

Abstract
We applied global gene expression arrays, quantitative real-time PCR, immunostaining, and functional assays to untangle the role of High Mobility Groups proteins (HMGs) in human osteoarthritis (OA)-affected cartilage. Bioinformatics analysis showed increased mRNA expression of Damage-Associated Molecular Patterns (DAMPs): HMGA, HMGB, HMGN, SRY, LEF1, HMGB1, MMPs, and HMG/RAGE-interacting molecules (spondins and S100A4, S100A10, and S100A11) in human OA-affected cartilage as compared with normal cartilage. HMGB2 was down-regulated in human OA-affected cartilage. Immunohistological staining identified HMGB1 in chondrocytes in the superficial cartilage. Cells of the deep cartilage and subchondral bone showed increased expression of HMGB1 in OA-affected cartilage. HMGB1 was expressed in the nucleus, cytosol, and extracellular milieu of chondrocytes in cartilage. Furthermore, HMGB1 was spontaneously released from human OA-affected cartilage in ex vivo conditions. The effects of recombinant HMGB1 was tested on human cartilage and chondrocytes in vitro. HMGB1 stimulated mRNA of 2 NFκB gene enhancers (NFκB1 and NFκB2), 16 CC and CXC chemokines (IL-8, CCL2, CCL20, CCL3, CCL3L1, CCL3L3, CCL4, CCL4L1, CCL4L2, CCL5, CCL8, CXCL1, CXCL10, CXCL2, CXCL3, and CXCL6) by ≥10-fold. Furthermore, HMGB1 and IL-1β and/or tumor necrosis factor α (but not HMGI/Y) also significantly induced inducible nitric oxide synthase, NO, and interleukin (IL)-8 production in human cartilage and chondrocytes. The recombinant HMGB1 utilized in this study shows properties that are similar to disulfide-HMGB1. The differential, stage and/or tissue-specific expression of HMGB1, HMGB2, and S100A in cartilage was associated with regions of pathology and/or cartilage homeostasis in human OA-affected cartilage. Noteworthy similarities in the expression of mouse and human HMGB1 and HMGB2 were conserved in normal and arthritis-affected cartilage. The multifunctional forms of HMGB1 and S100A could perpetuate damage-induced cartilage inflammation in late-stage OA-affected joints similar to sterile inflammation. The paracrine effects of HMGB1 can induce chemokines and NO that are perceived to change cartilage homeostasis in human OA-affected cartilage.
Introduction
The pathogenesis in osteoarthritis (OA) is observed in the joints, cartilage, and subchondral bone (Haseeb and Haqqi, 2013). Human OA-affected cartilage as compared with normal cartilage is an active site for chondrocyte hypertrophy, angiogenesis and chondrogenesis, inflammation, inflammation resolution, and extracellular matrix repair and remodeling (Pelletier et al., 2001; Attur et al., 2002a; Haseeb and Haqqi, 2013). The chondrocytes in different zones (I to IV) and subcondral regions are phenotypically and functionally distinct (Haseeb and Haqqi, 2013). Increased amounts of NFκB activity and catabolic activity by interleukin (IL)-1β, tumor necrosis factor α (TNFα), nitric oxide (NO), matrix metalloproteinase (MMPs), and eicosanoids in OA are associated with dysfunctional cartilage homeostasis (Pelletier et al., 2001; Attur et al., 2002a; Haseeb and Haqqi, 2013). Unlike the overt inflammation exhibited in rheumatoid arthritis (RA), OA displays “covert inflammation” (Attur et al., 2002a). We and others have previously shown that human OA-affected cartilage (as compared with normal cartilage) exhibits an inflammatory response devoid of the cardinal signs of inflammation (redness and swelling with heat and pain—rubor et tumor cum calore et dolor) due to unique architecture (avascular, aneural, and alymphatic) of the cartilage (Pelletier et al., 2001; Attur et al., 2002a). In spite of the lack of cardinal signs of inflammation in human OA-affected cartilage, a substantially increased level of mRNA and proteins representing several inflammatory mediators have been reported (Amin et al., 1996, 1999; Attur et al., 2001, 2002a; Pelletier et al., 2001; Haseeb and Haqqi, 2013). Human OA- and RA-affected PBMCs can be segregated based on an inflammatory response by using gene expression arrays (Amin et al., 2007).
