

Abstract
It is said that carbon, the most abundant element in organic matter, supplies life’s quantity, whereas nitrogen supplies its quality. It is therefore unsurprising that many natural products that contain basic nitrogens (alkaloids) are coveted for their benefit to human health. However, nitrogen is known to mire many chemical syntheses because of its basicity and susceptibility to oxidation. This challenge may be heightened by the presence of more than one nitrogen atom in a targeted complex alkaloid, but can be met by the selective introduction and removal of functional groups that mitigate basicity, as highlighted herein with the first chemical syntheses of citrinalin B and cyclopiamine B. The chemical connections that have been realized as a result of these syntheses, in addition to the isolation of both 17-hydroxycitrinalin B and citrinalin C through 13C feeding studies, supports the existence of a common bicyclo[2.2.2]diazaoctane containing biogenetic precursor to these compounds as has been proposed previously.
Introduction
The prenylated indole alkaloids are an emerging class of natural products typified by the presence of an indole ring, or derivatives thereof (i.e., spirooxindole or pseudoindoxyl), decorated by one or more prenyl groups or the vestige of a prenyl group. Isolates from this family of natural products include citrinalins A and B (1 and 2, see Figure 1) and cyclopiamines A and B (4 and 6), which are the focus of this Article. The modifications of the indole core in the prenylated indole alkaloid family, which occur by a reaction with dimethylallyl pyrophosphate (DMAPP)1, results in the introduction of a chromene unit as is found in (+) stephacidin A (10; see blue highlighted portion) or a bicyclo[2.2.2]diazaoctane core that is typical of many congeners including 11 and 12 (see red highlighted portion)2.
Figure 1. Selected prenylated indole alkaloids.

The prenylated indole alkaloid family encompasses over 80 natural products some of which contain a bicyclo[2.2.2]diazaoctane core as in 10, 11 and 12. Recently, several members of this family (e.g., 1 and 4) have emerged that do not possess this structural motif. Me, methyl.
Although structurally similar, the prenylated indole alkaloids display a diverse range of bioactivities including antitumor, insecticidal, anthelmintic, calmodulin-inhibition, and antibacterial properties3. The recent discovery of citrinadins A4 and B5 (7 and 8) and PF1270A–C6 (9a–c) has added an unprecedented dimension to the structural motifs afforded by the Penicillum strains as well as raised several questions as to the biogenesis of these structurally related alkaloids. Recently, elegant syntheses of citrinadins A and B have been achieved by the groups of Martin7 and Wood8. Particularly intriguing to us is a subset of this emerging subclass including citrinalins A and B (1 and 2) and cyclopiamines A and B (4 and 6), which like the citrinadins, lack the bicyclo[2.2.2]diazaoctane framework and, remarkably, possess an alkyl nitro group. Cyclopiamines A and B (4 and 6) were discovered first (in 1979) by Steyn and coworkers9 from a toxinogenic strain of P. cyclopium, whereas citrinalins A and B (1 and 2), were discovered by Berlinck and coworkers in 2010 from a strain of P. citrinum10. While natural products that possess aryl nitro groups are known, those that contain aliphatic nitro groups are extremely rare11. As such, the citrinalins and cyclopiamines, which also possess three nitrogen atoms in chemically distinct environments, are rather unusual and therefore attractive targets for synthesis. The synthetic studies described herein have culminated in the total syntheses of ent-citrinalin B (ent-2; ent, enantiomer) and cyclopiamine B (6) and, along with 13C feeding studies that have resulted in the isolation of two new citrinalins, provide support for a proposed biogenesis of the subset of prenylated indole alkaloids that lack the bicyclo[2.2.2]diazaoctane core.
