

Abstract
Insulin is secreted into blood vessels from β cells of pancreatic islets in response to high blood glucose levels. Insulin stimulates an array of physiological responses in target tissues, including liver, skeletal muscle, and adipose tissue, thereby reducing the blood glucose level. Insulin-dependent glucose uptake in skeletal muscle and adipose tissue is primarily mediated by the redistribution of the glucose transporter type 4 from intracellular storage sites to the plasma membrane. Evidence for the participation of the Rho family GTPase Rac1 in glucose uptake signaling in skeletal muscle has emerged from studies using cell cultures and genetically engineered mice. Herein, recent progress in understanding the function and regulation of Rac1, especially the cross-talk with the protein kinase Akt2, is highlighted. In addition, the role for another Rho family member TC10 and its regulatory mechanism in adipocyte insulin signaling are described.
Keywords: GLUT4, Rac1, TC10, adipose tissue, exocyst complex, exocytosis, glucose uptake, insulin, skeletal muscle
Introduction to Insulin Action
Insulin is a key hormone in the regulation of metabolism, organizing the use of energy sources, such as carbohydrate and fat, for either oxidation or storage. Insulin is secreted predominantly from β cells in pancreatic islets. Secretion of insulin from β cells is triggered by increased blood glucose levels, and also regulated by hormones and neurotransmitters.1-3 Glucose enters β cells via glucose transporters, and ATP is generated by glycolysis. The rise in the ATP/ADP ratio causes the ATP-sensitive potassium channel to close up, leading to depolarization of the cell. Depolarization activates voltage-gated calcium channels, which allow calcium ions to flow inward. The increase in the level of intracellular calcium ions triggers the release of insulin from its storage granules into blood vessels by exocytosis. Incretins such as glucose-dependent insulinotropic peptide (also known as gastric inhibitory polypeptide) and glucagon-like peptide-1 increase the intracellular cAMP level, and ultimately potentiate insulin secretion.
Insulin targets various tissues, including the liver, skeletal muscle, and fat tissue. Insulin generally acts as an anabolic hormone; it promotes glycogen synthesis and inhibits glycogenolysis and gluconeogenesis.4 Insulin also stimulates lipogenesis and protein synthesis. These diverse effects are achieved through the activation or inactivation of metabolic enzymes. For instance, phosphorylation-dependent inactivation of glycogen synthase kinase 3 occurs following insulin stimulation, leading to the accumulation of the unphosphorylated active form of glycogen synthase. On the other hand, insulin inactivates glycogen phosphorylase, the rate-limiting enzyme in glycogenolysis. Inactivation of glycogen phosphorylase allows phosphoprotein phosphatase 1 to act on glycogen synthase, leading to its dephosphorylation and activation further. In skeletal muscle and fat tissue, insulin also stimulates the uptake of glucose from the blood, contributing to the decline of the blood glucose level.
GLUT4 in Insulin-Stimulated Glucose Uptake
Insulin-regulated glucose uptake in skeletal muscle and adipose tissue is dependent on the redistribution of glucose transporter type 4 (GLUT4) from intracellular storage sites to the plasma membrane.5-7 Once embedded within the plasma membrane, GLUT4 permits circulating glucose to move into the cell down its concentration gradient. This glucose transport mechanism across the plasma membrane is ATP-independent, and is called facilitated diffusion. GLUT4 contains 12 transmembrane domains with both N- and C-termini located inside the cell. Unique amino acid sequence motifs in N- and C-terminal cytoplasmic domains direct endocytosis and exocytosis of this protein in continuously recycling membrane trafficking systems.
GLUT4 is localized in various membrane structures, recycling dynamically between the plasma membrane and intracellular compartments (Fig. 1).5-7 GLUT4 is sequestered into a specialized intracellular compartment called GLUT4 storage vesicles (GSVs) in the absence of insulin. GSVs are segregated from the endosomal recycling compartment (ERC) and biosynthetic compartments including the trans-Golgi network (TGN). Insulin accelerates an exocytic pathway that takes GLUT4 directly to the plasma membrane from GSVs, thereby accumulating GLUT4 on the cell surface. Multiple steps of exocytosis, including trafficking, tethering, docking, and fusion are enhanced following insulin stimulation. In addition, insulin attenuates endocytic processes of cell surface GLUT4 molecules in adipocytes. The interchange between membranes from ERC, TGN, and GSV is also subjected to insulin regulation. Recently, insulin has been shown to regulate GLUT4 vesicle formation, the earliest step of GLUT4 recycling, as well.8 Such pleiotropic and intricate effects of insulin contribute to a net accumulation of GLUT4 in the plasma membrane.
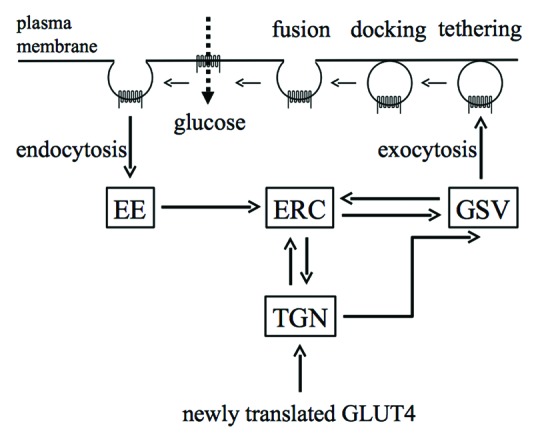
Figure 1. Intracellular trafficking of GLUT4. Intracellular compartments in which GLUT4 is localized are shown as squares. Intracellular translocation of GLUT4 between compartments is shown by thick arrows. In unstimulated cells, GLUT4 is sequestered in specialized compartments termed GSVs. GLUT4 is redistributed and becomes accumulated in the plasma membrane via various pathways when cells are stimulated with insulin. EE, early endosome; ERC, endosomal recycling compartment; GSV, GLUT4 storage vesicle; TGN, trans-Golgi network.