High Mobility Groups proteins (HMGs) are chromosomal proteins that can be subdivided into three super families, namely HMGA, HMGB, and HMGN, which contain an AT-hook domain, an HMG–box domain, and nucleosomal binding domain, respectively (Sparvero et al., 2009; Chen and Nuñez, 2010; Harris et al., 2012). It should be noted that the HMGN protein family of HMGs reduces the compactness of the chromatin fiber in the nucleosomes, thereby augmenting transcription. HMG proteins are involved in functions associated with transcription, replication, recombination, DNA repair, inflammation, cell differentiation, development, autophagy, and hypertrophy (Adjaye et al., 2005; Sparvero et al., 2009; Chen and Nuñez, 2010; Harris et al., 2012). Recently, a new class of molecules, Damage-Associated Molecular Pattern (DAMPs), which includes HMGBs, SRY, LEF1, S100, AGE, and extracellular matrix proteins (e.g. heparin sulfate), has been identified (Sparvero et al., 2009; Chen and Nuñez, 2010). These DAMPs have unique properties to initiate and perpetuate inflammatory responses via their receptors in the absence of infections (Sparvero et al., 2009; Chen and Nuñez, 2010). This is termed sterile inflammation (Sparvero et al., 2009; Chen and Nuñez, 2010). Many DAMPs such as HMGB1 (high-mobility group box 1) and heat-shock proteins are cytosolic and or nuclear proteins, which can also be released into the extracellular milieu after tissue injury (Sparvero et al., 2009; Chen and Nuñez, 2010; Harris et al., 2012). HMGB1, which can bind and bend DNA during transcription, can also function as a leaderless secretory protein. The predominant nuclear HMGB1 can translocate to the cytosol, where it undergoes post-translational acetylation. The cytosolic HMGB1 is subsequently secreted into the extracellular milieu. The post-translational process also includes modification of redox states of HMGB1, which partially dictates the bioactivity of extracellular HMGB1 in biological processes by interacting with DNA, several co-ligands, and receptors (Yang et al., 2005, 2010; Tang et al., 2012; Janko et al., 2014). For example, all-thiol HMGB1 generated in vitro is engaged in chemotaxis and leukocyte recruitment. Disulfide-HMGB1 produced in vitro induces cytokines and inflammation, whereas the oxidized HMGB1 exhibits no immune responses and may be involved in inflammation resolution (Tang et al., 2012; Yang et al., 2012). Furthermore, HMGB1 released by apoptotic cells or necrotic or dead cells are terminally oxidized and unable to induce cytokines and chemokines (Urbonaviciute et al., 2009; Venereau et al., 2012; Yang et al., 2012). The all-thiol HMGB1 and disulfide HMGB1 may be readily interchangeable (by reactive oxygen species [ROS] or reactive nitrogen species [RNS]) depending on the presence of an electron donor or acceptable in the environment (Venereau et al., 2012; Janko et al., 2014). For example, LPS-stimulated cells induced both all-thiol and disulfide HMGB1 (Venereau et al., 2012; Tang et al., 2012). All-thiol HMGB1 is prevalent immediately after (in vitro) muscle injury, but it switches to disulfide HMGB1 after a few hours (Venereau et al., 2012). Finally, the sustained ROS/RNS production eventually generates the terminal oxidation of HMGB1 “oxidized HMGB1” (Janko et al., 2014). This extracellular HMGB1 can trigger and prolong inflammatory responses via its cell-surface receptors (Toll-Like Receptors [TLR2, TLR4], Mucin domain 3 [TIM3], and Receptor for Advanced Glycation End products [RAGE]) and intracellular proteins/receptors such as spondins and S100 calcium binding proteins (Sparvero et al., 2009; Chen and Nuñez, 2010; Harris et al., 2012).
HMGB1 serum levels significantly correlated with disease activity scores (DAS-28) in patients with arthritis (Goldstein et al., 2007). One of the key characteristics of OA-affected cartilage was chondrocyte hypertrophy and increased synthesis of type X collagen, which could be driven in vitro by RAGE receptor of HMGB1 (Cecil et al., 2005). Animal models of OA (STR/ort mice) and collagen-induced arthritis showed increased levels of HMGB1 in the synovial membrane, cartilage, meniscus, and ligaments (Palmblad et al., 2007; Kyostio-Moore et al., 2011). The multifunctional activity of HMGs and its functions in tissue injury in other systems prompted us to analyze their expression in one of the target tissues in human OA. The present study shows differentially expressed HMGs and S100As in normal and human OA-affected cartilage using a genomic approach and the functional effect of HMGB1 in chondrocytes and cartilage. The discussion highlights the similarities in HMG expression of cartilage in man and mouse and the ability of HMGB1 to position itself to change cartilage homeostasis.
Materials and Methods
Reagents
All cell culture media, FCS, cytokines, and PCR primers were obtained from Gibco BRL, Pepto. Tech. Inc., and Yamanuchi Pharmaceuticals. IL-8 ELISA kits were purchased from R and D Systems. Rabbit anti-HMGB1 specific antibodies were a gift from Dr. Kevin Tracy (The Feinstein Institute for Medical Research-LIJ School of Medicine, NY).
Generation of native HMGB1
The native recombinant HMGB1 was cloned, expressed, and purified as previously reported (Wang et al., 1999; Li et al., 2004). Briefly, a full-length cDNA was cloned and expressed in Escherichia coli, and the native recombinant HMGB1 was generated. The protein was affinity purified and tested for endotoxin contamination by chromogenic Limulus amebocyte lysate assay and other methods as previously reported (Li et al., 2004; Rao et al., 2011; Yang et al., 2012). The HMGB1 in every batch was also tested for its ability to induce inflammatory mediators (such as Nitric Oxide or PGE2) in cells before it was used in experiments to ascertain its proinflammatory activity. It should be noted that the medium used in this study was devoid of any reducing or oxidizing agents. The purified native recombinant HMGB1 was provided by Dr. Kevin Tracy's Laboratory.
Procurement of human cartilage and demographics
Normal and OA-affected cartilage from human knees was procured as previously reported (Amin et al., 1997; Attur et al., 2000; Dave and Amin, 2013). The clinical samples represent a broad ethnic and age group, where OA is significantly susceptible (Haseeb and Haqqi, 2013). Briefly, knee OA-affected cartilage was obtained from donors who were undergoing knee replacement surgery under the guidelines of the Institution Review Board for use of human tissues in translational research. Nonarthritic knee cartilage was mostly obtained from patients with fractures or from accident victims after knee amputation or from the National Development and Research Institute (NDRI). The cartilage samples were scored based on the histology in the slides, pathology, and clinical records. The demographics of donors (including males, females, age, source, and ethnic backgrounds) are described in Supplementary Table S1(Supplementary Data are available online at www.liebertpub.com/dna) and (Dave and Amin, 2013). The classification of cartilage was based on OARSI scores (Pritzker et al., 2006; Dave and Amin, 2013).