Results and Discussion
Biosynthetic connections
As was proposed by Steyn and coworkers9, a stimulating connection may be drawn between cyclopiamine A and B via the intermediacy of nitronate iminium ion 5 (see Figure 1). The interconversion of 4 and 6 was in fact demonstrated by Steyn et al. by heating either compound in dioxane/water or dimethylformamide (DMF) 9. This led to a proposal that 6, which is the more stable of the two isomers (we have computed 6 to be 9.6 kcal/mol lower in energy as compared to 4 in a DMF solvent model, see the Supporting Information), may in fact be an isolation artifact. Given the likelihood that the citrinadins, citrinalins and cyclopiamines are all oxidative degradation products of a precursor containing a bicyclo[2.2.2]diazaoctane ring, such as marcfortine A (11; in the case of the citrinadins) or stephacidin A (10; in the case of the citrinalins and cyclopiamines), we wondered whether the citrinalins could be transformed to the cyclopiamines. On the basis of this assumption, it is particularly baffling that unlike cyclopiamines A and B, which are related by an aza-Henry (or nitro-Mannich) reaction as shown in Figure 1 (4⇔6, via 5), citrinalin A and the originally proposed structure of citrinalin B (3) would be related not by the formal epimerization of the C22 stereocenter but rather by the nature of the relative configuration of the C14 carbon (highlighted in 2 and 3). On the basis of the connection between cyclopiamine A and B as demonstrated by Steyn, we intuited that the structure of citrinalin B may be better represented by 2. To support this proposal, we undertook a computational simulation of the 1H and 13C NMR spectra that would be expected for the neutral and salt forms of citrinalins A and B (see the Supporting Information for details). As has been convincingly demonstrated by Tantillo and coworkers in numerous cases, this method provides an accurate prediction of the structures of complex natural products12. We found that the computed and empirical data for the trifluoroacetic acid (TFA) salt form of citrinalin A is in good agreement with those reported by Berlinck and workers, who isolated these compounds. The corrected mean absolute deviation (CMAD) in the 1H and 13C NMR resonances is 0.21 and 2.0 ppm, respectively (largest outliers are 1.0 and 5.2 ppm, respectively). On the other hand, the computed data for the TFA salt form of 3 (the originally proposed structure of citrinalin B) differs significantly from that recorded using the naturally occurring material (CMAD = 0.45 and 2.0 ppm; largest outliers = 2.3 and 9.6 ppm for 1H and 13C, respectively). The best match to the reported spectral data was found to correspond to 2 in its neutral form (CMAD = 0.12 and 1.6 ppm; largest outliers = 0.38 and 4.4 ppm for 1H and 13C, respectively), which corroborates a potentially similar biosynthetic connection as has been established for the cyclopiamines (outlined in Figure 1). As a result, we chose to proceed with the hypothesis that 2 most likely represents the correct structure of citrinalin B. Ultimately, a reanalysis of the NMR data of citrinalin B, collected in MeOH-d4 (see Supporting Information for details), corroborates the assignment of 2 as the true structure of citrinalin B.
Synthesis
As outlined in Figure 2, cyclopiamine B (6) can be obtained from the enantiomer of citrinalin B (ent-2) by employing a chromanone rearrangement to forge the tetrahydroquinolone structural moiety found in the cyclopiamines. In turn, ent-2 could be taken back using an ‘indole to spirooxindole’ transform to fused hexacycle 13. Fused indole 13 would arise from tricycle 14, which may be prepared from diene 15, the tert-butyldimethylsilyl (TBS) variant of which was first prepared by Rawal and Jewett13, and tetrahydroindolizinone 16 (unprecedented prior to this report) that would ultimately arise from D-proline (17).
Figure 2. Retrosynthetic analysis plan for cyclopiamine B and citrinalin B.
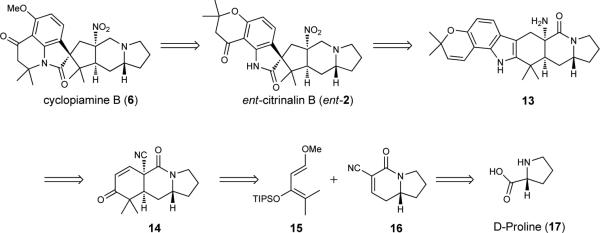
The syntheses of natural products, 2, and 6 is expected to arise from common intermediate 13. TIPS, triisopropylsilyl; Me, methyl.
We initiated our synthetic studies with the tert-butoxycarbonyl (Boc)-protection of D-proline (Figure 3), which was followed by the reduction of the carboxylic acid group and Swern oxidation of the resulting hydroxyl to afford aldehyde 1814. Alkynylative homologation of the aldehyde group of 18 using the Ohira-Bestmann method15, followed by removal of the Boc group and acylation with 2-cyanoacetyl chloride gives alkyne 19, which serves as a substrate for an unprecedented formal cycloisomerization likely proceeding via a metal vinylidene intermediate (generated using the method of Grotjahn)16, anti-Markovnikov hydration, and Knoevenagel condensation to give tetrahydroindolizinone 16. At this stage, a SnCl4-catalyzed Diels-Alder [4+2] reaction17 between 16 and diene 15 and a subsequent basic workup affords an enone (not shown), which is iodinated to yield iodoenone 2018. A mild hydrolysis of the nitrile group of 20 is achieved using Pt-complex 21 under the conditions introduced by Ghaffar and Parkins19 to afford the corresponding carboxamide, which serves as a substrate for a Hofmann rearrangement that is effected with phenyliodosylbistrifluoroacetate (PIFA) to yield carbamate 2220. Suzuki cross-coupling of 22 with known boronic ester 2321 gives adduct 24, which is efficiently converted to fused indole 25 using two sequential reductions – all in accord with the effective protocols established by Herzon and Myers22.