Various studies using mouse models with genetic manipulations have shown that GLUT4 plays an important role in whole-body glucose homeostasis.6 Skeletal muscle- or adipose tissue-specific deficiency of GLUT4 in mice caused impaired responsiveness to insulin and diabetic phenotypes.9,10 Conversely, transgenic mice with high level GLUT4 expression in skeletal muscle or adipose tissue were highly sensitive to insulin and showed enhanced glucose disposal.11-13 Hence, it is important to understand how insulin regulates the overall activity of GLUT4, thereby enhancing glucose uptake in skeletal muscle and adipose tissue.
The Involvement of the Serine/Threonine Kinase Akt2 in Insulin-Dependent Glucose Uptake
Circulating insulin binds to its receptor that is highly expressed on the surface of target cells. Following the binding of insulin, the insulin receptor becomes activated, triggering downstream signal transduction by phosphorylating target proteins, including the adaptor protein IRS1 (Fig. 2). The regulatory subunit of class I phosphoinositide 3-kinase (PI3K) recognizes specific phosphotyrosines in IRS1, forming a signaling complex consisting of the insulin receptor, IRS1, PI3K, and other signaling molecules such as Grb2 and SHP2. PI3K catalyzes phosphorylation in position 3 of the inositol ring of phosphoinositides, producing lipid second messengers, such as phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3).14,15 PtdIns(3,4,5)P3 in turn recruits downstream protein kinases PDK1 and Akt2 to the plasma membrane, in which Akt2 is activated through phosphorylation of specific residues by PDK1 and the mammalian target of rapamycin complex 2.16
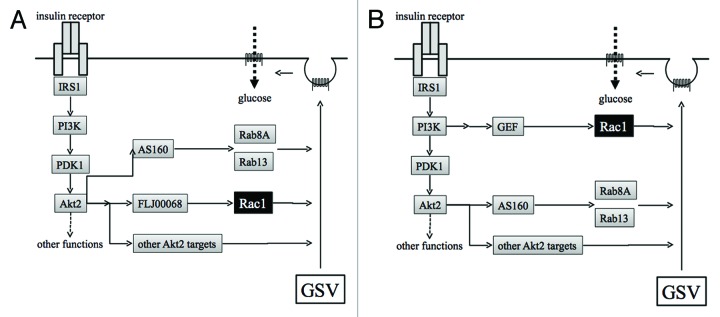
Figure 2. Proposed models for insulin-dependent signal transduction that stimulates exocytosis of GLUT4 from GSVs to the plasma membrane in skeletal muscle. (A) The insulin receptor, when occupied by insulin, stimulates a downstream kinase cascade composed of PI3K, PDK1, and Akt2. Akt2 acts as a master switch that regulates various signaling pathways necessary for GLUT4 translocation. Rac1 is also regulated downstream of Akt2. (B) Akt2 and Rac1 act independently in pathways bifurcated downstream of PI3K. GSV, GLUT4 storage vesicle; PI3K, phosphoinositide 3-kinase.
Akt2 plays a central role in the maintenance of insulin-dependent glucose homeostasis. Akt2 knockout mice exhibited hyperglycemia and glucose intolerance, and total body insulin-dependent glucose disposal was reduced in these mice.17,18 In fact, glucose uptake in extensor digitorum longus and soleus muscles isolated from akt2 knockout mice on exposure to a low concentration of insulin was impaired.17,18 Hence, it is likely that redistribution of GLUT4 from intracellular storage compartments to the cell surface highly depends on Akt2. However, the mechanism whereby Akt2 directs GLUT4 translocation in response to insulin remains incompletely understood.
Role of Akt2 Targets in Insulin-Stimulated GLUT4 Translocation
Given that Akt2 is critically involved in GLUT4 translocation in skeletal muscle and adipose tissue, it is important to identify its substrates that are responsible for the regulation of glucose uptake. A well-characterized Akt substrate that regulates GLUT4 translocation is a TBC domain-containing Rab-GTPase-activating protein (GAP) termed AS160 (also termed TBC1D4).19 In the human genome, over 60 Rab family small GTPases have been identified, being implicated in the regulation of consecutive steps in intracellular membrane trafficking, such as vesicle budding, delivery, and fusion with the target (acceptor) membrane.20,21 Rab8A, Rab10, and Rab13 are involved in the regulation of insulin-dependent GLUT4 trafficking in skeletal muscle (Rab8A and Rab13) and adipose tissue (Rab10), respectively, and are characterized as substrates of AS160.22,23 Phosphorylation of AS160 by Akt2 following insulin stimulation caused the dissociation of AS160 from GLUT4 vesicles and attenuated Rab-GAP activity.24-26 Subsequently, the activated GTP-bound form of Rab proteins may increase in GLUT4-containing vesicles. Direct binding of 14-3-3 proteins to AS160 occurred in a phosphorylation-dependent manner, which may negatively affect Rab-GAP activity of AS160.27 TBC1D1, a close relative of AS160, is also a Rab-GAP that is phosphorylated by Akt2. TBC1D1 has also been shown to regulate GLUT4 translocation in response to insulin stimulation.28
The C2 domain-containing phosphoprotein CDP138 is another substrate of Akt2.29 This protein was indeed required for insulin-stimulated GLUT4 translocation to the plasma membrane, particularly the fusion step, in adipocytes.29 A guanine nucleotide exchange factor (GEF) for ARF6, termed Grp1, is also phosphorylated by Akt2, thereby regulating insulin-dependent GLUT4 translocation.8
Although the role of several Akt2 targets has been clarified as described above, precise mechanisms whereby Akt2 directs glucose uptake in response to insulin stimulation remain incompletely understood. Still unidentified signaling molecules presumably exert their critical function downstream of Akt2.