Human OA-cartilage explant assay for soluble HMGB1
The OA-cartilage ex vivo assay was performed as previously reported in our laboratory (Amin et al., 1996; Attur et al., 2002b). The assay for the detection of extracellular HMGB1 from human cartilage was similar to that reported for mouse chondrocytes (Liu-Bryan and Terkeltaub, 2010). Briefly, knee articular cartilage from OA-affected joints of donors was cut into discs and placed in a 24-well plate. Articular cartilage from different parts of the joint was pooled for the experiment. The cartilage was incubated in Ham's F-12 medium (2 mL), and 1% endotoxin free human albumin (and devoid of any oxidizing or reducing agent) in the presence and absence of different modulators. The medium from each well was aspirated and analyzed for HMGB1 or NO as described. Soluble extracellular-cartilage HMGB1 was evaluated by loading 50 μL of supernatants onto a thin 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel in the presence of a positive control (1 μg of recombinant human HMGB1), and Western blotted to follow the specific immuno-reactivity to 29 kDa HMGB1 and the corresponding molecular weight. Bound antibody was detected by a secondary antibody conjugated with horseradish peroxidase, and developed using the enhanced chemiluminescence system (Amersham Corp.) on Kodak Xomatic X-ray film. The presence of a 29 kDa immune-reactive protein was identified in the cartilage supernatant that co-migrated with the internal positive control. Nitric Oxide was estimated as their stable end product, nitrite, from the cartilage or cell supernatant using a Griess assay (Amin et al., 1996).
Isolation and cultivation of human chondrocytes
Human chondrocytes were isolated and cultivated as previously reported (Dave and Amin, 2013). Chondrocytes were released from cartilage, and equal amounts of cells (1×105 cells per well) were cultivated in 24-well plates as formerly described (Amin et al., 1996; Attur et al., 2002b). They were subject to various experimental conditions in the presence and absence of different modulators.
Western blotting of inducible nitric oxide synthase
Primary human chondrocytes were released from cartilage, and equal amounts of cells were cultivated (Dave and Amin, 2013). The medium was changed, and the cells were challenged with different modulators for an additional 24 h. Cells were harvested and washed with PBS. Total protein was extracted from the cells and estimated by BCA reagent (Pierce). Cell extracts were loaded onto the gel with molecular-weight markers and subjected to Western blot analysis (Amin et al., 1996). The blots were probed with specific anti-inducible nitric oxide synthase (iNOS) monoclonal antibodies as described by BD Transduction Laboratories and developed using ECL Western blot system (Amersham Corp.), and Kodak Xomatic X-ray film.
Quantitative real-time PCR analysis
Primer and probe were designed using the Primer Express Algorithm (PE Applied Biosystems). The quantitation was performed by quantitative real-time PCR (qPCR) with a flourogenic probe. qPCR was performed for specific transcripts (gene) using RNA from a minimum of three normals (N) and six to seven OA-affected cartilage donor samples (OA). The corresponding primer probes were utilized for each gene analyzed. Amplitaq Gold DNA polymerase was used for amplification in a Prism7700 System (PE Applied Biosystems). To exclude PCR amplification of contaminating genomics DNA, we performed PCR (RT minus control) with RNA, which was not reverse transcribed into DNA. In these experiments, RT minus controls were negative. The values for HMGB1 were presented as units of relative expression after normalizing the expression with GAPDH in each cartilage sample. The rest of the qPCR data was presented as units of relative expression after normalization with the expression of GAPDH and after conversion of the values into percent normal before they were calculated for mean±SD and p-values (Attur et al., 2002c; Rao et al., 2011).
Histological analysis of cartilage
Histological evaluation was performed on sagittal sections of normal and OA-affected cartilage (Attur et al., 2009). After dissection of cartilage samples, specimens were fixed in buffered formalin and embedded in paraffin blocks. Sections were stained with specific anti-HMGB1-antibodies. The HMGB1 was stained in brown, and the nucleus was stained in blue.
Gene expression arrays and bioinformatics
RNA extraction from cartilage, sample pooling, and gene expression arrays were performed as previously reported in our laboratory (Attur et al., 2001, 2002b, 2009). Briefly, RNA was extracted from normal (OARSI score: 0–1) and OA-affected cartilage (OARSI score: 3–5). Equal amounts of total RNA were pooled into six different cohorts (N1 to N6) of normal and (OA1 to OA6) of OA, respectively. Each cohort of normal or OA-affected cartilage groups consisted of RNA that represented a minimum of 5 and a maximum of 12 donors. Probes were prepared from the pooled cartilage samples, hybridized to U133A gene chips as described (www.affymetrix.com), and previously reported for normal and OA-affected cartilage (Attur et al., 2002c). In the 2nd set of gene expression arrays, the primary human chondrocytes were cultivated for 24 h, after which the medium was aspirated, and fresh medium was added to the cells with (5 μg/mL of HMGB1) and without HMGB1 (Wt) for an additional 24 h. The chondrocytes were harvested, and the RNA was isolated as described earlier. Probes were prepared from Wt, and HMGB1-treated cells, and hybridized to U95 gene chips as described (www.affymetrix.com) and previously reported (Attur et al., 2002c). Gene expression microarray raw data (CEL files) was background corrected and normalized using the Bioconductor package “affy” with “RMA” method; and differential expression and statistical analysis was performed using Linear Models (Smyth, 2004). Probes to EnsEMBL genes (version 70) were annotated using Biomart database. When more than one probe was annotated to the same gene, average expression was taken. Significances of differentially expressed genes were considered if the multiple-tests corrected (FDR) p-value was ≤0.05. Heatmap was produced on median-centered expression value (difference of expression of each gene from the median expression value of all samples of that gene) using Gitools (Perez-Llamas and Lopez-Bigas, 2011). Stable housekeeping genes used in these analyses were selected from evidence-based selection and a meta-analysis of 13, 629 human gene arrays (De Jonge et al., 2007).
