diverse strategies have been developed to boost the contiguity of de novo genome assemblies from short reads. These include end sequencing of fosmid clones6, fosmid clone dilution pool sequencing9,10, optical mapping11–14 and genetic mapping with restriction site associated DNA (RAD) tags15. However, each of these strategies has important limitations. Fosmid libraries and optical mapping are technically challenging and provide only mid-range contiguity. Genetic maps are more powerful but are costly or impractical to generate for many species. Particularly as initiatives such as the 10K Genome Project16 gain momentum, the genomics field is in need of scalable, broadly accessible methods enabling chromosome-scale de novo genome assembly.
Hi-C and related protocols use proximity ligation and massively parallel sequencing to probe the three-dimensional architecture of chromosomes within the nucleus, with interacting regions captured to paired-end reads17,18. In the resulting datasets, the probability of intrachromosomal contacts is on average much higher than that of interchromosomal contacts, as expected if chromosomes occupy distinct territories. Moreover, although the probability of interaction decays rapidly with linear distance, even loci separated by >200 Mb on the same chromosome are more likely to interact than loci on different chromosomes17.
We speculated that genome-wide chromatin interaction datasets, such as those generated by Hi-C, might provide long-range information about the grouping and linear organization of sequences along entire chromosomes. In exploring this, we developed LACHESIS (ligating adjacent chromatin enables scaffolding in situ), a computational method that exploits the signal of genomic proximity in Hi-C datasets for ultra-long-range scaffolding of de novo genome assemblies. LACHESIS works in three steps (Fig. 1) – first, clustering contigs or scaffolds to chromosome groups; second, ordering contigs or scaffolds within each chromosome group; and finally, assigning relative orientations to individual contigs or scaffolds. We demonstrate the effectiveness of this approach by combining shotgun fragment and short insert mate-pair (<3 Kb) sequences with Hi-C data to generate reasonably accurate chromosome-scale de novo assemblies of the H. sapiens, M. musculus and D. melanogaster genomes. We also show that Hi-C data can be used to validate chromosomal rearrangements in cancer genomes.
Figure 1.
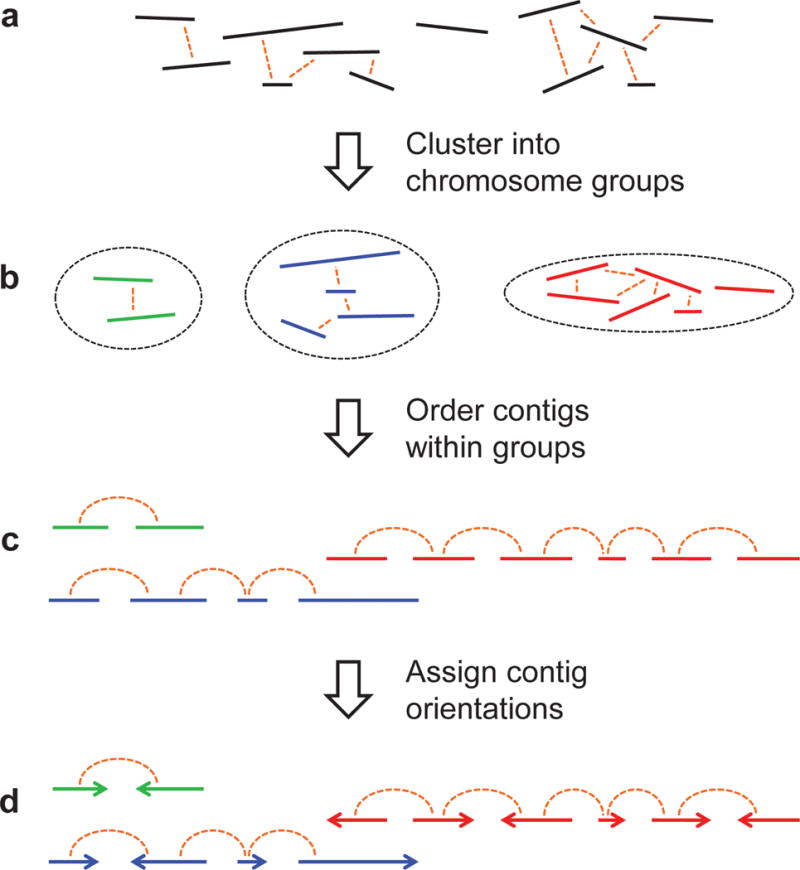
A schematic of the LACHESIS scaffolding method. (a) The input consists of a set of contigs (or scaffolds) from a draft assembly and a set of genome-wide chromosome interaction data, e.g., Hi-C links. (b) Contigs on the same chromosome tend to have more Hi-C links between them, relative to contigs on different chromosomes. LACHESIS exploits this to cluster the contigs into groups that largely correspond to individual chromosomes. (c) Within a chromosome, contigs in close proximity tend to have more links than contigs that are distant. LACHESIS exploits this to order the contigs within each chromosome group. (d) Lastly, LACHESIS uses the exact position of links between adjacent contigs to predict the relative orientation of each contig.
Results
The input to LACHESIS consists of a set of contigs or scaffolds (the term ‘contig’ is used in this description of the method to indicate both possibilities), such as are generated by de Bruijn graph–based de novo assemblers5,6, and a genome-wide chromatin interaction dataset, such as is generated by the Hi-C method17,18 The Hi-C reads are aligned to the contigs, and the number of Hi-C read pairs linking each pair of contigs is tabulated (Figure 1a). In a first step, LACHESIS uses hierarchical agglomerative clustering to group contigs that are likely to derive from the same chromosome, exploiting the fact that intrachromosomal contacts are on average more probable than interchromosomal contacts in Hi-C datasets17 (Fig. 1b; Supplementary Fig. 1). An average-linkage metric19 is used for this clustering, with linkage defined as the normalized density of Hi-C read-pairs linking any given pair of contigs. The final number of groups is pre-specified, ideally set to the expected number of chromosomes.