Figure 3. Preparation of fused hexacycle 25.

The use of a Diels-Alder reaction involving a proline-derived indolizidinone dienophile affords a key tricycle that is advanced to hexacycle 25 by Suzuki coupling to boronic ester 23. Reagents and conditions are as follows. a. di-tert-butyl dicarbonate (Boc2O), NaHCO3, H2O/tetrahydrofuran (THF), room temperature (RT = 23 °C). b. BH3•THF, THF, 0 °C to RT. c. (COCl)2, dimethylsulfoxide (DMSO), CH2Cl2, diisopropylethylamine (DIPEA), − 78 °C. d. Dimethyl (diazomethyl)phosphonate, K2CO3, MeOH, 0 °C to RT. e. 4N HCl/Dioxane, 0 °C to RT. f. 2-cyanoacetylchloride, Et3N, CH2Cl2, 0 °C to RT. g. acetonitrile bis[2-diphenylphosphino-6-t-butylpyridine] cyclopentadienylruthenium(II) hexafluorophosphate (8 mol%), acetone/H2O, 70 °C. h. 15, SnCl4, −78 °C to −42 °C. i. I2, 4-dimethylaminopyridine (DMAP), pyridine/CCl4, 60 °C. j. 21 (20 mol%), EtOH/H2O, RT. k. phenyliodosylbistrifluoroacetate (PIFA), MeOH, RT. l. dppfPdCl2 (10 mol%), K3PO4, dimethylformamide (DMF), 40 °C. m. Zn dust, NH4Cl, HCO2NH4, p-TsOH, MeOH, RT; n. NaCNBH3, 1 N aq. HCl, 0 °C to RT. dppf, diphenylphosphinoferrocene; Me, methyl.
The face selective oxygenation of C2/C3-fused indoles is a well-established route to hydroxyindolenines, which serve as precursors to the corresponding spirooxindoles23. As such, we envisioned the oxygenation of indole 25 (Figure 4A) as a path to the spirooxindole structural moiety found in 1, 2, 4 and 6. On the basis of related precedent from Sorensen24, Williams25, and Martin7 for heteroatom-directed oxygenation, we expected the carbamate group of 25 to direct oxygenation to the alpha face and provide 28. Surprisingly, the use of the oft-employed Davis’ oxaziridine26 (29, 3.0 equiv) leads to 26 and trace amounts of both hydroxyindolenine 28 and spirooxindole 27 (spirooxindole 27 arises via the intermediacy of hydroxyindolenine 26). A survey of other oxaziridines including 30 and 31 leads, at best (using 31), to a 1:1 ratio of the desired hydroxyindolenine 28 and both hydroxyindolenine 26 and spirooxindole 27. Because the inherent face selectivity for the oxygenation of 25 is poor, attention was turned to the use of reagent control to achieve the desired diastereoselective oxygenation. In this regard, we were drawn to the peptide-derived catalysts developed by Miller and coworkers27. Following an investigation of a focused library of peptide catalysts developed in the Miller laboratory for oxygenations, 32 (Figure 4B) emerged as the superior catalyst (20 mol% loading) and provided hydroxyindolenine 28 in 83% yield from 25. Hydroxyindolenine 28 rearranges with heating using Sc(OTf)3 over 2 hours to afford pseudoindoxyl 33 (Figure 4C) instead of the desired spirooxindole. The equilibrium between pseudoindoxyls and spirooxindoles is well recognized and has been studied for the migration of C2 alkyl substituents by Borschberg28 and recently for C2 aryl substituents by Movassaghi and coworkers29. However, despite prolonged heating, further rearrangement of pseudoindoxyl 33 to the desired spirooxindole was not observed. It is possible that an intramolecular hydrogen bond stabilizes pseudoindoxyl 33 toward further rearrangement (a bond distance of 2.24Å is computed for the pseudoindoxyl carbonyl group and N-H proton of the carbamate group in 33; see the Supporting Information for more details). A possible stabilizing intramolecular hydrogen bond in 33 is supported by the observation that hydroxyindolenine 26 (prepared by oxidation of 25 with Davis’ oxaziridine) rearranges readily at room temperature in the presence of mild acid to spirooxindole 27; a pseudoindoxyl generated from 26 would lack the analogous stabilizing hydrogen bond. However, the possibility exists that 26 proceeds to an epoxide intermediate (see A in inset in Figure 4C) that rearranges to 27. The recalcitrance of pseudoindoxyl 33 to undergo further rearrangement caused us to consider alternative tactics that would produce the desired spirooxindole structural moiety of the citrinalins and cyclopiamines.