Identification of the Rho Family GTPase Rac1 as a Critical Regulator of Insulin-Induced Glucose Transport in Skeletal Muscle Cells
The role of Rho family proteins in insulin-stimulated glucose uptake seems to be cell-type specific. Although, in adipocytes, TC10 has been identified as a regulator of glucose uptake in a PI3K-independent pathway (see below), this protein does not participate in insulin signaling in muscle cells.30 Instead of TC10, another Rho family GTPase Rac1 has been implicated in insulin-stimulated, PI3K-dependent glucose uptake signaling in muscle cells.30-34 On the other hand, it was reported that Rac1 is not involved in insulin signaling in adipocytes.35 However, the role of Rac1 in adipocytes remains controversial considering a recent finding that a GEF for Rac1 termed P-Rex1 participates in glucose uptake in adipocytes (see below).36
Rac1 was in fact activated following insulin stimulation in muscle cell culture.30,34 Furthermore, insulin-dependent GLUT4 translocation to the plasma membrane was almost completely blocked by siRNA-based knockdown of Rac1 or overexpression of a dominant inhibitory Rac1 mutant.30,32-34 On the other hand, ectopic expression of an activated mutant of Rac1 induced GLUT4 translocation.34,37 These results altogether lead to the conclusion that Rac1 is crucial for insulin-dependent GLUT4 translocation in muscle cells.
Evidence that supports an important role of Rac1 has been provided not only by cell culture studies, but also by studies using mouse and human mature skeletal muscle. The activation of Rac1 was actually observed in mouse skeletal muscle following intravenous injection of insulin38 and in isolated skeletal muscle after incubation with insulin.39 In addition, a hyperinsulinemic euglycemic clamp induced Rac1 activation in human vastus lateralis muscle.39 An exofacial epitope-tagged GLUT4 reporter40 was ectopically expressed in mouse skeletal muscle, and its redistribution following intravenous injection of insulin was monitored by confocal immunofluorescent microscopy.38 The results clearly demonstrated that insulin-induced GLUT4 translocation to the sarcolemma was largely suppressed in gastrocnemius muscle of muscle-specific rac1 knockout mice. Immunogold electron microscopic analysis of endogenous GLUT4 provided similar results.38 Furthermore, glucose uptake was measured in soleus and extensor digitorum longus muscle isolated from wild-type and muscle-specific rac1 knockout mice.39 As expected, insulin-stimulated glucose uptake was reduced in rac1 knockout mouse muscle.39 Pharmacological inhibition of Rac1 in isolated skeletal muscle also decreased insulin-dependent glucose uptake.39 Moreover, a constitutively activated Rac1 mutant, when expressed in skeletal muscle, induced GLUT4 translocation as observed in myoblast cell culture.38 Taken together, these results highlight a pivotal role of Rac1 in insulin-stimulated glucose uptake in mature skeletal muscle.
Given that Rac1 is critical for insulin-dependent glucose uptake, it is reasonable to think that whole body glucose metabolism may be altered in rac1 knockout mice. Actually, muscle-specific rac1 knockout mice displayed decreased glucose tolerance, and plasma insulin concentrations following glucose injection were higher in muscle-specific rac1 knockout mice.39 Moreover, the activation of the serine/threonine kinase PAK, an effector of Rac1, was impaired in insulin resistant human and mouse skeletal muscle.39 These findings provide in vivo evidence that Rac1 is a key regulator of insulin-dependent glucose uptake in skeletal muscle.
In contrast to skeletal muscle, both constitutively activated and dominant-negative mutants of Rac1 were reported not to affect GLUT4 translocation, and therefore, Rac1 has not been implicated in GLUT4 translocation in adipocytes.35,41 However, a more recent study suggested a role of Rac1 also in adipocytes. A Rac1-specific GEF termed P-Rex1 enhanced insulin-induced GLUT4 translocation and membrane ruffle formation downstream of PI3K in adipocytes.36 In fact, the action of P-Rex1 was mediated by Rac1. In addition, knockdown of P-Rex1 inhibited insulin-induced glucose uptake. Hence, it is likely that P-Rex1 and its substrate Rac1 are involved in insulin-dependent actin cytoskeletal remodeling and GLUT4 translocation.36 Of particular note, the P-Rex1 gene has been mapped to a type 2 diabetes susceptibility locus, reinforcing the possibility that P-Rex1 is involved in insulin signaling.42
Regulatory Mechanisms Underlying Rac1 Activation in Insulin Signaling
Studies using a specific inhibitor indicate that PI3K mediates the activation of Rac1 in response to insulin stimulation.30,34 However, precise mechanisms by which PI3K induces Rac1 activation remain obscure. Recently, possible involvement of Akt2 in insulin-stimulated activation of Rac1 has been proposed based on the observation that insulin-dependent activation of Rac1 was sensitive to downregulation of Akt2 by a chemical inhibitor or siRNAs (Fig. 2A).43 Rac1 activation by constitutively activated PI3K was also inhibited by knockdown of Akt2.44 Moreover, GLUT4 translocation induced by a constitutively activated mutant of Akt2 was totally sensitive to knockdown of Rac1 in both cultured myoblasts and mouse gastrocnemius muscle.44 Taken together, these results are supportive of a critical role for Akt2 in insulin-dependent regulation of Rac1. The observation that phosphorylation of the activation segment of Akt in gastrocnemius muscle after intravenous administration of insulin was totally unaffected by muscle-specific rac1 knockout indicates that Rac1 is not involved in Akt activation, and is consistent with the notion that Rac1 acts downstream of Akt2.38
Another model in which Rac1 and Akt2 act independently in pathways bifurcated downstream of PI3K has also been proposed (Fig. 2B).31,37,45 In this model, Rac1 is primarily involved in the regulation of cytoskeletal reorganization, whereas Akt2 promotes GLUT4 vesicle trafficking through phosphorylation of various target molecules such as AS160. Both Akt2 and Rac1 are thought to be activated by the PI3K product PtdIns(3,4,5)P3 although the detailed mechanism for PtdIns(3,4,5)P3-dependent Rac1 activation remains unknown.31,37,45 Insulin-independent activation of Akt2 was triggered by Rac1 superactivation through the production of PtdIns(3,4,5)P3, which may account for the observation that constitutively activated Rac1, when ectopically expressed alone, can stimulate glucose uptake.37 The bifurcated Rac1 and Akt2 pathways are also supported by mouse studies, in which it was shown that Rac1 and Akt2 inhibitors additively lowered insulin-dependent glucose uptake in soleus and extensor digitorum longus muscles.45
FLJ00068 (also termed PLEKHG4 and puratrophin-1) is a GEF that has been reported to be involved in insulin-dependent Rac1 activation in cultured muscle cells.34 Overexpression of this GEF, in fact, enhanced insulin-stimulated Rac1 activation and GLUT4 translocation. A constitutively activated mutant of FLJ00068, when ectopically expressed, induced Rac1 activation and GLUT4 translocation. Moreover, insulin-dependent GLUT4 translocation was significantly suppressed when the expression of FLJ00068 was reduced by specific siRNAs. Thus, FLJ00068 is a plausible candidate for the regulator that acts immediate upstream of Rac1 in an insulin signaling cascade in skeletal muscle. Recently, we confirmed that a constitutively activated FLJ00068 mutant can induce GLUT4 translocation to the sarcolemma in wild-type, but not muscle-specific rac1 knockout, mouse skeletal muscle (Takenaka et al., unpublished results). In addition to FLJ00068, the Rac1 GEF Tiam1 was reported to induce GLUT4 translocation when overexpressed in muscle cells.37
FLJ00068 harbors DH/PH domains in its C-terminal portion, and isolated DH/PH domains exhibit constitutive GEF activity.34 Therefore, the N-terminal region may have an inhibitory role in the regulation of GEF activity. It remains unclear how Akt2 conveys the insulin signal to FLJ00068, inducing the conformational change of FLJ00068 for its activation. Direct phosphorylation of FLJ00068 by Akt2 seems unlikely because FLJ00068 does not have any consensus sequence for Akt substrates. A still unidentified Akt2 substrate may modulate FLJ00068 upon insulin stimulation.