In a second step, LACHESIS orders contigs linearly within each chromosome group by taking advantage of the higher Hi-C link densities expected between closely located contigs (Fig. 1c; Supplementary Fig. 2). For each chromosome group, a graph is built with vertices representing contigs and edge weights corresponding to the inverse of the normalized Hi-C linkage density between pairs of contigs. A minimum spanning tree (MST) is found in this graph, and the longest path in the MST is extracted as the ‘trunk’, an incomplete but high-confidence ordering of contigs within each chromosome group. To generate a full ordering, contigs excluded from the trunk are reinserted into it at sites that maximize the amount of linkage between adjacent contigs.
In a third step, the ordered contigs are oriented with respect to one another by taking into account precisely where the Hi-C reads map on each contig (Fig. 1d; Supplementary Fig. 3). For each chromosome group, a weighted directed acyclic graph (WDAG) is built representing all possible ways to orient the contigs, given the predicted order. The weights are calculated as the log-likelihood of the observed Hi-C links between adjacent contigs in a given combined orientation, assuming that the probability of a link connecting two reads at a genomic distance of x decays as 1/x for x ≥ ~100 Kb17. The maximum likelihood path through this graph yields a predicted orientation for each contig.
Chromosome-scale assembly of mammalian genomes
We sought to evaluate the effectiveness of this approach for the chromosome-scale de novo assembly of mammalian genomes. We focused on human and mouse as test cases because of the availability of the necessary datasets and the high quality of these reference genomes as gold standards for comparison. For human, we used ALLPATHS-LG to assemble previously generated6 shotgun fragment and short jump (~2.5 Kb) mate-pair sequences to an N50 scaffold length of 437 Kb and a total length of 2.74 Gb. We refer to this below as the ‘shotgun assembly.’ We intentionally excluded fosmid end sequencing data6 because libraries of this type require cloning and are laborious to generate. Furthermore, we hoped that the chromatin interaction data would effectively substitute for the ~40 Kb fosmid links while also providing even longer-range contiguity.
After aligning Hi-C read-pairs from a human male ESC line20 to this shotgun assembly, we applied LACHESIS to cluster the scaffolds into 23 chromosome groups (the libraries used to generate the shotgun assembly were derived from female DNA6), and then to order and orient the scaffolds within each chromosome group (Fig. 2, 3; Table 1; Supplementary Fig. 4; Supplementary Tables 1, 2). Most scaffolds (n = 13,528, comprising 98.2% of the length of the shotgun assembly) were clustered into one of the 23 groups (Fig. 2a). Nearly all of these groups corresponded to individual chromosomes, with the exceptions of the X chromosome, whose two arms were split in separate groups (Fig. 3u,v); one chimeric group containing very little sequence from many chromosomes (6.5 Mb total; Supplementary Fig. 4w); chromosomes 19 and 22, which were “fused” into a single group (Fig. 3s); and chromosomes 20 and 21, also fused into a single group (Fig. 3t). The fusions are probably due to the greater density of interchromosomal links observed between short chromosomes in Hi-C data17,21. Apart from these errors, 98.6% of clustered scaffolds (comprising 99.86% of their sum length) were correctly grouped (Table 1), suggesting that Hi-C data are highly informative for the clustering of sequences derived from individual chromosomes, including across centromeres.
Figure 2.
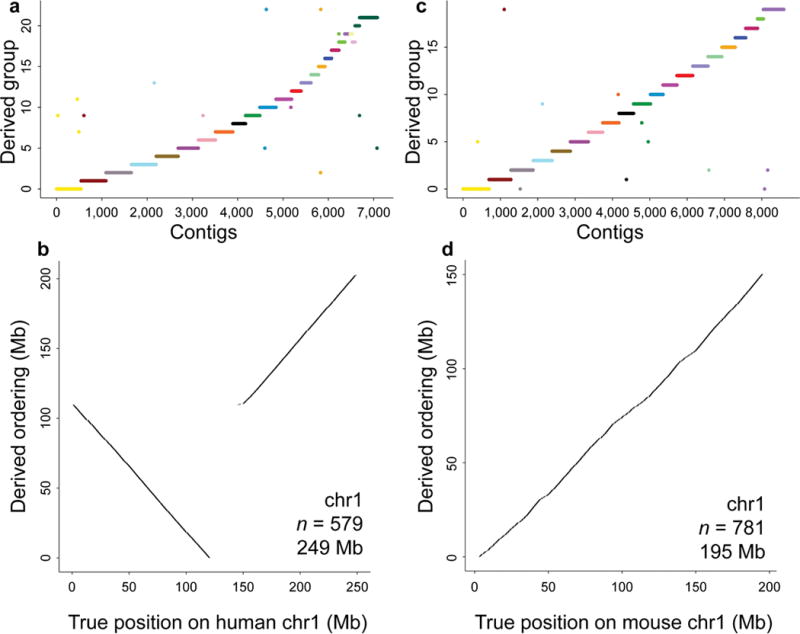
Clustering and ordering mammalian sequences with LACHESIS. (a) The results of LACHESIS clustering on the de novo human assembly. Shown on the x-axis are the 7,083 scaffolds (total length: 2.49 Gb) that are large (≥25 AAGCTT restriction sites) and not repetitive (Hi-C link density less than 2 times average), which LACHESIS uses as informative for clustering. The y-axis shows the 23 groups created by LACHESIS, with the order chosen for the purposes of clarity. The color scheme is the standard SKY (spectral karyotyping) color scheme for human. (b) The results of LACHESIS ordering and orienting of 579 scaffolds within the group from a corresponding to human chromosome 1. On the x-axis is the true position of these scaffolds along human chromosome 1. On the y-axis is the order in which LACHESIS has placed these scaffolds. Also listed in the panel are the chromosome name, the number of scaffolds in the derived ordering and the reference length of this chromosome. (c) The results of LACHESIS clustering on the de novo mouse assembly. Shown on the x-axis are the 8,594 scaffolds (total length: 1.94 Gb) that are large and not repetitive, which LACHESIS uses as informative for clustering. The y-axis shows the 20 groups created by LACHESIS, with the order chosen for the purposes of clarity. The color scheme is as in a. (d) The results of LACHESIS ordering and orienting of 781 scaffolds within the group from c corresponding to mouse chromosome 1. The plotting is as in b.
Figure 3.