Figure 4.

A. Oxidative rearrangement studies of fused indole 25 with a range of oxaziridines leads predominantly to the undesired, epimeric, hydroxyindolenine (26) and spirooxindole (27). Figure 4B. Use of indole oxidation peptide catalyst 32 to effect oxidation yields the desired hydroxyindolenine (28). Figure 4C. The desired hydroxyindolenine 28 rearranges to an undesired pseudoindoxyl (33) whereas the epimeric hydroxyindolenine (26) affords the corresponding spirooxindole (27). Reagents and conditions are as follows. a. oxaziridine (29, 30, or 31), CH2Cl2, room temperature (RT = 23 °C). b. 32 (20 mol%), 4-dimethylaminopyridine (DMAP), diisopropylcarbodiimide (DIC), H2O2, CHCl3, 4 °C. c. Sc(OTf)3, toluene, 110 °C. d. 23 mM HCl, CH2Cl2, RT. Me, methyl; Bu, butyl; Bn, benzyl; Cbz, carboxybenzyl; Ph, phenyl.
Amino compound 35 (Figure 5) was prepared on the basis of a hypothesis that an amino group or some oxidized derivative thereof (e.g., the corresponding hydroxylamine) could serve as a hydrogen bond donor to effect stereoselective oxygenation of the indole C2–C3 bond and then, by further oxidation to a nitroso or nitro group, remove the presumed intramolecular hydrogen bond that may stabilize the pseudoindoxyl form (as in 33). It appeared reasonable that this sequence would facilitate the eventual conversion of 35 to nitro spirooxindole compound 36. Initial experiments established that epoxidation of the chromene ring was a competing reaction that occurred under various oxygenation conditions. As such, we opted to effect a Wacker oxidation30 of 25 to afford chromanone 34 (Figure 5), which would be advantageous as the chromanone unit is found in the citrinalins and cyclopiamines. Remarkably, treatment of 35 (following removal of the methoxycarbonyl group in 34) with an excess of dimethyldioxirane (DMDO) (formed in situ from acetone and Oxone®) affords spirooxindole 36 as the major product (4:1 d.r., diastereomeric ratio) where the spiro center is as desired and the nitro group has been installed. It is possible that spirooxindole 36 arises from epoxide B (see inset in Figure 5) on the basis of studies by Foote and co-workers for DMDO oxidations of indoles to spirooxindoles31. Therefore, it is possible that the introduction of the chromanone diminishes the participatory role of the indole nitrogen lone pair leading, after rearrangement (see direction of arrow in B), to 3632. With spirooxindole 36 in hand, what remained was a selective removal of the tertiary amide carbonyl group by reduction, which had to be accomplished in the presence of the chromanone and secondary amide carbonyl groups as well as the newly introduced nitro group. After extensive investigation, this task was effectively accomplished using a modification of a procedure developed by Borch33 by treating 36 with a variant of Meerwein's salt (Me3OBF4), which likely leads to a methylated amidinium intermediate that is cleanly reduced with sodium cyanoborohydride to give ent-citrinalin B (ent-2) in 66% yield (79% brsm; based on recovered starting material). The spectroscopic data for the neutral form of ent-2 are fully consistent with the data reported by Berlinck and coworkers for the compound believed to be citrinalin B (corroborating the computational predictions and reanalysis in MeOH-d4), except for the sign of optical rotation, which is opposite. The structure of ent-2 was unambiguously confirmed by X-ray crystallographic analysis of its HCl salt. ent-Citrinalin B is easily converted to cyclopiamine B (6) upon treatment of ent-2 with sodium hydride and heating (to effect the chromanone to tetrahydroquinolone conversion) and subsequent methylation of the resulting phenol. The structure of cyclopiamine B (6) was also unambiguously confirmed by X-ray crystallographic analysis. Thus, the synthesis of ent-2 and its conversion to 6 conclusively supports ent-2 as the true structure of citrinalin B, albeit the enantiomer of the naturally occurring material.
Figure 5. Completion of the syntheses of ent-citrinalin B and cyclopiamine B.

The total synthesis of 2 and 6 required the identification of conditions that accomplished the oxidation of the amino group and spirooxindole formation in one pot as well as unique conditions to selectively reduce the tertiary amide carbonyl group. The rearrangement of ent-citrinalin B (2) to cyclopiamine B (6) was also demonstrated. Reagents and conditions are as follows. a. Pd(OAc)2 (40 mol%), benzoquinone, H2SO4, MeCN/H2O, room temperature (RT = 23 °C). b. Me2S, methanesulfonic acid (MsOH), 40 °C. c. Oxone® (10 equiv), NaHCO3, acetone/H2O, °0 C to RT. d. Me3OBF4, CH2Cl2, 4 Å MS, 45 °C; then e. NaCNBH3, MeOH, 0 °C. f. NaH, dimethylformamide (DMF), 60 °C. g. MeI, K2CO3, acetone, 60 °C. Oxone®, potassium peroxymonosulfate; MS, molecular sieves; Me, methyl; brsm, based on recovered starting material.