Statistical analysis and data presentation
The Student's t-test was employed for statistical analysis as previously reported (Rao et al., 2011; Dave and Amin, 2013). The data (from a minimum of triplicates) are presented as mean±standard deviation. The p-value≤0.05 was considered significant.
Results
Gene expression arrays and qPCR of normal and OA-affected cartilage
A systems biology approach to human OA using genomics, proteomic, and bioinformatics approaches was initiated as described (Attur et al., 2002c). Normal and OA-affected cartilage was harvested from a broad spectrum (age and ethnic background) of donors as shown in Supplementary Table S1. Total RNA was isolated from cartilage and stored for further use. Five to twelve RNA samples were pooled in equal amounts and generated six different cohorts of normal and six cohorts of OA-affected cartilage. Gene expression arrays, normalization, statistical, and bioinformatics analysis were performed and each cohort represented data generated from a separate gene chip (n=6) as shown in Figure 1. The heatmap showed a significant difference in expression between HMGB1 and HMGB2. HMGB1 increased and HMGB2 decreased in human OA-affected cartilage as compared with normal cartilage (Fig. 1). The differential expression of HMGB1 and two of its pseudogenes (HMGBP5 and HMGBP10) were statistically significant. The expression of nucleosomal HMGN1 and HMGN3 was also increased in four of the six cohorts of human OA-affected cartilage as compared with (none) in normal cartilage (Fig. 1). Similar to HMGB1, S100 proteins interacted with RAGE, promoting cellular recruitment to tissues (Sims et al., 2010). Gene expression arrays showed a statistically significant increase in mRNA expression of S100A4, S100A10, and S100A11 in human OA-affected cartilage as compared with normal cartilage. A detailed list of genes, fold changes, and p-values for each transcript is described in Supplementary Table S2 in the addendum. Several proteins (such as MMP-2, -9, and -13, osteoglycin and spondins) that are known to be up-regulated in human OA-affected cartilage as compared with normal cartilage (Pelletier et al., 2001; Attur et al., 2002a, 2009; Haseeb and Haqqi, 2013) also showed an increase in the expression of their respective mRNA in these gene arrays (Fig. 1). The changes in the gene expression arrays were further validated by qPCR analysis of selected transcripts. qPCR analysis of HMGB1 from a separate set of three normal and three OA-affected cartilage samples is shown in Figure 2. Other transcripts were validated using qPCR and another set of clinical samples (three to seven donors) of normal cartilage (N) and OA-affected cartilage (OA) as described in the “Materials and Methods” section. For example, Thrombospondin 2 (TSP2), (N: 207±129, vs. OA: 803±274), Fibroblasts Growth Factor-18 (FGF-18) (N: 220±104, vs. OA: 475±175), Osteoglycin (OGN), (N: 678±653, vs. OA: 2146±537), and vascular endothelial growth factor (VEGF) (N: 91±18, vs. OA: 30±4.0) were also observed to be differentially expressed and statistically significant (p≤0.05) similar to those observed in the heatmaps in Figure 1. Human HMGB2, as compared with HMGB1, showed significant and differential expression of transcripts in both normal and OA-affected cartilage (Fig. 3).
FIG. 1.

Gene expression array of Damage-Associated Molecular Patterns (DAMPs) and High Mobility Groups proteins (HMGs) in normal and OA-affected cartilage. Heatmap shows up- and down-regulated transcripts from normal (N) and OA-affected (OA) cartilage. Gene expression arrays were performed from cohorts of six pools of normal and six pools of OA. Gene expression profiles are shown in rows. Color toward red indicates higher expression, and color toward green indicates lower expression as compared with median expression as shown on the scale. The statistical analysis is described on the left-hand side of the figure in the form of a heat map in yellow and gray, where n=6. The significant p-values (p≤0.05) are represented in yellow. The detailed characteristics of clinical samples are described in Supplementary Table S1. Details about the fold changes and statistical values for each gene are described in Supplementary Table S2. Color images available online at www.liebertpub.com/dna
FIG. 2.

qPCR of mRNA of HMGB1 in normal and OA-affected cartilage. Total RNA was isolated and quantitated from three normals and three OA-affected cartilage samples from different donors. The data are presented normalizing the values with GAPDH mRNA (housekeeping gene) and SD.
FIG. 3.

Bioinformatics and statistical analysis of HMGB1 and HMGB2 in normal and OA-affected cartilage. HMGB1 and HMGB2 share 80% homology at the amino-acid level (Sparvero et al., 2009). The gene expression of HMGB1 and HMGB2 was compared in both normal and OA-affected cartilage to examine their expression during the disease process. The figure describes the ORASI score of the cartilage in normal and OA-affected cartilage and the corresponding expression of HMGB1 and HMGB2 with their p-values.