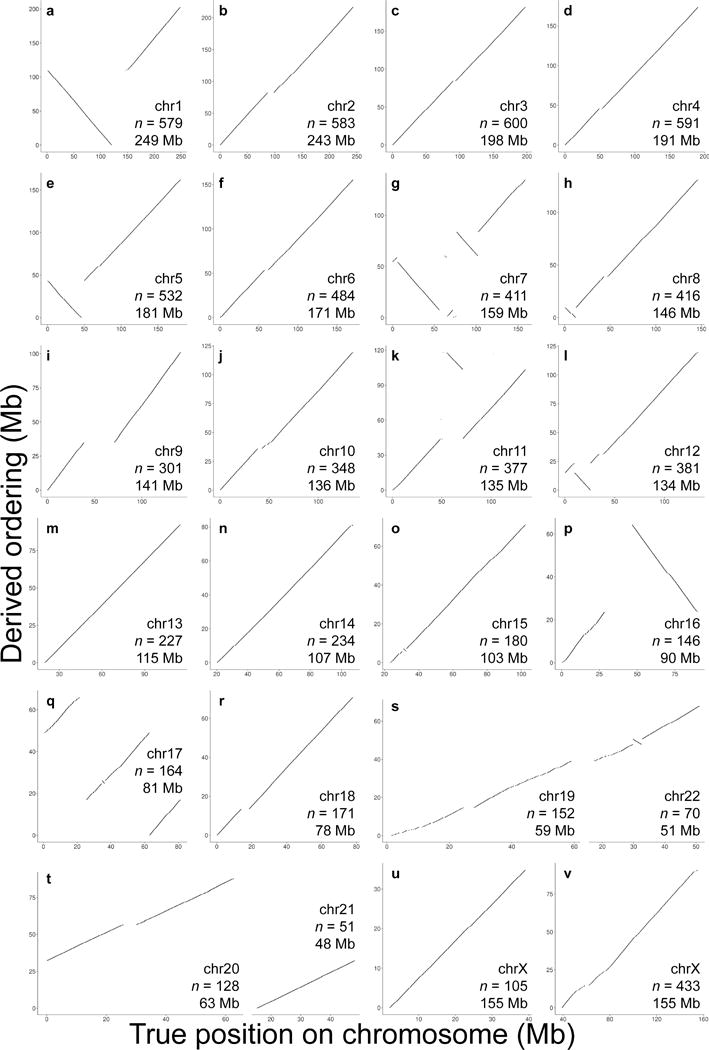
LACHESIS ordering of scaffolds in a de novo human assembly. (a–v) The results of LACHESIS ordering and orienting on 22 of the 23 chromosome groups in the de novo human assembly. For each ordering, only the scaffolds on the “dominant chromosome” (the chromosome containing the plurality of aligned sequence) are shown. The exceptions are two groups that correspond to fusions of small chromosomes (19 and 22 (s); 20 and 21 (t)) (see Supplementary Table 2). Within each of these fused groups, the two chromosomes were well separated by ordering (s,t). The X chromosome clustered into two separate groups (u,v). Not shown is one very small chimeric group (length = 6.5 Mb; see Supplementary Fig. 4w). Also listed in each panel are the identity of the dominant chromosome, the number of scaffolds in the derived ordering and the reference length of the dominant chromosome.
Table 1. Metrics for LACHESIS-based scaffolding of shotgun assemblies.
The human and mouse shotgun assemblies are based on read-pairs from short-insert and ~2.5 Kb jumping libraries, while the Drosophila shogun assembly is based solely on read-pairs from short-insert libraries6. The human and mouse shotgun assemblies consist of scaffolds, whereas the Drosophila shotgun assembly consists of contigs. LACHESIS places scaffolds or contigs into groups and then orders and orients them within each group. An ordering error means that a contig or scaffold’s position is out of the expected order with respect to its neighbors. An orientation error means that its orientation is not the orientation implied by its position with respect to its immediate predecessor. ‘High-quality predictions’ refers to a subset of contigs or scaffolds whose position and orientation in their ordering is deemed more certain; the threshold for high quality is chosen for convenience for each assembly.
| Metric | De novo assemblies | ||
|---|---|---|---|
| Human | Mouse | Drosophila | |
| Shotgun assembly metrics | |||
| Total assembly length, including gaps (Mb) | 2,739 | 2,370 | 127 |
| Number of contigs or scaffolds | 18,921 | 25,964 | 7,109 |
| N50 contig or ungapped scaffold size (Kb) | 437 | 224 | 68 |
| Clustering | |||
| % sequence (% contigs) clustered into groups | 98.2% (71.5%) | 98.0% (87.8%) | 81.2% (64.3%) |
| % clustered sequence (% contigs) mis-clustered | 0.14% (1.4%) | 0.24% (0.5%) | 3.4% (10.5%) |
| Ordering | |||
| % clustered seq. (% contigs) ordered | 94.4% (55.3%) | 86.7% (42.7%) | 82.0% (24.5%) |
| % ordered seq. (% contigs) w/ ordering errors | 0.5% (0.8%) | 0.5% (1.1%) | 4.6% (5.2%) |
| % ordered seq. (% contigs) w/ orientation errors | 1.2% (2.5%) | 1.9% (4.6%) | 4.1% (6.1%) |
| High-quality predictions | |||
| % ordered seq. (% contigs) w/ high quality | 92.8% (79.0%) | 93.3% (82.9%) | 94.1% (88.1%) |
| % high-quality seq. (% contigs) w/ ordering errors | 0.3% (0.4%) | 0.3% (0.7%) | 3.3% (3.4%) |
| % high-quality seq. (% contigs) w/ orientation errors | 0.4% (0.5%) | 0.5% (1.0%) | 2.5% (2.7%) |
Within each chromosome group, the vast majority of the length of the clustered scaffolds was successfully ordered and oriented by LACHESIS (94.4% or 2.55 Gb; Table 1). The predicted orderings are highly concordant with the reference human genome (GRCh37), including across most megabase-scale centromere gaps, except for the occasional rearrangement of large segments within which nearly all scaffolds were well-ordered (Fig. 3; Supplementary Fig. 4). For example, scaffolds corresponding to the long and short arms of chromosome 1 are grouped together and respectively very well-ordered, but the reconstructed arms are joined incorrectly (Fig. 2b). To quantify local errors, we defined ordering errors as instances where a contig or scaffold is not in the expected order with respect to its immediate neighbors, and orientation errors as instances where a contig or scaffold is not in the expected orientation implied by its immediate predecessor in the ordering. By these definitions, 99.2% of clustered scaffolds, representing 99.5% of the sum length, were correctly ordered; 97.5% of clustered scaffolds, representing 98.8% of the sum length, were correctly oriented.