Regulation of Cytoskeletal Rearrangements by Rac1
Cytoskeletal rearrangements are critical events for insulin-stimulated glucose uptake in adipocytes and skeletal muscle.31,46-48 In fact, reorganized microtubules and actin polymers serve as filaments along which GLUT4 vesicles are conveyed by motor proteins kinesins (KIF3 and KIF5b) and myosins (Myo1c, Myo5a, and Myo5b), respectively.49-54 Reorganization of microtubules and actin filaments is often regulated by Rho family GTPases in many types of cells,55,56 and therefore it is likely that one important function of Rac1 in GLUT4 translocation in muscle cells is the regulation of the cytoskeleton (Fig. 3).31 IQGAP1 and PAK1 are Rac1 targets that are reported to regulate reorganization of microtubules, whereas other targets of Rac1, Sra-1, and IRSp53, in addition to PAK1, participate in the regulation of actin cytoskeletal reorganization (Fig. 3).55,56 Thus, these molecules may be involved in cytoskeletal rearrangements and GLUT4 vesicle movement also in skeletal muscle.
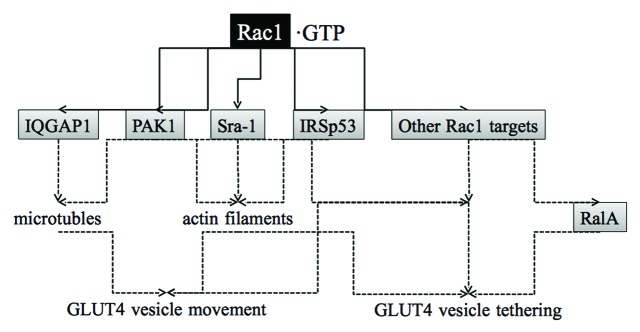
Figure 3. Signaling proteins that may act downstream of Rac1 to stimulate GLUT4 translocation to the plasma membrane in skeletal muscle. IQGAP1, PAK1, Sra-1, and IRSp53 are Rac1 targets that regulate cytoskeletal remodeling. PAK1 may have other roles for GLUT4 vesicle movement and tethering. Still unknown Rac1 targets may also be implicated in the regulation of GLUT4 translocation. RalA participates in the regulation of GLUT4 vesicle tethering downstream of Rac1, but a Rac1 target that links Rac1 and RalA remains unidentified.
In muscle cells, GLUT4 vesicle trafficking along cortical actin filaments is directed by the myosin motor Myo5b.51,57 Furthermore, cortical actin remodeling is required for the retention of GLUT4 vesicles beneath the plasma membrane and subsequent fusion events.58 As predicted, Rac1 was shown to be involved in these processes.58 The motor protein Myo1c may serve as a link between the GLUT4 vesicle and actin filaments in the tethering step.49,57,59 The idea that Rac1 regulates glucose uptake by inducing actin cytoskeletal rearrangements is also supported by a pharmacological study in mature skeletal muscle.45
Signaling Molecules that Act Downstream of Rac1 for the Induction of Glucose Uptake
Given that ectopic expression of constitutively activated Rac1 induces GLUT4 translocation in muscle cultured cells and mouse skeletal muscle,34,37,38 Rac1 presumably participates not only in the regulation of cytoskeletal rearrangements, but also in other processes necessary for GLUT4 translocation. A key downstream player that is responsible for Rac1 function other than the regulation of cytoskeletal remodeling may be the Ras family small GTPase RalA (Fig. 3).60 The involvement of RalA in GLUT4 vesicle transport was first proposed in adipocytes.59,61 RalA is localized in GLUT4 vesicles and activated following insulin stimulation (Fig. 4).59 Activated RalA interacts with Sec5 and Exo84 subunits of the exocyst complex, thereby tethering the GLUT4 vesicle to the plasma membrane.59,62,63 RalA also binds to the Myo1c motor protein, which links the GLUT4 vesicle to the actin filament.49,59 These RalA-dependent mechanisms may also be employed downstream of Rac1 in muscle cells considering that insulin-dependent activation of RalA is also observed in muscle cells.64 In fact, ectopic expression of constitutively activated Rac1 lead to RalA activation and induced the redistribution of activated RalA to the membrane ruffling area.64 Furthermore, knockdown of endogenous RalA suppressed constitutively activated Rac1-induced GLUT4 translocation.64 Therefore, it is reasonable to consider that RalA may be responsible for GLUT4 vesicle tethering to the plasma membrane downstream of Rac1 in muscle cells. Detailed mechanisms for RalA activation by Rac1, however, remain unclear.