Biosynthetic considerations
The total syntheses of ent-citrinalin B (ent-2; 19 steps from D-proline, 5.5% overall yield) and cyclopiamine B (6; 21 steps from D-proline, 4.3% overall yield) not only unambiguously establish the structures of these metabolites, but also provide possible insight into the biogenesis of these natural products (especially as to the possible formation of the cyclopiamines from the citrinalins).
The citrinalins, and in turn the cyclopiamines likely arise from a bicyclo[2.2.2]diazaoctane precursor. However, such a precursor was unknown prior to the findings that are reported herein (vide infra). Consistent with numerous biosynthetic studies of the prenylated indole alkaloids, the structural features of 1, 2, 4 and 6 suggest that tryptophan, proline and two isoprene units are biosynthetic precursors to these compounds. While no biosynthetic studies on 1 and 2 or 4 and 6 or the related citrinadins and PF1270 alkaloids has appeared, a hypothesis suggesting they are derived from bicyclo[2.2.2]diazaoctane precursors that suffer the “loss” of one diketopiperazine carbonyl group has been advanced by Kobayashi and coworkers5. Through the isolation of 17-hydroxycitrinalin B (37, Figure 6A) and more importantly citrinalin C (38) following a series of stable isotope labeling experiments (summarized in Figure 6B; see the Supporting Information for more details), we have now obtained support for the possible biogenesis of the citrinalins and cyclopiamines from a precursor bearing the bicyclo[2.2.2]diazaoctane moiety.
Figure 6.

A. Structures of 17-hydroxycitrinalin B and citrinalin C. Two additional citrinalins, 37 and 38, were isolated upon refractionation and reanalysis of secondary metabolites from P. citrinum F53. Figure 6B. Summary of the 13C labeling studies. 13C incorporation studies of P. citrinum F53 reveal that glucose (pink), anthranilic acid (blue) and ornithine (red) are biosynthetic precursors to the citrinalins.
The nuclear magnetic resonance (NMR) and mass spectroscopy (MS) characterization data for 37 is fully consistent with the assigned structure. Moreover, the assigned relative configuration fully corroborates the revised structure of citrinalin B (2). By analogy to citrinalin B (2), the absolute configuration of 37 was assigned as 1S,14R,16R,17R,22R. 17-Hydroxycitrinalin B (37) was initially isolated from P. citrinum F53 grown in a nitrogen depleted culture medium. Stable isotope feeding studies with [U-13C]anthranilic acid and [1-13C]glucose gave significant 13C labeling (see the Supporting Information). High levels of [U-13C]ornithine were also incorporated into 37, while additional feeding studies with [U-13C]proline gave almost undetectable labeling. Ornithine is a well-known biosynthetic precursor to proline, but to our knowledge has never been reported as a efficient substrate for isotopic labeling of the putative proline-derived atoms in the biosynthesis of prenylated indole alkaloids of fungal origin bearing the bicyclo[2.2.2]diazaoctane moiety. The labeling investigations suggest that, 17-hydroxycitrinalin B (37) might arise from either 3-hydroxyl ornithine, 3-hydroxy proline, or by the late-stage oxygenation of the citrinalin A, B or C skeleton.
Citrinalin C (38), isolated as a minor component from the culture medium of P. citrinum F53, gives NMR and MS data (see Supporting Information, Table S4) that is fully consistent with the relative and absolute configuration illustrated for this natural product. The isolation of 38, along with the congeners lacking the bicyclo[2.2.2]diazaoctane structural moiety from P. citrinum F53, lends support to a bicyclo[2.2.2]diazaoctane-containing precursor, which arises from a committed intramolecular Diels-Alder (IMDA) cycloaddition step as has been studied in detail for other congeners by Williams and Sherman34. In accordance with the proposal of Kobayashi, hydrolysis of the amide bridge of citrinalin C (38), followed by decarboxylation, and amino group oxidation to the nitro group, as proposed in the biosynthesis of the structurally related citrinadin B5, would then yield citrinalin A. These latter steps are the subject of current biosynthesis studies. A question that remained at this stage concerned the biogenesis of citrinalin B. On the basis of the observations of Steyn in the cyclopiamine series (see 4 → 6, Figure 1), we anticipated that citrinalin A (1) might be converted to citrinalin B (2) via a nitronate/iminium intermediate analogous to 5. In the event, heating a solution of a naturally occurring sample of citrinalin A (1) in DMF-d7 at 100 °C for 20 hours leads to a 1:1 ratio of 1 and 2 (with complete conversion to citrinalin B (2) after 60 hours, see the Supporting Information, Figure S22), confirming the connection of these metabolites presumably by the same aza-Henry/nitro-Mannich epimerization sequence established for the cyclopiamines by Steyn and coworkers. However, we have observed some key differences. First, the epimerization in the citrinalin series occurs at a qualitatively slower rate (likely due to a non-productive proton transfer from the vinylogous imide N–H to the tertiary amine) and higher temperature. In addition, we have not been able to achieve any observable conversion of citrinalin B to citrinalin A even at elevated temperatures (165 °C) over prolonged periods (24 h). Our current efforts are focused on gaining a deeper understanding of these differences and exploring the biosynthetic conversion of citrinalin C to citrinalin A.