Immunohistological staining of HMGB1 in normal and OA cartilage and release of HMGB1 from cartilage
Going forward, we focused our attention on an archetypical alarmin (HMGB1) involved in noninfectious inflammation whose mRNA expression was up-regulated in human OA-affected cartilage. The tissue-specific and sub-cellular localization of HMGB1 is well documented (Sparvero et al., 2009; Chen and Nuñez, 2010). We, therefore, examined the expression of HMGB1 protein in normal and OA-affected cartilage. Immunohistological staining of normal and OA-affected cartilage showed HMGB1-positive cells in both normal and OA-affected cartilage (Fig. 4). Kim et al. (2000) reported that although there were apoptotic cells in OA-affected cartilage, apoptotic cells were rarely seen in normal cartilage. The staining (with variable intensity) was more pronounced near the surface and deep region of the normal and OA-affected cartilage as compared with the mid-region. The deep region of the OA-affected cartilage showed an increased number of HMGB1-positive cells as compared with the normal cartilage. As expected, Western blotting of cartilage extracts from normal and OA-affected cartilage showed the presence of HMGB1 (unpublished data) similar to those observed in normal and arthritis-affected chondrocytes from monkeys (Loeser et al., 2005). The cartilage exhibited tissue-specific expression of HMGB1 (within the cartilage) with a variable intensity of HMGB1-positive cells in two different regions. In summary, immunostaining showed differential expression of HMGB1 with distinct functions in different zones of the cartilage.
FIG. 4.

Immunohistological staining of HMGB1 in normal and OA-affected cartilage. The figure shows the surface, middle, and deep region of the normal and OA-affected cartilage. Cartilage samples were stained (brown) with anti-HMGB1 antibodies+chromphore. The nuclear staining is represented in blue. Some cells show both blue and brown staining of nuclear HMGB1 as indicated by the red arrow. The white arrow shows the HMGB1-positive cells with cytoplasmic staining The black arrow shows HMGB1 staining in the extracellular milieu. Color images available online at www.liebertpub.com/dna
All nucleated cells harbor HMGB1 in the nucleus. There are typically ∼106 HMGB1 molecules per cell. Some chondrocytes showed increased staining in the nucleus as indicated by the red arrow. The presence of HMGB1 in human chondrocytes was also observed in both the cytosol and extracellular region (lacunae) of the cells as marked in Figure 4. The HMGB1-positive cells in normal and OA-affected cartilage appeared with a large nucleus, suggesting the presence of healthy cells. Extracellular HMGB1 is known to serve as an alarmin and/or a proinflammatory cytokine, by damaged/necrotic cells and/or activate/inflammatory cells (Yang and Tracey, 2005; Yang et al., 2005; Sparvero et al., 2009; Chen and Nuñez, 2010). In view of the immunostaining of cartilage described earlier, damaged, necrotic, and hypertrophic chondrocytes in OA-affected cartilage, we applied our human cartilage ex vivo assay to examine the spontaneous release of HMGB1 outside the cartilage (Amin et al., 1996, 1997). The OA-affected cartilage was washed with sterile saline, diced, and incubated in a 24-well plate in triplicate in ex vivo conditions to evaluate the spontaneous accumulation of HMGB1 in the serum-free medium containing 1% endotoxin-free human albumin. The assay for the HMGB1 was performed by taking an equal volume of the supernatants from the medium and loading them onto SDS-PAGE gel after 24 and 48 h. The recombinant HMGB1 was used as the positive control for Western blotting to examine the release of HMGB1 from cartilage into the supernatant. Our analysis showed a 29 kDa immune-reactive protein present at 24 and 48 h (similar to recombinant 29 kDa HMGB1) that was spontaneously released by OA-affected human cartilage in ex vivo conditions in the cartilage milieu (Fig. 5). IL-1β is known to augment inflammatory mediators in OA-affected cartilage, and it increases apoptotic cells (Zhou et al., 2008). Preliminary experiments were performed by incubating the OA-affected cartilage in the presence and absence of IL-1β. There was about 50% decrease in the accumulation of HMGB1 in the medium as evaluated by western blotting by IL-1β-treated cells (data not presented). These observations and those described earlier rule out the possibility of apoptotic cells in normal cartilage as a major source of extracellular/cartilage HMGB1.
FIG. 5.

Spontaneous release of HMGB1 by OA-affected cartilage. Human OA-affected cartilage was incubated in ex vivo conditions. The spontaneous release of HMGB1 was determined at 24 and 48 h. An equal amount of supernatant was loaded onto the gel. One lane of the recombinant HMGB1 that was loaded onto the gel is shown on the right-hand side of the figure. Western blot of HMGB1 was performed as described in the “Materials and Methods” section. A 29 kDa signal of HMGB1 corresponding to the positive control in the Western blot analysis was observed as shown by an arrow.