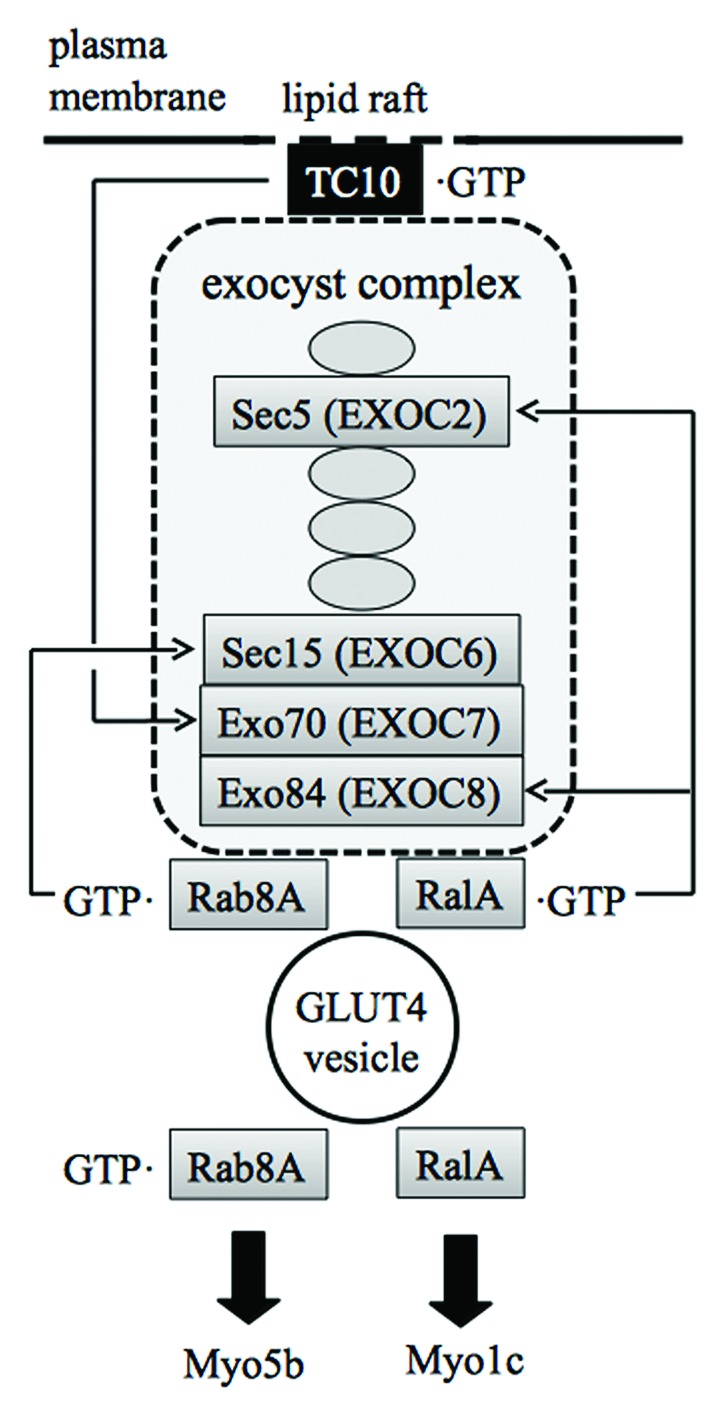
Figure 4. Interaction of the GULT4 vesicle with the exocyst complex and myosin motor proteins mediated by small GTPases. TC10 is localized in lipid raft microdomains and anchors the exocyst complex beneath the plasma membrane. Rab8A and RalA connect the GLUT4 vesicle to the exocyst complex. Rab8A and RalA also act as a link between the GLUT4 vesicle and Myo5b and Myo1c motors, respectively. The interaction between RalA and Myo1c is independent of the nucleotide status of RalA.
Although diverse targets of the activated form of Rac1 have been identified in many types of cells, targets (other than the regulators of the cytoskeleton) which are implicated in glucose uptake in skeletal muscle remain elusive. One candidate for such Rac1 targets is the serine/threonine kinase PAK1 (Fig. 3).65,66 Actually, pak1 knockout mice exhibited peripheral insulin resistance coupled to this defect.66 Moreover, insulin-dependent PAK activation was reduced in both acute and chronic insulin resistant states in humans.39 More recently, the involvement of PAK1 in AMP-activated protein kinase (AMPK)-mediated glucose uptake in muscle cells was reported.67 Collectively, PAK1 may have a pivotal role as a target of Rac1 in glucose uptake. However, the activation of PAK1 may not be sufficient for the induction of glucose uptake, because constitutively activated Cdc42, another activator of PAK1, was not able to stimulate GLUT4 translocation.34
As mentioned above, roles of PI3K and Akt2 in constitutively activated Rac1-induced GLUT4 translocation remain controversial. Ueda et al.34 reported that the phosphorylation level of Akt was not increased when constitutively activated Rac1 was transiently expressed in myoblasts, supporting the notion that Rac1 is not involved in Akt activation. On the other hand, inhibition of PI3K diminished constitutively activated Rac1-induced GLUT4 translocation.34 Therefore, basal, but not insulin-stimulated, activity of PI3K may be required. Considering a recent report that GLUT4 translocation by a constitutively activated Rac1 mutant was not affected by inhibition of Akt2,43 it is likely that the critical target of PI3K with the basal activity is a molecule other than Akt2.
To the contrary, the involvement of Akt in constitutively activated Rac1-dependent GLUT4 translocation has recently been reported.37 In this study, phosphorylation of Akt and its substrate AS160 was detected following ectopic expression of constitutively activated Rac1.37 An Akt inhibitor was reported to reduce constitutively activated Rac1-stimulated GLUT4 translocation significantly.37 Furthermore, constitutively activated Rac1 yielded PtdIns(3,4,5)P3 in the plasma membrane, which may be responsible for the activation of Akt.37 The reason for the apparent discrepancy between results from different groups is currently unknown.