Conclusion
We have achieved the first total syntheses of the prenylated indole alkaloids ent-citrinalin B and cyclopiamine B. Our results secure unambiguously the identity of citrinalin B both through synthesis, a reanalysis of the naturally isolated material, and by an X-ray crystallographic study. Our studies on the isolation of metabolites from P. citrinum support a bicyclo[2.2.2]diazaoctane-containing metabolite such as citrinalin C (38) as an intermediate in the biogenesis of citrinalins A (1) and B (2) (Fig. 7). The extension of the synthetic methods reported herein to the syntheses of other prenylated indole alkaloids are ongoing and will be reported in due course.
Figure 7. Biosynthetic proposal for citrinalins.
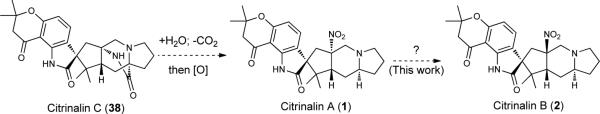
Consistent with previous reports on the bicyclo[2.2.2]diazaoctane congeners, the citrinalins likely arise through an intramolecular Diels-Alder reaction to form citrinalin C (38), which is followed by a decarboxylation event and amine group oxidation to the nitro group.
METHODS SUMMARY
All reactions were performed under a nitrogen atmosphere using dry solvents under anhydrous conditions, unless otherwise noted. Dry tetrahydrofuran, toluene, methanol, triethylamine, benzene and diethyl ether were obtained by passing the commercially available, oxygen free, solvents through activated alumina columns from GlassContour®. Dichloromethane was distilled over calcium hydride under a nitrogen atmosphere. Yields refer to materials purified using silica gel column chromatography. Full experimental details and characterization data for all new compounds (1H NMR, 13C NMR, mass spectrometry, infrared, Rf value), including 14–36, 2 and 6, appears in the Supporting Information. Crystallographic data were collected on a MicroSTAR-H APEX II (ChexStar: RUA # 1091) instrument and the Bruker SAINT and SADABS software programs were used for integrating and scaling the data, respectively. Computational analyses were conducted following conformational searches using the MMFF94 force field (Spartan’10). DFT calculations were performed with GAUSSIAN09 (B3LYP/6-31+G(d,p) level of theory). Full details are included in the Supporting Information.
Supplementary Material
Acknowledgements
R.S. and P.G.R thank the USA National Institutes of Health (NIGMS RO1 086374) for financial support. R.S. is a Camille Dreyfus Teacher Scholar. E.V.M.M. acknowledges the National Science Foundation (USA) for a graduate fellowship (GRFP). S.R., E.F.P., and R.G.S.B are grateful to the National Counsel of Technological and Scientific Development (CNPq, Brazil, Grant 470643/2010-2) and the São Paulo Research Foundation (FAPESP, Brazil, Grant 2012/50026-3) for funding. D.E.W. and R.J.A. thank NSEPC for funding. M.W.L. and D.J.T. acknowledge support from the US National Science Foundation (CHE-0957416 and supercomputing resources through a grant from the XSEDE program: CHE-030089). S.J.M. is grateful for the NIH for support (GM096403). We thank Dr. Antonio DiPasquale (UC Berkeley) for solving the crystal structures of ent-2•HCl, 6, 27 and 36 (supported by NIH Shared Instrumentation Grant S10 RR027172) and acknowledge the CYLView program (developed by Prof. Claude Y. Legault, Dept. of Chemistry, Université Sherbrooke) for X-ray depictions. We would like to thank Dr. Terry Lebold (currently at Jannsen Pharmaceuticals), Professors Robert M. Williams (Colorado State University) and David Sherman (University of Michigan) for helpful discussions.