Most ordering errors involve the inversion of local segments consisting of one or several contiguous scaffolds (Supplementary Fig. 4). Compared to correctly ordered scaffolds, incorrectly ordered scaffolds are short and are enriched for segmental duplications and simple repeats (Supplementary Fig. 5; Supplementary Table 3). This suggests that complexities in the primary sequence are the source of many ordering errors, possibly via inaccuracies in the shotgun assembly or by confounding the mapping of Hi-C read-pairs. Other errors appear to be associated with the non-uniform distribution of biological interactions, e.g., chromatin domains at various scales (Supplementary Fig. 6). To address this in part, we calculated a quality score for ordering and orientation, defined as the relative log-likelihood of the contig’s predicted orientation to its opposite orientation in the WDAG. Local accuracy was better for scaffolds with high quality scores (Table 1). For scaffolds with high quality scores occurring within the assembly trunk, which comprise 2.09 Gb or 76.4% of the overall shotgun assembly, 99.9% of sequence is correctly ordered and 99.7% correctly oriented (Supplementary Table 1).
We also attempted the chromosome-scale de novo assembly of the mouse genome by an identical approach. We first used ALLPATHS-LG to assemble previously generated6 shotgun fragment and short jump (~2.2 Kb) mate-pair sequences to an N50 scaffold length of 224 Kb and a total length of 2.37 Gb. After aligning Hi-C read-pairs from a mouse ESC line20 to this shotgun assembly, we applied LACHESIS to cluster the scaffolds into 20 chromosome groups, and then to order and orient the scaffolds within each chromosome group (Fig. 2c,d; Table 1; Supplementary Fig. 7; Supplementary Tables 1, 4). Most scaffolds (n = 22,802, comprising 98.0% of the length of the shotgun assembly) were clustered into one of the 20 groups (Fig. 2c). There was a clear 1-to-1 correspondence between these groups and bona fide chromosomes (GRCm38), although a small part of mouse chromosome 10 (2.6 Mb) was erroneously clustered with chromosome 8 (Supplementary Table 4). Of the clustered scaffolds, 99.5% (comprising 99.76% of their sum length) were correctly grouped (Table 1). The majority of the length of the clustered scaffolds was ordered and oriented by LACHESIS (86.7% or 2.02 Gb; Table 1). Almost all (99.5%) of clustered scaffolds, representing 98.9% of the sum length, were correctly ordered; 95.4% of scaffolds, representing 98.1% of the sum length, were correctly oriented. Overall, the results for chromosome-scale de novo assembly of the mouse and human genomes are highly consistent.
Chromosome-scale assembly of the fruit fly genome
To further evaluate the generality of this method, we next applied it to the de novo assembly of Drosophila melanogaster, for which a high-quality reference genome is also available as a gold standard for comparison. We first used ALLPATHS-LG to assemble shotgun fragment sequences (without jumping libraries) to an N50 contig length of 68 Kb and a total length of 127 Mb6,22. We then aligned Hi-C read-pairs derived from D. melanogaster23 to this shotgun assembly and used LACHESIS to cluster the contigs into 4 chromosome groups. Most contigs (81.2% of the length of the shotgun assembly) were clustered into one of the 4 groups (Supplementary Fig. 8). This proportion is lower than that for the assemblies described above (≥98% for human and mouse), most likely because of the lower contiguity of the shotgun assembly (N50 contig size of 68 Kb for Drosophila versus N50 scaffold size of 437 Kb and 224 Kb for human and mouse, respectively). Nonetheless, the four groups corresponded well to the four Drosophila chromosomes (X, 2, 3 and 4), even though chromosome 4 is minuscule compared to the others (1.4 Mb or ~1% of the reference genome). Of the clustered scaffolds, 89.5% (comprising 96.6% of their sum length) were correctly grouped.
We next applied LACHESIS to order and orient the Drosophila contigs within each of the 4 chromosome groups (Supplementary Table 5; Supplementary Fig. 9). A lower proportion of the shotgun assembly was ordered (82.0% by length for fly versus 94.4% for human), again likely because the Drosophila assembly has shorter contigs than the mammalian shotgun assemblies used above. The predicted order corresponded well with the actual order based on contig alignments to the D. melanogaster reference genome (FB2013_02, euchromatic sequences only), and the R and L arms of chromosomes 2 and 3 were well-separated (Supplementary Fig. 9). Once again, a subset of the chromosome groups contained rearrangements of large segments within which nearly all contigs were well-ordered. At a local scale, 94.8% of clustered contigs (95.4% of sum length) were correctly ordered, and 93.9% of clustered contigs (95.9% of sum length) were correctly oriented (Table 1).
Robustness to contig size and Hi-C data quantity
Our results for chromosome-scale scaffolding of the human, mouse and fly genomes were based on initial de novo assemblies with reasonably high N50s, i.e., 437 Kb, 224 Kb and 68 Kb, respectively. To evaluate the power of this approach as a function of the contiguity of this initial assembly, we sought to reassemble simulated contigs of varying size derived from the human reference genome (GRCh37). In each iteration, we split the human reference genome into equally sized contigs (10, 20, 50, 100, 200, 500 or 1,000 Kb) and mapped Hi-C read-pairs20 to these simulated shotgun assemblies. We then used LACHESIS to cluster, order and orient the simulated contigs (results for 100 Kb simulated contigs are shown in Supplementary Fig. 10, 11). As shown in Table 2, the performance of the method with respect to completeness and local accuracy is robust above an initial N50 of 50 Kb, but degrades rapidly below this point.
Table 2. Metrics for LACHESIS-based scaffolding of simulated assemblies.