Genomic analysis of the effect of HMGB1 on chemokines, IL-8, and iNOS in human chondrocytes
Sterile injury (in the absence of an infection) has been reported to induce and promote the autocrine and paracrine effects of HMGB1 (Klune et al., 2008). Previous studies have shown that E. coli-derived native recombinant HMGB1 can induce inflammatory mediators (Wang et al., 1999; Li et al., 2004; Liu-Bryan and Terkeltaub, 2010; Venereau et al., 2012) and also promote chemotaxis (Taniguchi et al., 2007, 2011; Kew et al., 2012). This suggests that the recombinant HMGB1 harbors at least both: all-thiol HMGB1 and disulfide HMGB1 in unknown proportions which may be interchanged (Venereau et al., 2012; Yang et al., 2012). The recombinant HMGB1 may provoke an inflammatory response and/or migration of stem cells toward the inflamed region to promote repair and regeneration (Klune et al., 2008). Since the functional role of HMGB1 in human chondrocytes remains indefinable in human OA-affected cartilage, we performed gene expression arrays to identify the role of soluble HMGB1 in human chondrocytes, which are the governing cells in the cartilage. Primary human chondrocytes from OA-affected cartilage were incubated with 5 μg/mL of HMGB1 and assayed by gene expression arrays to mimic the paracrine effects in human chondrocytes. In the present communication, we examined the expression of chemokines, IL-8, and iNOS. The expression of each transcript was represented as a ratio of HMGB1-treated cells with uninduced cells (Wt). The changes (ratio) in the basal expression of four housekeeping transcripts in HMGB1/Wt were approximately 1.0. These housekeeping genes did not show any appreciable difference in the presence of HMGB1. Table 1 shows 17 gene transcripts that were identified based on (1) increased mRNA expression (by >10-fold) in HMGB1-treated cells as compared with untreated cells; (2) their potential role in inflammation, chemotaxis, and/or cartilage homeostasis. These included 16 different chemokines (of CC and CXC family) whose average expression was increased by 57-fold (Table 1). This included the CC (CCL2, CCL20, CCL3, CCL3L1, CCL3L3, CCL4, CCL4L1, CCL4L2, CCL5, and CCL8) chemokines. These transcripts also represented gene products representing pro and/or anti-inflammatory activity, apoptosis and anti-apoptosis functions, cell growth/differentiation, cell–cell, and cell–matrix adhesion as described in Table 1. Although most of these highly induced transcripts show pleiotropic functions of chemokines that bind to more than one receptor (Allen et al., 2007), some of the general functions of these transcripts in inflammation and cell homeostasis are described via a hyperlink to Gene Expression Atlas or Wikigene or Genecard or Wikipedia based on the maximum information available from each site. Commercially available recombinant HMGB1 (Zhang et al., 2013) of native recombinant HMGB1 used in this study (Wang et al., 1999) or chemically modified HMGB1: disulfide HMGB1 (but not all-thiol HMGB1) induced activation of the NF-kB pathway, and increased levels of cytokines and chemokines (Kew et al., 2012; Venereau et al., 2012). Our gene expression arrays using the native HMGB1 showed activation of two members of NFκB family members (Hayden and Ghosh, 2012) via increased expression of mRNA for NFκB light chain enhancers (NFκB1 by 16.3-fold and NFκB2 by 3.6-fold) as shown in Table 1. Both NFκB1 and NFκB2 promote the NFκB pathways during inflammation, UV radiation, and generation of free radicals (Hayden and Ghosh, 2012).
Table 1.
Increased Expression of Chemokine, iNOS, and NFκB Enhancer mRNA by HMGB1-Treated Human Chondrocytes
| No. | Gene symbol | Gene description (and related function) | Fold change ratio HMGB1/Wt |
|---|---|---|---|
| 1 | NOS2 | Nitric oxide synthase 2, inducible (inflammation and oxidative stress) | 104 |
| 2 | IL8 | Interleukin 8 (chemotaxis and proinflammatory) | 188 |
| 3 | CCL2 | Chemokine (C-C_motif )_ligand_2 (chemotaxis, osteoclast differentiation) | 53 |
| 4 | CCL20 | Chemokine (C-C_motif )_ligand_20 (MIP3A, pro-inflammatory) | 173 |
| 5 | CCL3 | Chemokine (C-C_motif )_ligand_3 (MIP-1α, macrophage & inflammation) | 30 |
| 6 | CCL3L1 | Chemokine (C-C_motif )_ligand_3-like_1 (pro-inflammatory activity) | 30 |
| 7 | CCL3L3 | Chemokine (C-C_motif )_ligand_3-like_3 (pro-inflammatory activity) | 30 |
| 8 | CCL4 | Chemokine (C-C_motif )_ligand_4 (MIP-1β, macrophage & inflammation) | 13 |
| 9 | CCL4L1 | Chemokine (C-C_motif )_ligand_4-like_1 (chemotaxis, pro-inflammatory) | 13 |
| 10 | CCL4L2 | Chemokine (C-C_motif )_ligand_4-like_2 (pro-inflammatory activity) | 13 |
| 11 | CCL5 | Chemokine (C-C_motif )_ligand_5 (RANTES, pro-inflammatory activity) | 228 |
| 12 | CCL8 | Chemokine (C-C_motif )_ligand_8 (MCP-2, chemotaxis, inflammation) | 57 |
| 13 | CXCL1 | chemokine_(C-X-C_motif )_ligand_1_(GRO1 growth and oncogene) | 38 |
| 14 | CXCL10 | Chemokine (C-X-C_motif )_ligand_10 (chemotaxis, colony formation) | 11 |
| 15 | CXCL2 | Chemokine (C-X-C_motif )_ligand_2 (MIP-α2, macrophage & inflammation) | 40 |
| 16 | CXCL3 | Chemokine (C-X-C_motif )_ligand_3 (GRO 3 oncogene, adhesion, migration) | 28 |
| 17 | CXCL6 | Chemokine (C-X-C_motif )_ligand_6 (chemotaxis) | 23 |
| 18 | NFκB1 | Nuclear factor NF-kappa-B p105 subunit (inflammation, UV, free radicles) | 16 |
| 19 | NFκB2 | Nuclear factor NF-kappa-B p100 subunit (inflammation, UV, phagocytosis) | 4 |
| 20 | RPL24 | Ribosomal protein-L24 (housekeeping gene) | 0.6 |
| 21 | RPS26 | Ribosomal protein-S26 (housekeeping gene) | 1.0 |
| 22 | RRP1B | Ribosomal RNA processing 1 homolog B (housekeeping gene) | 1.0 |
| 23 | LDHA | Lactate dehydrogenase A (housekeeping gene) | 0.9 |
Human chondrocytes were stimulated with 5 μg/mL of HMGB1 for 24 h, and the cells were harvested as described in the “Materials and Methods” section. The total RNA from control (Wt) cells and HMGB1-treated cells were subjected to gene expression array. The signal was normalized as described in the “Materials and Methods” section, and the data were represented as a percent increase as compared with control cells. The table shows selected transcripts (Chemokines and iNOS) up-regulated by ≥1000% as compared with normal unstimulated cells. The variations in NFκB are presented as changes in Log2. The terms in the parentheses represent the general function of the gene related to the pathophysiology of OA as described in Gene Expression Atlas or Wikigene or Genecard or Wikipedia based on the maximum information available on each site.
iNOS, inducible nitric oxide synthase.