Footnotes
Author Contributions R.S. conceived and directed the synthetic aspects of the research, as well as composed the majority of the manuscript (with input from all authors) except the section on biosynthesis, which was contributed by S.R., E.F.P., D.E.W., R.J.A. and R.G.S.B. The synthetic plan was designed by R.S. with input from E.V.M.M. and P.G.R. who also executed the plan under the supervision of R.S. Oxidation catalyst 32 was provided by D.K.R. and S.J.M., who along with P.G.R, E.V.M.M. and R.S. designed the oxidation studies of 25, which were executed by P.G.R. The computational NMR predictions for 1, 2 and 3 were designed and executed by M.W.L. and D.J.T. with input from P.G.R., E.V.M.M. and R.S. Biosynthetic studies were designed and conducted by S.R., E.F.P. and R.G.S.B. who also isolated and characterized 3, 37, and 38. D.E.W. and R.J.A. provided facilities and contributed to the purification, data analysis and structural analysis of 3, 37, and 38.
Author Information P. citrinum F53 is deposited at the Brazilian Collection of Environmental and Industrial Microorganisms (CBMAI) under the accession code CBMAI 1186. CCDC 984477, 984478, 984480 and 984479 contains the supplementary crystallographic data for crystal structures ent-2•HCl, 6, 27 and 36, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.naturecom/nature.
References
- 1.Stocking EM, Sanz-Cervera JF, Williams RM. Reverse versus normal prenyl transferases in paraherquamide biosynthesis exhibit distinct facial selectivities. Angew Chem. Int. Ed. 1999;38:786–789. doi: 10.1002/(SICI)1521-3773(19990315)38:6<786::AID-ANIE786>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 2.Finefield JM, Frisvad JC, Sherman D,H, Williams RM. Fungal origins of the bicyclo[2.2.2]diazaoctane ring system of prenylated indole alkaloids. J. Nat. Prod. 2012;75:812–833. doi: 10.1021/np200954v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KA, Williams RM. Synthetic approaches to the bicyclo[2.2.2]diazaoctane ring system common to the paraherquamides, stephacidins and related prenylated indole alkaloids. Chem. Soc. Rev. 2009;38:3160–3174. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsuda M, et al. Citrinadin A, a novel pentacyclic alkaloid from marine-derived fungus Penicillium citrinum. Org. Lett. 2004;6:3087–3089. doi: 10.1021/ol048900y. [DOI] [PubMed] [Google Scholar]
- 5.Mugishima T, et al. Absolute stereochemistry of citrinadins A and B from marine-derived fungus. J. Org. Chem. 2005;70:9430–9435. doi: 10.1021/jo051499o. [DOI] [PubMed] [Google Scholar]
- 6.Kushida N, et al. PF1270A, B and C, novel histamine H3 receptor ligands produced by penicillium waksmanii PF1270. J. Antibiot. 2007;60:667–673. doi: 10.1038/ja.2007.85. [DOI] [PubMed] [Google Scholar]
- 7.Bian Z, Marvin CC, Martin SF. Enantioselective total synthesis of (−)-citrinadin A and revision of its stereochemical structure. J. Am. Chem. Soc. 2013;135:10886–10889. doi: 10.1021/ja405547f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kong K, et al. An enantioselective total synthesis and stereochemical revision of (+)-citrinadin B. J. Am. Chem. Soc. 2013;135:10890–10893. doi: 10.1021/ja405548b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bond RF, Boeyens JCA, Holzapfel CW, Steyn PS. Cyclopiamines A and B, novel oxindole metabolites of penicillium cyclopium westling. J. Chem. Soc., Perkin Trans. 1979;1:1751–1761. [Google Scholar]
- 10.Pimenta EF, et al. Use of experimental design for the optimization of the production of new secondary metabolites by two penicillium species. J. Nat. Prod. 2010;73:1821–1832. doi: 10.1021/np100470h. [DOI] [PubMed] [Google Scholar]
- 11.Parry R, Nishino S, Spain J. Naturally-occurring nitro compounds. Nat. Prod. Rep. 2011;28:152–167. doi: 10.1039/c0np00024h. [DOI] [PubMed] [Google Scholar]
- 12.Lodewyk MW, Siebert MR, Tantillo DJ. Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 13.Jewett JC, Rawal VH. Total synthesis of pederin. Angew. Chem. Int. Ed. 2007;46:6502–6504. doi: 10.1002/anie.200701677. [DOI] [PubMed] [Google Scholar]