Simulated assemblies were created by breaking up the human reference genome into simulated contigs of varying sizes, and then using LACHESIS to cluster, order and orient the simulated contigs. The simulated contigs’ expected order and orientation are derived from their true position in the reference genome. Ordering and orientation errors are defined as in Table 1.
| Metric | Simulated contig size | ||||||
|---|---|---|---|---|---|---|---|
| 10 Kb | 20 Kb | 50 Kb | 100 Kb | 200 Kb | 500 Kb | 1 Mb | |
| Number of contigs | 309,579 | 154,794 | 61,927 | 30,970 | 15,489 | 6,206 | 3,113 |
| % sequence clustered into groups | 30.1% | 74.2% | 91.9% | 92.7% | 92.9% | 93.1% | 93.4% |
| % clustered sequence mis-clustered | 1.6% | 0.47% | 0.41% | 0.46% | 0.66% | 0.66% | 0.26% |
| % clustered sequence ordered | 48.5% | 79.9% | 98.9% | 99.8% | 99.97% | 99.93% | 99.98% |
| % ordered sequence w/ ordering errors | 37.2% | 18.0% | 4.4% | 2.2% | 1.4% | 0.8% | 0.8% |
| % ordered sequence w/ orientation errors | 44.8% | 28.7% | 7.7% | 2.6% | 1.2% | 0.8% | 0.7% |
In a separate analysis, we down-sampled the sequencing depth of Hi-C data and attempted chromosome-scale scaffolding of the human shotgun assembly (N50 = 437 Kb; Supplementary Table 6). Although clustering is robust to marked reductions in the amount of Hi-C data, accurate ordering and orienting of scaffolds within chromosome groups requires ~400M read-pairs. Nonetheless, we note that even the full amount of Hi-C data used here is less than 20% of the amount of sequencing data used to generate the initial shotgun assembly (59 Gb vs. 303 Gb).
Validating translocations in cancer genomes
We also speculated that the strong intrachromosomal signal observed in Hi-C data might enable the global discovery or validation of interchromosomal rearrangements in cancer genomes, many of which are challenging to detect with methods other than karyotyping because the breakpoints occur in repetitive regions. For example, recent studies combined several mate-pair sequencing strategies to detect rearranged marker chromosomes in the aneuploid HeLa cancer cell line24,25, but such methods were only successful for a small proportion of rearrangements, and for none of the rearrangements involving centromeric sequences. Of note, the 4C method was previously used to detect chromosomal breakpoints in cancer genomes, but in a targeted rather than global fashion26.
To test this, we constructed a Hi-C library from HeLa cells and sequenced it to high depth (154 M unique read-pairs). These data were used to generate a matrix of link densities between ~1 Mb windows in the human reference genome. Visual examination of the matrix revealed off-diagonal patches of strong linkage with asymmetric decay, consistent with interchromosomal rearrangements (Fig. 4). Most of these corresponded well to previously described marker chromosomes27, although we also observed strong evidence for two novel marker chromosomes (der(2;7)(q36;q10), “U1” and der(3;20)(q25;q10), “U2”). We implemented a rearrangement-calling method that successfully identified all of the suspected marker chromosomes, albeit with limited specificity (Supplementary Fig. 12). Using chromatin interaction data in this way may enable the validation of candidate chromosomal rearrangements, or the detection of chromosomal rearrangements in heterogeneous cancer cell populations that might not be detected by karyotyping of limited numbers of cells.
Figure 4.
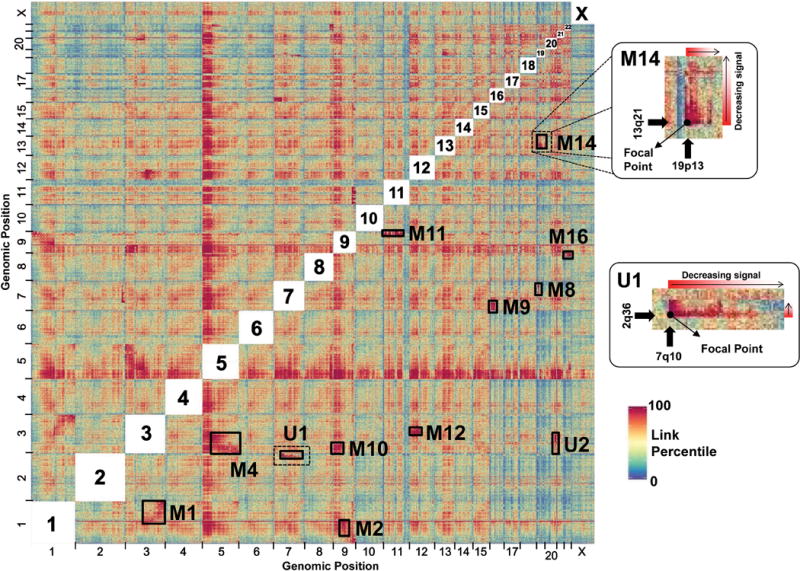
Detection of chromosome fusions in HeLa S3 using Hi-C data. Normalized interchromosomal links for a HeLa S3 Hi-C library between megabase windows were derived as described in Online Methods and are represented as an all-by-all heatmap. For visualization purposes, link weights were ranked and converted to a percentile. Previously identified marker chromosomes were identified (M1, M2, M4, M8, M9, M10, M11, M12, M14 and M16) as well as two additional peaks representing previously undescribed marker chromosomes (U1: der(2;7)(q36;q10) and U2: der(3;20)(q25;q10)). Two rearrangements are highlighted (M14 and U1) to demonstrate the signal focal point at the location of the fusion event with asymmetrical signal decay outward in the direction of the sequence contained in the chromosome fusion, thus allowing breakpoint identification as well as orientation.
Discussion
Here we demonstrate that genome-wide chromatin interaction datasets, such as those generated by Hi-C, are a rich source of long-range information for assigning, ordering and orienting genomic sequences to chromosomes, including across megabase-scale centromere gaps, as well as for validating chromosomal translocations in cancer genomes. There are a number of avenues for the potential improvement of this approach, both experimentally and computationally.