Regulation of CXC chemokine and IL-8 by HMGB1
The human OA-affected chondrocytes showed >10-fold increased expression of CXC (CXCL1, CXCL10, CXCL2, CXCL3, and CXCL6) chemokines. Injury to cartilage is a recognized sequela of neutrophil activation and chemotaxis by IL-8 in arthritic joints (Lotz et al., 1992). Recently, in vitro studies also showed the ability of IL-8 to promote chondrocyte hypertrophy (Yammani, 2012). We have previously shown that the spontaneous release of IL-8 and nitric oxide was from human OA-affected cartilage in ex vivo condition, but unlike nitric oxide, IL-8 was not regulated by endogenous IL-1β (Attur et al., 1998), but IL-8 could be induced in vitro by IL-1β in human chondrocytes (Amin and Abramson, 1998; Haseeb and Haqqi, 2013). The mRNA expression of IL-8 was induced much higher (188-fold increase) as compared with the average expression of other chemokines shown in Table 1. We, therefore, compared the modulation of IL-8 in human chondrocytes in the presence of IL-1β and two different concentrations of HMGB1 as shown in Figure 6. Although we have previously reported significantly increased levels of IL-8 in OA-affected cartilage as compared with normal cartilage (Attur et al., 1998), there was a much higher increase in the level of IL-8 by IL-1β. However, a tenfold increase in levels of IL-8 was observed in the presence of 5 μg/mL of HMGB1 when compared with IL-1β. These observations show the ability of HMGB1 to super-induce IL-8 and other related chemokines in human chondrocytes as shown in Table 1 and Figure 6.
FIG. 6.

Regulation of IL-8 by HMGB1 and IL-1β in human chondrocytes. Human OA-affected chondrocytes were seeded in triplicates for 24 h as described in the “Materials and Methods” section. One μg/mL of IL-1β or 1–5 μg/mL of HMGB1 was added to the cells. The production of IL-8 was estimated at the end of 72 h. Data are expressed as mean±SD (n=3). The p-values for IL-8 are compared with the nonstimulated cells, where *p≤0.05.
Regulation of iNOS by HMGB1
Human OA-affected cartilage has been reported to spontaneous release NO (Abramson et al., 2001a, 2001b). Hypoxia and or oxidative stress (similar to that observed in OA-affected cartilage) is known to induce HMGB1 in nonimmune cells (Klune et al., 2008). The instinctively released HMGB1 by cells under stress can act as danger molecules by signaling neighboring cells of ongoing damage and might also trigger an inflammatory signaling cascade (Yang and Tracey, 2005; Harris et al., 2012). We, therefore, examined whether human chondrocytes were sensitive to soluble recombinant HMGB1, using nitric oxide as another model multifunctional inflammatory mediator that is also induced under stressful conditions. Nitric oxide is also known to be modulated by IL-1β and TNFα (Attur et al., 1998; Patel et al., 1998). The up-regulation of iNOS mRNA was about 104-fold in HMGB1-treated cells as compared with basal levels in untreated cells (Table 1). Primary chondrocytes from OA-affected cartilage were stimulated with IL-1β. TNFα, HMGB1, and HMGI/Y (negative control) and the levels of nitric oxide were examined at 48 h. IL-1β, TNFα, and HMGB1 (but not HMGI/Y) showed significantly increased levels of NO production as compared with nontreated or HMGI/Y-treated cells. Western blot analysis of human chondrocytes stimulated with IL-1β, TNFα, HMGB1, and HMGI/Y showed induction of 130 kDa iNOS in IL-1β, TNFα, and HMG1-stimulated cells but not in HMGI/Y or unstimulated cells. The increase in NO was consistent with the induction of iNOS mRNA and protein in human chondrocytes (Fig. 7).
FIG. 7.