Role of Rac1 in Contraction-Stimulated Glucose Uptake
AMPK is a serine/threonine kinase with a heterotrimeric structure consisting of a catalytic α subunit and regulatory β and γ subunits.68 AMPK is known to be activated by an increase in the intracellular AMP:ATP ratio, acting as a cellular energy sensor. Although the detailed mechanism underlying AMP-dependent activation remains elusive, AMP binding to the γ-subunit is suggested to induce a conformational change, leading to allosteric activation of the enzyme. Phosphorylation on a specific threonine residue of the α subunit by the serine/threonine kinase LKB1 is also required for maximal kinase activity.69
AMPK inhibits anabolic pathways, such as fatty acid synthesis, thus conserving ATP, and also stimulates catabolic pathways that generate ATP.68 AMPK also stimulates glucose uptake via GLUT4 translocation to the plasma membrane in skeletal muscle.68 However, the signaling mechanism for this effect seems different from that utilized by insulin. The reason why particular attention has been devoted to understanding the role of AMPK in glucose uptake in skeletal muscle is that this enzyme is shown to be activated by exercise in rat and humans.68 The activation of AMPK following contraction depends not only on the increased intracellular AMP:ATP ratio, but also on LKB1, as demonstrated by a study using knockout mice.70
A pivotal role of Rac1 in contraction-stimulated glucose uptake in skeletal muscle has been clarified by using muscle-specific rac1 knockout mice.71 The activation of Rac1 was observed after exercise in both mice and humans, and contraction-stimulated glucose uptake was abrogated in rac1 knockout mouse skeletal muscle. Thus, Rac1 has a critical role also in the regulation of glucose uptake in response to contraction. However, the mechanisms underlying Rac1 activation may be different from those in insulin-stimulated signal transduction. For instance, a GEF other than FLJ00068 may be responsible for contraction-stimulated Rac1 activation. Additionally, it was reported that Rac1 activation in response to contraction is not mediated by AMPK.71 This may be an unexpected result, taking into consideration that AMPK has been implicated in contraction-dependent glucose uptake as described above, and AMPK can stimulate Rac1 (see below).
The biguanide metformin is used for the treatment of type 2 diabetes, and known to activate AMPK. Recently, the involvement of Rac1 in metformin/AMPK-stimulated glucose uptake in muscle cells was reported.67 Metformin and another AMPK activator compound 5-aminoimidazole-4-carboxy-amide-1-D-ribofuranoside markedly stimulated the expression of the Rac1-specific GEF Tiam-1.67 Furthermore, knockdown of Tiam-1 inhibited metformin-induced increase in glucose uptake.67 Therefore, AMPK may employ the Tiam-1-Rac1 signaling cascade to promote glucose uptake.
An Adipocyte-Specific, Akt2-Independent Pathway that Involves the Rho Family GTPase TC10
Two signal transduction pathways are activated upon engagement of the insulin receptor in adipocytes.72 One is PI3K-dependent, and the other is PI3K-independent. The PI3K-dependent pathway is common between adipose tissue and skeletal muscle, leading to the activation of the downstream effector kinase Akt2. The PI3K-independent pathway is adipocyte-specific, and involves the Rho family GTPase TC10.72,73 In adipocytes, insulin exploits these two distinct signaling pathways to elicit translocation of GLUT4 to the plasma membrane.
In response to insulin stimulation, a complex composed of Cbl, CAP, CrkII, and C3G is formed through tyrosine phosphorylation of Cbl, leading to the activation of TC10 (Fig. 5).73 This complex is specifically localized in caveolin-enriched lipid raft microdomains, in which TC10 is also accumulated. The activated TC10 in turn interacts with the exocyst component Exo70, recruiting the exocyst complex to the plasma membrane (Fig. 4).74 The recruited exocyst complex is required for GLUT4 vesicle tethering to appropriate sites of the plasma membrane, preparing for the subsequent fusion step.

Figure 5. Role of TC10 downstream of the insulin receptor in adipocytes. Insulin stimulates the formation of a signaling complex consisting of CAP, Cbl, CrkII, and the GEF C3G. Activated TC10 recruits Gapex-5, a GEF for Rab31, to the plasma membrane through the interaction with CIP4. Rab31 in turn fails to be activated, and GLUT4 trafficking from ERC to GSV is released from the negative regulation by Rab31. TC10 is also implicated in the regulation of GLUT4 vesicle tethering to the plasma membrane through the interaction with the exocyst complex and actin cytoskeletal rearrangements. ERC, endosomal recycling compartment; GSV, GLUT4 storage vesicle.
Another mechanism by which TC10 enhances GLUT4 translocation involves Rab31 (Fig. 5).75 Rab31 is a member of the Rab5 subfamily of GTPases, negatively regulating the escape of GLUT4 from the retention in ERCs to GSVs. In fact, dominant-negative Rab31 increased the accumulation of GLUT4 in the plasma membrane, whereas constitutively activated Rab31 blocked insulin-stimulated GLUT4 translocation.75 Furthermore, knockdown of Rab31 enhanced insulin-stimulated GLUT4 translocation.75 Insulin stimulates the formation of the GTP-bound form of TC10 in the plasma membrane. The GTP-bound form of TC10 in turn recruits a protein termed CIP4, which is complexed with a GEF for Rab31, Gapex-5. In this way, insulin sequesters the CIP4/Gapex-5 complex away from its substrate Rab31, thereby inhibiting the activation of Rab31. The failure of Rab31 activation results in the escape of GLUT4 from the retention in ERCs.75
TC10 is also involved in the regulation of actin cytoskeletal rearrangements like other Rho family GTPases. TC10 differentially regulates cortical and perinuclear actin polymerization in adipocytes as shown by studies using constitutively activated and dominant-interfering mutants.76
Conclusions and Perspectives
Participation of Rho family GTPases in insulin action in target tissues, such as skeletal muscle and adipose tissue, has been demonstrated by studies using cultured cell lines and genetically modified mouse models. To date, however, compelling evidence that dysfunction of Rho family GTPases in target tissues is actually associated with type 2 diabetes in humans has not been provided. Precise mechanisms by which insulin and other stimuli modulate Rho family GTPases also remain obscure. Moreover, there are still many questions to be answered surrounding signal transduction pathways downstream of Rho family GTPases. Nevertheless, it is desirable to explore the possibility that signaling cascades involving Rho family GTPases may become novel therapeutic targets for type 2 diabetes. In that case, a regulatory molecule with high specificity for each signal, such as the GEF, may be a better choice for the drug target.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Glossary