Although the experimental methods for Hi-C are straightforward, current protocols require a large amount of material (106–108 cells). As such, reducing the input requirements is an important technical goal. To date, global chromatin interaction datasets have been generated on organisms including yeast18, human17,20,21, mouse20, fruit fly23 and Arabidopsis thaliana28. This is consistent with broad applicability, but demonstrating these protocols on an even more diverse range of organisms is imperative. On a related point, as the success of this approach depends on chromosomes occupying distinct territories in the nucleus, it will be important to further validate LACHESIS in diverse species to confirm that this is ubiquitously the case. We also note that using multiple restriction enzymes (or developing new methods that avoid restriction digestion altogether) will likely improve performance, particularly for smaller contigs or scaffolds. Along the same lines, even if this approach broadly enables chromosome-scale scaffolding, the contiguity required for the initial de novo assembly (~50 Kb) may be challenging to achieve for many organisms. As such, there will remain a strong need for methods delivering “intermediate” contiguity information in a highly cost-effective and scalable manner.
Computationally, a substantial limitation of our current algorithm is that the clustering step requires the number of chromosomal groups to be specified a priori. We assessed whether the scoring metric used during clustering enables reliable inference of chromosome number, but this is not the case (Supplementary Fig. 13). One potential solution is to order contigs or scaffolds prior to determining chromosome groups, but this is computationally difficult with large numbers of contigs or scaffolds. Alternatively, statistical methods for predicting the optimal number of clusters may prove useful29,30.
Ordering and orientation errors were associated with short scaffolds, segmental duplications and simple repeats (Supplementary Table 3). It is possible that our full exclusion of ambiguously mapping reads may be introducing ‘gaps’ in contiguity information that increase the probability of errors in such regions. Alternatively, these errors may be secondary to flaws in the initial shotgun assembly. Consistent with the latter, we also ran LACHESIS on a human ‘shotgun assembly’ that has higher contiguity because it used fosmid end-pair data6 (N50 scaffold length 11.5 Mb versus 437 Kb). We achieved chromosome-scale scaffolding of this assembly as well, but with lower accuracy owing to a small fraction of incorrectly joined scaffolds in the input to LACHESIS (Supplementary Table 1). This suggests that conservative de novo assembly prior to using chromatin interaction mapping for long-range scaffolding may be optimal. Lastly, we note that our use of chromatin interaction data for long-range scaffolding (via LACHESIS) was entirely separate from the initial assembly of contigs/scaffolds (by ALLPATHS-LG). We anticipate that a more integrated approach might improve accuracy.
Starting from draft human and mouse genome assemblies, each consisting of tens of thousands of scaffolds, we were able to cluster nearly all scaffolds into groups that overwhelmingly corresponded to individual chromosomes. A high fraction of these assignments were correct (comprising >99% of the sum length of clustered scaffolds). We were further able to order and orient contigs within each chromosome group, including scaffolding across megabase-scale centromere gaps, with surprisingly few errors. As such, we achieved reasonably accurate de novo mammalian genome assemblies with chromosome-scale contiguity using just three types of libraries, all generated by in vitro methods and sequenced as short read-pairs on a single platform (for human, shotgun fragment (161 Gb); ~2.5 Kb short jump (142 Gb); and Hi-C (59 Gb)). Although its broad applicability beyond the genomes assembled here has still to be demonstrated, our approach may enable a new generation of de novo genome assemblies that do not sacrifice the high standards for contiguity set by the HGP.
Online Methods
Input datasets
In the Hi-C procedure17, DNA in a nucleus is crosslinked, then cut with a restriction enzyme, leaving pairs of distally located but physically associated DNA molecules attached to one another. The sticky ends of these fragments are biotinylated and then ligated to each other to form chimeric circles. Biotinylated circles are enriched for, sheared again, and then processed to sequencing libraries in which individual templates are chimeras of the physically associated DNA molecules from the original cross-linking.
Four Hi-C datasets were used, corresponding to human cells, mouse cells, Drosophila melanogaster tissue and HeLa cells. The human dataset was produced from human embryonic stem cells (hESCs)20. The hESC replicates 1 and 2 were used (NCBI SRA accessions: GSM862723, GSM892306) for a total of 734 M read-pairs. The mouse dataset was produced from mouse embryonic stem cells (mESCs)20. The mESC replicates 1 and 2 were used (NCBI SRA accessions: GSM862720, GSM862721) for a total of 806 M read-pairs. The D. melanogaster dataset was produced from embryos23 and includes 363 M read-pairs (NCBI SRA accession: SRX111555). The HeLa dataset was produced as part of this study (see “Chromosome Fusion Detection in HeLa”, below) and includes 305 M read-pairs.
Two types of shotgun assemblies were created as inputs to LACHESIS. First, we created shotgun assemblies for human, mouse and Drosophila melanogaster by downloading the appropriate sequence libraries from SRA and assembling them with ALLPATHS-LG. Table 1 shows statistics for these three assemblies. Second, simulated shotgun assemblies were made by breaking up the human reference genome into contigs of varying sizes, ranging from 10 Kb to 1 Mb. Table 2 shows statistics for these assemblies.
Shotgun assemblies
To create the human shotgun assembly, we downloaded the sequence files6 corresponding to the fragment library and two short jumping libraries for individual NA12878 from the NCBI Short Read Archive (NCBI SRA accession SRA024407). The files were converted from sra to fastq format, and formatted as required by the ALLPATHS-LG assembler using the PrepareAllPathsInputs.pl script included with the ALLPATHS-LG distribution. The reads were assembled using the ALLPATHS-LG assembler6 (version r41985) with the following parameters (the rest being default): HAPLOIDIFY = TRUE, MAX_MEMORY_GB = 400, THREADS = 32, EVALUATION = STANDARD. Insert size estimates (mean and standard deviation) for each library were specified based on the values provided previously6. Scaffolds in this assembly were treated as contigs by LACHESIS. Because we intentionally excluded fosmid end sequencing data, this assembly had far less mid-range contiguity than the full de novo assembly produced previously6 (N50 scaffold length 437 Kb versus 11.5 Mb), and thus it more closely represents a typical de novo assembly created exclusively from in vitro libraries.