- 14.Omura K, Swern D. Oxidation of alcohols by “activated” dimethyl sulfoxide. A preparative, steric and mechanistic study. Tetrahedron. 1978;34:1651–1660. [Google Scholar]
- 15.Ohira S. Methanolysis of dimethyl (1-diazo-2-oxopropyl) phosphonate: generation of dimethyl (diazomethyl) phosphonate and reaction with carbonyl compounds. Synth. Commun. 1989;19:561–564. [Google Scholar]
- 16.Grotjahn DB, Lev DA. A general bifunctional catalyst for the anti-markovnikov hydration of terminal alkynes to aldehydes gives enzyme-like rate and selectivity enhancements. J. Am. Chem. Soc. 2004;126:12232–12233. doi: 10.1021/ja046360u. [DOI] [PubMed] [Google Scholar]
- 17.Kishi Y, et al. Synthetic approach towards tetrodotoxin. I. Diels-Alder reaction of alpha-oximinoethylbenzoquinones with butadiene. Tetrahedron Lett. 1970;11:5127–5128. doi: 10.1016/s0040-4039(00)96956-9. [DOI] [PubMed] [Google Scholar]
- 18.Johnson CR, et al. Direct alpha-iodination of cycloalkenones. Tetrahedron Lett. 1992;33:917–918. [Google Scholar]
- 19.Ghaffar T, Parkins AW. A new homogeneous platinum containing catalyst for the hydrolysis of nitriles. Tetrahedron. Lett. 1995;36:8657–8660. [Google Scholar]
- 20.Moriarty RM, Chany CJ, II, Vaid RK, Prakash O, Tuladhar SM. Preparation of methyl carbamates from primary alkyl- and arylcarboxamides using hypervalent iodine. J. Org. Chem. 1993;58:2478–2482. [Google Scholar]
- 21.Herzon SB, Myers AG. Enantioselective synthesis of stephacidin B. J. Am. Chem. Soc. 2005;127:5342–5344. doi: 10.1021/ja0510616. [DOI] [PubMed] [Google Scholar]
- 22.Myers AG, Herzon SB. Identification of a novel michael acceptor group for the reversible addition of oxygen- and sulfur-based nucleophiles. Synthesis and reactivity of the 3-alkylidene-3H-indole 1-oxide function of avrainvillamide. J. Am. Chem. Soc. 2003;125:12080–12081. doi: 10.1021/ja0372006. [DOI] [PubMed] [Google Scholar]
- 23.Marti C, Carreira E. Construction of spiro[pyrrolidine-3,3′-oxindoles] − recent applications to the synthesis of oxindole alkaloids. Eur. J. Org. Chem. 2003:2209–2219. [Google Scholar]
- 24.Guerrero CA, Sorensen EJ. Concise, stereocontrolled synthesis of citrinadin B core architecture. Org. Lett. 2011;13:5164–5167. doi: 10.1021/ol2020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grubbs AW, Artman GD, III, Tsukamoto S, Williams RM. A concise total synthesis of the notoamides C and D. Angew. Chem. Int. Ed. 2007;46:2257–2261. doi: 10.1002/anie.200604377. [DOI] [PubMed] [Google Scholar]
- 26.Davis FA, Stringer OD. Chemistry of oxaziridines. 2. Improved synthesis of 2-sulfonyloxaziridines. J. Org. Chem. 1982;47:1774–1775. [Google Scholar]
- 27.Kolundzic F, et al. Chemoselective and enantioselective oxidation of indoles employing aspartyl peptide catalysts. J. Am. Chem. Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Güller R, Borschberg H-J. A stereoselective transformation of pseudoindoxyls into oxindoles in a single operation. Tetrahedron Lett. 1994;35:865–868. [Google Scholar]
- 29.Movassaghi M, Schmidt MA, Ashenhurst JA. Stereoselective oxidative rearrangements of 2-aryl tryptamine derivatives. Org. Lett. 2008;10:4009–4012. doi: 10.1021/ol8015176. [DOI] [PubMed] [Google Scholar]
- 30.Miller DG, Wayner DDM. Improved method for the wacker oxidation of cyclic and internal olefins. J. Org. Chem. 1990;55:2924–2927. [Google Scholar]
- 31.Zhang X, Foote CS. Dimethyldioxirane oxidation of indole derivatives. Formation of novel indole-2,3-epoxides and a versatile synthetic route to indolinones and indolines. J. Am Chem. Soc. 1993;115:8867–8868. [Google Scholar]
- 32.We thank a reviewer for this alternative explanation.
- 33.Borch RF. A new method for the reduction of secondary and tertiary amides. Tetrahedron Lett. 1968;9:61–65. [Google Scholar]
- 34.Ding Y, et al. Genome-based characterization of two prenylation steps in the assembly of the stephacidin and notoamide anticancer agents in a marine-derived aspergillus sp. J. Am. Chem. Soc. 2010;132:12733–12740. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.