Abbreviations:
- AMPK
AMP-activated protein kinase
- EE
early endosome
- ERC
endosomal recycling compartment
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GLUT4
glucose transporter type 4
- GSV
GLUT4 storage vesicle
- PI3K
phosphoinositide 3-kinase
- PtdIns(3,4,5)P3
phosphatidylinositol-3,4,5-triphosphate
- TGN
trans-Golgi network
References
- 1.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–79. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 2.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–42. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 3.Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev. 1981;61:914–73. doi: 10.1152/physrev.1981.61.4.914. [DOI] [PubMed] [Google Scholar]
- 4.Kahn CR. The molecular mechanism of insulin action. Annu Rev Med. 1985;36:429–51. doi: 10.1146/annurev.me.36.020185.002241. [DOI] [PubMed] [Google Scholar]
- 5.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem. 2012;81:507–32. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- 6.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–52. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Malaby AW, Famulok M, Sabe H, Lambright DG, Hsu VW. Grp1 plays a key role in linking insulin signaling to glut4 recycling. Dev Cell. 2012;22:1286–98. doi: 10.1016/j.devcel.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–33. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 10.Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–8. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 11.Shepherd PR, Gnudi L, Tozzo E, Yang H, Leach F, Kahn BB. Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J Biol Chem. 1993;268:22243–6. [PubMed] [Google Scholar]
- 12.Tozzo E, Shepherd PR, Gnudi L, Kahn BB. Transgenic GLUT-4 overexpression in fat enhances glucose metabolism: preferential effect on fatty acid synthesis. Am J Physiol. 1995;268:E956–64. doi: 10.1152/ajpendo.1995.268.5.E956. [DOI] [PubMed] [Google Scholar]
- 13.Tsao TS, Burcelin R, Katz EB, Huang L, Charron MJ. Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes. 1996;45:28–36. doi: 10.2337/diab.45.1.28. [DOI] [PubMed] [Google Scholar]
- 14.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–88. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 15.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 16.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 18.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB β. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–8. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 20.Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci U S A. 2006;103:11821–7. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–17. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 22.Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5:293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci U S A. 2010;107:19909–14. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006;281:31478–85. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- 25.Larance M, Ramm G, Stöckli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–13. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 26.Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 27.Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–80. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- 28.Roach WG, Chavez JA, Mîinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J. 2007;403:353–8. doi: 10.1042/BJ20061798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie X, Gong Z, Mansuy-Aubert V, Zhou QL, Tatulian SA, Sehrt D, Gnad F, Brill LM, Motamedchaboki K, Chen Y, et al. C2 domain-containing phosphoprotein CDP138 regulates GLUT4 insertion into the plasma membrane. Cell Metab. 2011;14:378–89. doi: 10.1016/j.cmet.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.JeBailey L, Rudich A, Huang X, Di Ciano-Oliveira C, Kapus A, Klip A. Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol. 2004;18:359–72. doi: 10.1210/me.2003-0294. [DOI] [PubMed] [Google Scholar]
- 31.Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal. 2011;23:1546–54. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 32.JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56:394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- 33.Khayat ZA, Tong P, Yaworsky K, Bloch RJ, Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci. 2000;113:279–90. doi: 10.1242/jcs.113.2.279. [DOI] [PubMed] [Google Scholar]
- 34.Ueda S, Kataoka T, Satoh T. Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol Cell. 2008;100:645–57. doi: 10.1042/BC20070160. [DOI] [PubMed] [Google Scholar]
- 35.Marcusohn J, Isakoff SJ, Rose E, Symons M, Skolnik EY. The GTP-binding protein Rac does not couple PI 3-kinase to insulin-stimulated glucose transport in adipocytes. Curr Biol. 1995;5:1296–302. doi: 10.1016/S0960-9822(95)00256-9. [DOI] [PubMed] [Google Scholar]
- 36.Balamatsias D, Kong AM, Waters JE, Sriratana A, Gurung R, Bailey CG, Rasko JE, Tiganis T, Macaulay SL, Mitchell CA. Identification of P-Rex1 as a novel Rac1-guanine nucleotide exchange factor (GEF) that promotes actin remodeling and GLUT4 protein trafficking in adipocytes. J Biol Chem. 2011;286:43229–40. doi: 10.1074/jbc.M111.306621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiu TT, Sun Y, Koshkina A, Klip A. Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance. J Biol Chem. 2013;288:17520–31. doi: 10.1074/jbc.M113.467647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ueda S, Kitazawa S, Ishida K, Nishikawa Y, Matsui M, Matsumoto H, Aoki T, Nozaki S, Takeda T, Tamori Y, et al. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 2010;24:2254–61. doi: 10.1096/fj.09-137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sylow L, Jensen TE, Kleinert M, Højlund K, Kiens B, Wojtaszewski J, Prats C, Schjerling P, Richter EA. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes. 2013;62:1865–75. doi: 10.2337/db12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogan JS, McKee AE, Lodish HF. Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol Cell Biol. 2001;21:4785–806. doi: 10.1128/MCB.21.14.4785-4806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou JC, Shigematsu S, Crawford HC, Anastasiadis PZ, Pessin JE. Dual regulation of Rho and Rac by p120 catenin controls adipocyte plasma membrane trafficking. J Biol Chem. 2006;281:23307–12. doi: 10.1074/jbc.M603127200. [DOI] [PubMed] [Google Scholar]