To create the mouse shotgun assembly, we downloaded the sequence files6 corresponding to the fragment and three short jumping libraries from the NCBI Short Read Archive (NCBI SRA accession SRA009956). The libraries were assembled using the ALLPATHS-LG assembler6 (version r41985) with the following parameters (the rest being default): HAPLOIDIFY = TRUE, MAX_MEMORY_GB = 500, THREADS = 32. Insert size estimates (mean and standard deviation) for each library were specified based on the values provided previously6.
To create the Drosophila melanogaster shotgun assembly, we downloaded the sequence files for D. melanogaster (Drosophila Genomic Reference Panel22 corresponding to sequencing runs SRR516038 (Sample DGRP-348) and SRR516001 (Sample DGRP-821) from the NCBI Short Read Archive. SRR516038 served as the “fragment” library as per ALLPATHS-LG terminology. The ALLPATHS-LG assembler also requires a “jumping” library. We were unable to find a previously sequenced jumping library for D. melanogaster. As a work-around, we used a standard shotgun library with a slightly higher insert size (SRR516001) and artificially converted it into a jumping library by flipping the orientation of reads. All files were first converted from sra to fastq format, then formatted as required by the ALLPATHS-LG assembler using the PrepareAllPathsInputs.pl script included with the ALLPATHS-LG distribution. Insert size distributions for these libraries (mean = 205bp, standard deviation = 25bp for fragment library; mean = 320bp, standard deviation = 52bp for jumping library) were obtained by aligning a subset of reads to the D. melanogaster reference genome using BWA31. The reads were assembled using the ALLPATHS-LG assembler6 (version r41985) with the following parameters (the rest being default): HAPLOIDIFY = TRUE, MAX_MEMORY_GB = 300, THREADS = 16, VAPI_WARN_ONLY=True.
Aligning Hi-C reads
Hi-C reads were aligned to shotgun assemblies or reference genomes using BWA31 with default parameters. Reads were considered artifactual if they did not align within 500 bp of a restriction site, as recommended21. Non-uniquely aligning reads were assigned a mapping quality of 0 by BWA and were excluded from subsequent analysis. Additionally, read-pairs were considered for downstream analysis only if both reads in the pair aligned to contigs from the assembly.
Clustering contigs or scaffolds into chromosome groups
Contigs or scaffolds (the term ‘contig’ is used in this description of the method to indicate both possibilities) were placed into groups using hierarchical clustering (Supplementary Fig. 1). A graph was built, with each node initially representing one contig, and each edge between nodes having a weight equal to the number of Hi-C read-pairs linking the two contigs. The contigs were merged together using hierarchical agglomerative clustering with an average-linkage metric19, which was applied until the number of groups was reduced to the expected number of distinct chromosomes (counting only groups with more than one contig). Repetitive contigs (contigs whose average link density with other contigs, normalized by number of restriction fragment sites, was greater than two times the average link density) and contigs with too few restriction fragment sites (<5 for the simulated human assembly; <25 for the human and mouse de novo assemblies; <250 for the D. melanogaster assembly) were not clustered. However, after clustering, each of these contigs was assigned to a group if its average link density with that group was greater than four times its average link densities with any other group.
Ordering contigs or scaffolds within chromosome groups
Each group of contigs or scaffolds (the term ‘contig’ is used in this description of the method to indicate both possibilities) was ordered using the following algorithm (Supplementary Fig. 2). First, a graph was built as in the clustering step, but with the edge weights between nodes equal to the inverse of the number of Hi-C links between the contigs, normalized by the number of restriction fragment sites per contig. Short contigs (<5 restriction fragment sites for the simulated human assemblies; <20 sites for the human and mouse de novo assemblies; <20 Kb for the Drosophila de novo assembly) were excluded from this graph. A minimum spanning tree was calculated for this graph. The longest path in this tree, the “trunk”, was found. The spanning tree was then modified so as to lengthen the trunk by adding to it contigs adjacent to the trunk, in ways that kept the total edge weight heuristically low.
After a lengthened trunk was found for each group, it was converted into a full ordering as follows. The trunk was removed from the spanning tree, leaving a set of “branches” containing all contigs not in the trunk. These branches were reinserted into the trunk, the longest branches first, with the insertion sites chosen so as to maximize the number of links between adjacent contigs in the ordering. Short fragments (<5 restriction fragment sites for the simulated human assemblies; <20 sites for the human and mouse de novo assemblies; <40 Kb for the Drosophila de novo assembly) were not reinserted; as a result, many small contigs that were clustered were left out of the final LACHESIS assembly.
Orienting contigs or scaffolds
The orientation of each contig or scaffold (the term ‘contig’ is used in this description of the method to indicate both possibilities) within its ordering was determined by taking into account the exact position of the Hi-C link alignments on each contig (Supplementary Fig. 3). It was assumed that, as demonstrated in previous Hi-C studies17, the likelihood of a Hi-C link connecting two reads at a genomic distance of x is roughly 1/x for x ≥ ~100 Kb. A weighted directed acyclic graph (WDAG) was built representing all possible ways to orient the contigs in the given order. Each edge in the WDAG corresponded to a pair of adjacent contigs in one of their four possible combined orientations, and the edge weight was set to the log-likelihood of observing the set of Hi-C link distances between the two contigs, assuming they were immediately adjacent with the given orientation.
For each contig, a quality score for its orientation was calculated as follows. The log-likelihood of the observed set of Hi-C links between this contig, in its current orientation, and its neighbors, was found. Then the contig was flipped and the log-likelihood was calculated again. The first log-likelihood was guaranteed to be higher because of how the orientations were calculated. The difference between the log-likelihoods was taken as a quality score.
Validation
To determine the true position of the contigs or scaffolds in the shotgun assemblies, we aligned them to the human, mouse or D. melanogaster reference genome using BLASTn32 with parameters ‘-perc_identity 99 -evalue 100 -word_size50’. For each contig, a “truth placement” on reference was derived as follows. First, the chromosome was chosen containing the plurality of aligned sequence from the contig. Second, the single best alignment to this chromosome (measured by E-value) was used to “seed” a chromosomal region. Third, the other alignments to this chromosome were considered by descending E-value, and the region was extended to include as many of them as possible without exceeding the total length of the assembly contig.