- 42.Lewis JP, Palmer ND, Ellington JB, Divers J, Ng MC, Lu L, Langefeld CD, Freedman BI, Bowden DW. Analysis of candidate genes on chromosome 20q12-13.1 reveals evidence for BMI mediated association of PREX1 with type 2 diabetes in European Americans. Genomics. 2010;96:211–9. doi: 10.1016/j.ygeno.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nozaki S, Takeda T, Kitaura T, Takenaka N, Kataoka T, Satoh T. Akt2 regulates Rac1 activity in the insulin-dependent signaling pathway leading to GLUT4 translocation to the plasma membrane in skeletal muscle cells. Cell Signal. 2013;25:1361–71. doi: 10.1016/j.cellsig.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 44.Takenaka N, Izawa R, Wu J, Kitagawa K, Nihata Y, Hosooka T, Noguchi T, Ogawa W, Aiba A, Satoh T. A critical role of the small GTPase Rac1 in Akt2-mediated GLUT4 translocation in mouse skeletal muscle. FEBS J. 2014;281:1493–504. doi: 10.1111/febs.12719. [DOI] [PubMed] [Google Scholar]
- 45.Sylow L, Kleinert M, Pehmøller C, Prats C, Chiu TT, Klip A, Richter EA, Jensen TE. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal. 2014;26:323–31. doi: 10.1016/j.cellsig.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Brozinick JT, Jr., Hawkins ED, Strawbridge AB, Elmendorf JS. Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J Biol Chem. 2004;279:40699–706. doi: 10.1074/jbc.M402697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem. 2001;276:42436–44. doi: 10.1074/jbc.M108297200. [DOI] [PubMed] [Google Scholar]
- 48.Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem. 1994;269:29934–42. [PubMed] [Google Scholar]
- 49.Bose A, Guilherme A, Robida SI, Nicoloro SM, Zhou QL, Jiang ZY, Pomerleau DP, Czech MP. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature. 2002;420:821–4. doi: 10.1038/nature01246. [DOI] [PubMed] [Google Scholar]
- 50.Imamura T, Huang J, Usui I, Satoh H, Bever J, Olefsky JM. Insulin-induced GLUT4 translocation involves protein kinase C-λ-mediated functional coupling between Rab4 and the motor protein kinesin. Mol Cell Biol. 2003;23:4892–900. doi: 10.1128/MCB.23.14.4892-4900.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishikura S, Klip A. Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am J Physiol Cell Physiol. 2008;295:C1016–25. doi: 10.1152/ajpcell.00277.2008. [DOI] [PubMed] [Google Scholar]
- 52.Semiz S, Park JG, Nicoloro SM, Furcinitti P, Zhang C, Chawla A, Leszyk J, Czech MP. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J. 2003;22:2387–99. doi: 10.1093/emboj/cdg237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yip MF, Ramm G, Larance M, Hoehn KL, Wagner MC, Guilhaus M, James DE. CaMKII-mediated phosphorylation of the myosin motor Myo1c is required for insulin-stimulated GLUT4 translocation in adipocytes. Cell Metab. 2008;8:384–98. doi: 10.1016/j.cmet.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 54.Yoshizaki T, Imamura T, Babendure JL, Lu JC, Sonoda N, Olefsky JM. Myosin 5a is an insulin-stimulated Akt2 (protein kinase Bbeta) substrate modulating GLUT4 vesicle translocation. Mol Cell Biol. 2007;27:5172–83. doi: 10.1128/MCB.02298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol. 2003;15:590–7. doi: 10.1016/S0955-0674(03)00097-8. [DOI] [PubMed] [Google Scholar]
- 56.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Loubéry S, Coudrier E. Myosins in the secretory pathway: tethers or transporters? Cell Mol Life Sci. 2008;65:2790–800. doi: 10.1007/s00018-008-8350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Randhawa VK, Ishikura S, Talior-Volodarsky I, Cheng AW, Patel N, Hartwig JH, Klip A. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J Biol Chem. 2008;283:27208–19. doi: 10.1074/jbc.M804282200. [DOI] [PubMed] [Google Scholar]
- 59.Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell. 2007;13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 60.Feig LA. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 2003;13:419–25. doi: 10.1016/S0962-8924(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 61.Chen XW, Leto D, Xiao J, Goss J, Wang Q, Shavit JA, Xiong T, Yu G, Ginsburg D, Toomre D, et al. Exocyst function is regulated by effector phosphorylation. Nat Cell Biol. 2011;13:580–8. doi: 10.1038/ncb2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- 63.Sugihara K, Asano S, Tanaka K, Iwamatsu A, Okawa K, Ohta Y. The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nat Cell Biol. 2002;4:73–8. doi: 10.1038/ncb720. [DOI] [PubMed] [Google Scholar]
- 64.Nozaki S, Ueda S, Takenaka N, Kataoka T, Satoh T. Role of RalA downstream of Rac1 in insulin-dependent glucose uptake in muscle cells. Cell Signal. 2012;24:2111–7. doi: 10.1016/j.cellsig.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 65.Tsakiridis T, Taha C, Grinstein S, Klip A. Insulin activates a p21-activated kinase in muscle cells via phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:19664–7. doi: 10.1074/jbc.271.33.19664. [DOI] [PubMed] [Google Scholar]
- 66.Wang Z, Oh E, Clapp DW, Chernoff J, Thurmond DC. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J Biol Chem. 2011;286:41359–67. doi: 10.1074/jbc.M111.291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.You GY, Lee JO, Kim JH, Kim N, Lee SK, Moon JW, Jie S, Lee HJ, Kim SJ, Park SH, et al. Tiam-1, a GEF for Rac1, plays a critical role in metformin-mediated glucose uptake in C2C12 cells. Cell Signal. 2013;25:2558–65. doi: 10.1016/j.cellsig.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 68.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 69.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–20. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A, et al. Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes. 2013;62:1139–51. doi: 10.2337/db12-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanzaki M, Pessin JE. Insulin signaling: GLUT4 vesicles exit via the exocyst. Curr Biol. 2003;13:R574–6. doi: 10.1016/S0960-9822(03)00478-0. [DOI] [PubMed] [Google Scholar]
- 73.Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, Macara IG, Pessin JE, Saltiel AR. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature. 2001;410:944–8. doi: 10.1038/35073608. [DOI] [PubMed] [Google Scholar]
- 74.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–33. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 75.Lodhi IJ, Chiang SH, Chang L, Vollenweider D, Watson RT, Inoue M, Pessin JE, Saltiel AR. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 2007;5:59–72. doi: 10.1016/j.cmet.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanzaki M, Watson RT, Hou JC, Stamnes M, Saltiel AR, Pessin JE. Small GTP-binding protein TC10 differentially regulates two distinct populations of filamentous actin in 3T3L1 adipocytes. Mol Biol Cell. 2002;13:2334–46. doi: 10.1091/mbc.01-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]