Chromosome Fusion Detection in HeLa
A single, complex Hi-C library was constructed for the HeLa S3 cancer cell line (ATCC CCL2.2; grown in DMEM with 10% FBS and 1X Pen. Strep.) according to a published33 protocol. This library was sequenced on two lanes of Illumina HiSeq 2000, followed by read trimming to 50 bp to eliminate ligation-spanning reads that confound alignment. Reads were aligned to the human reference genome using BWA31 with default parameters, followed by removal of PCR duplicates. Reads were then assigned to genomic windows containing approximately one megabase of sequence (mean = 955,176 bp) that were determined by bases of unique mappability to the genome. Links between windows were normalized first to the number of HindIII restriction sites present in the window to account for biases inherent to restriction based library preparation, then to the total count of short pairs within the window (defined as pairs with an insert size ≤1 Kb) to account for the underlying copy number of the window.
Rearrangements were called by first identifying stretches of ≥10 consecutive windows within a row where ≥80% of windows have a link score ≥1 standard deviation above the mean of the entire row. Stretches of windows present in columns were called using the same parameters. Windows present in outlier stretches for both rows and columns were defined as outlier windows. These windows were then clustered with all proximal windows ≤2 windows away and the outlier window count and density within the outer borders of the cluster determined. Outlier spans and clusters are shown in Supplementary Fig. 12.
Software Availability
The LACHESIS software was written in C++ using Boost (http://www.boost.org/) and includes auxiliary scripts written in Perl. It runs in a Unix environment. A distribution of the LACHESIS source code is included as Supplementary Data File 1 (LACHESIS.tar.gz) and a documentation and user’s guide are included as Supplementary Data File 2 (README.txt). Both of these files are also freely available for public download at http://www.gs.washington.edu/~jnburton/LACHESIS/.Updated versions of the source code will also be made available there.
Supplementary Material
Acknowledgments
We thank F. Ay, J. Felsenstein, P.Green, W. Noble, R. Waterston and members of the Shendure Lab for helpful discussions. Some of the sequencing data used in this research was derived from a HeLa cell line. Henrietta Lacks, and the HeLa cell line that was established from her tumor cells without her knowledge or consent in 1951, have made significant contributions to scientific progress and advances in human health. We are grateful to Henrietta Lacks, now deceased, and to her surviving family members for their contributions to biomedical research. The HeLa associated genomic datasets described in this manuscript are available via the database of Genotypes and Phenotypes (dbGaP) as a substudy of the HeLa Cell Genome Sequencing Studies, phs000640. Our work was supported by grant HG006283 from the National Human Genome Research Institute (NHGRI; to J.S.); a graduate research fellowship DGE-0718124 from the National Science Foundation (to A.A. and J.O.K.); and grant T32HG000035 from the NHGRI (to J.N.B.).
Footnotes
Author Contributions
J.N.B., A.A., J.O.K. and J.S. conceived and designed the study. J.N.B. designed and wrote the LACHESIS software. J.N.B. and R.P.P. performed the de novo assemblies. R.Q. conducted the HeLa Hi-C experiments. A.A. analyzed the HeLa Hi-C data. J.N.B., A.A. and J.S. prepared the manuscript. J.S. supervised the study.
Competing Financial Interests
The authors are in the process of filing a provisional patent application on this method. J.S. is a member of the scientific advisory board or serves as a consultant for Adaptive Biotechnologies, Ariosa Diagnostics, Stratos Genomics, GenePeeks, Gen9, Good Start Genetics and Rubicon Genomics.
Accession Codes
The HeLa associated genomic datasets described in this manuscript are available via the database of Genotypes and Phenotypes (dbGaP) as a substudy of the HeLa Cell Genome Sequencing Studies, phs000640.
References
- 1.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:1–62. [Google Scholar]
- 2.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 3.Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26:1–11. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 4.Shendure J, Lieberman-Aiden E. The expanding scope of DNA sequencing. Nature Biotechnology. 2012;30:1084–94. doi: 10.1038/nbt.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Compeau P, Pevzner P, Tesler G. How to apply de Bruijn graphs to genome assembly. Nature Biotechnology. 2011;29:987–91. doi: 10.1038/nbt.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1–6. [Google Scholar]
- 7.Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Research. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:1–43. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 9.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nature Biotechnology. 2011;29:1–6. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang G, et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490:49–54. doi: 10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz DC, et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science. 1993;262:110–4. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, et al. The genome of Prunus mume. Nature Communications. 2012;3:1318. doi: 10.1038/ncomms2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Y, et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus) Nature Biotechnology. 2013;31:135–41. doi: 10.1038/nbt.2478. [DOI] [PubMed] [Google Scholar]
- 14.Lam E, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nature Biotechnology. 2012;30:771–6. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baird NA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genome 10K Community of Scientists. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. The Journal of Heredity. 2009;100:659–74. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan Z, et al. A three-dimensional model of the yeast genome. Nature. 2010;465:363–7. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisen M, Spellman P, Brown P, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon J, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nature Genetics. 2011;43:1059–65. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 22.Mackay T, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–8. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sexton T, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–72. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Landry J, et al. The genomic and transcriptomic landscape of a HeLa cell line. Genes | Genomes | Genetics. 2013:1–27. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adey A, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simonis M, et al. High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nature Methods. 2009;6:837–42. doi: 10.1038/nmeth.1391. [DOI] [PubMed] [Google Scholar]
- 27.Macville M, et al. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Research. 1999;59:1–11. [PubMed] [Google Scholar]
- 28.Moissiard G, et al. MORC family ATPases required for heterochromatin condensation and gene silencing. Science. 2012;336:1448–51. doi: 10.1126/science.1221472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fraley C, Raftery AE. How Many Clusters? Which Clustering Method? Answers Via Model-Based Cluster Analysis. The Computer Journal. 1998;41:578–588. [Google Scholar]
- 30.Jung Y, Park H, Du DZ, Drake B. A decision criterion for the optimal number of clusters in hierarchical clustering. Journal of Global Optimization. 2003:1–21. [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.van Berkum NL, et al. Hi-C: a method to study the three-dimensional architecture of genomes. Journal of Visualized Experiments. 2010:1–7. doi: 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.