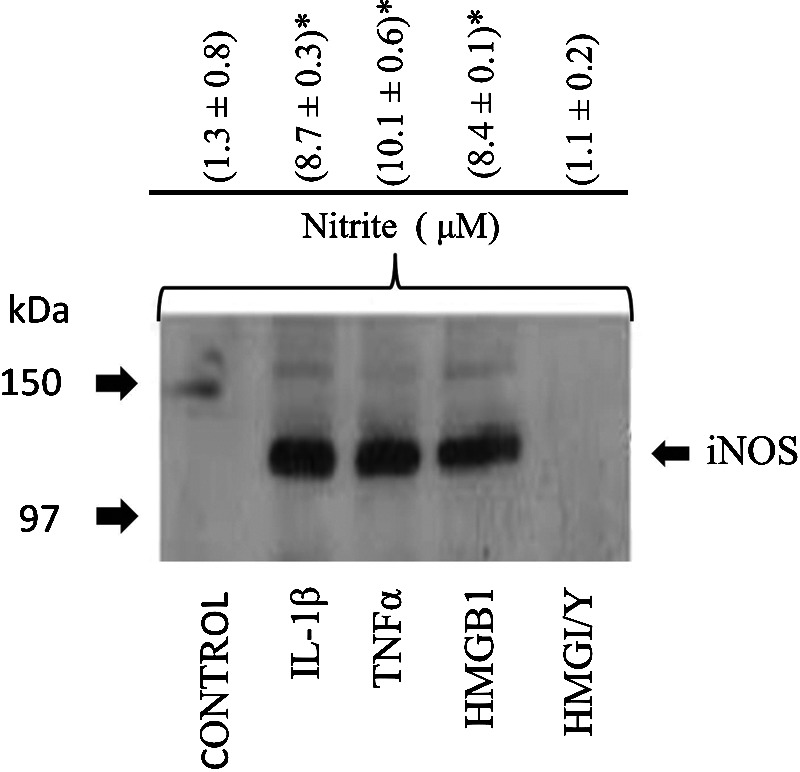
Effect of HMGB1, IL-1β, TNFα, and HMGI/Y on NO in human chondrocytes. Human OA-affected chondrocytes were grown for 24 h before they were stimulated with IL-1β (10 ng/mL), TNFα (1000 U/mL), HMGB1, or HMGI/Y (2.5 μg/mL) for an additional 24 h. Cell extracts were prepared, and an equal amount of total protein was analyzed for expression of iNOS by western blotting as previously reported. The 130 kDa iNOS is shown by an arrow. In a similar experiment, NO levels were estimated at 48 h in triplicate as shown in the upper panel. Significant levels of NO were calculated using Student's t-test. (*p≤0.01) where n=3, and the values were compared with the control cells.
Paracrine effects of HMGB1 in human OA-affected cartilage
The earlier experiments demonstrated that HMGB1 generated within the cartilage cells could seep out of the cartilage in ex vivo conditions. The paracrine effects of inflammatory mediators in synovial fluids have been reported to influence chondrocyte functions (Haseeb and Haqqi, 2013). Human arthritis-affected synovial fluid harbors 1–10 μg/mL of HMGB1 (Kokkola et al., 2002). We next examined whether the paracrine effects of HMGB1 in organ cultures of OA-affected cartilage could influence the levels of nitric oxide within the cartilage. Human OA-affected cartilage was incubated with different concentrations of IL-1β and HMGB1 in two different experiments. There was a significant increase in the accumulation of NO in IL-1β- and HMGBI-treated cartilage as compared with nontreated cartilage (Fig. 8). Similar to IL-1β, these experiments show that 1–5 μg/mL of HMGB1 can exert paracrine effects in human OA-affected cartilage.
FIG. 8.

Paracrine effect of HMGB1 on NO in human cartilage in vitro. Human OA-affected cartilage was incubated in triplicate in ex vivo conditions in two separate experiments (A, B) in the presence of IL-1β and HMGB1 for 48 hrs. The levels of NO were estimated in the form of nitrites, where n=3. The p (*)-values between control-unstimulated cells and IL-1β or HMGB1-treated cartilage was≤0.01.
Discussion
Differential expression of several members of the HMG group of transcripts was observed in normal and diseased cartilage with sterile injury. Furthermore, it unveiled the role of HMGB1 in “covert or silent” inflammation in chondrocytes and cartilage during the pathogenesis of human OA. The mRNA expression showed a distinct pattern of transcripts that correlated with the OARSI histological (grade 3–5) of the OA-affected cartilage and grade 0–1 of normal cartilage. The experimental protocol of pooling mRNA from similar clinically defined samples revealed consistency in gene expression among different cohorts. Furthermore, the log2 (two- to four-fold) changes in mRNA expression (between normal and OA-affected cartilage) could be independently validated using an alternate technology such as qPCR. Indeed, some of these differentially expressed transcripts observed in our arrays were previously reported throughout the literature (Amin et al., 1999; Pelletier et al., 2001; Attur et al., 2002a, 2002c, 2009; Haseeb and Haqqi, 2013).
The expression of HMGB1 and HMGB2 (which also binds to DNA) was tightly coordinated toward common functions in cartilage (Muller et al., 2001; Sparvero et al., 2009), but they exhibit distinct expression in normal and diseased cartilage. Both the HMGs exhibit pronounced expression in the superficial and deep zone of normal and OA-affected cartilage. The decreased expression of HMGB2 was associated with cartilage erosion and aging in mice (Taniguchi et al., 2007, 2009). Further studies in mice showed that HMGB2 expression was inversely correlated with differentiation of mesenchymal stem cells (MSC), and overexpression by transfection of HMGB2 gene into MSCs suppressed chondrogenesis (Taniguchi et al., 2009, 2011). Collectively, the age-related loss of HMGB2 or HMGB1's gain in articular cartilage may represent a trend for the decline in adult cartilage stem cell populations and cartilage erosion (Taniguchi et al., 2007, 2011, 2009; Kyostio-Moore et al., 2011). HMGN1 HMGN3, and HMGB1 were reported to reduce the compactness of the chromatin fiber and augment transcription (Bustin, 2010). In addition, HMGN1 is preferentially localized to DNAase I hypersensitive sites, promoters, functional enhancers, and transcription factor binding sites (Cuddapah et al., 2011).
The present study shows the simultaneous increase of TSP2, OGN, FGF-18, and S100 and decrease in VEGF, which are relevant to the pathophysiology of OA-affected cartilage and repair (Palmblad et al., 2007; Sparvero et al., 2009; Li et al., 2013). For example, TSP2 regulated the ratio of cartilage to bone during fracture healing (Taylor et al., 2009), and TSP2−/− mice showed abnormalities in collagen fibrils (Kyriakides et al., 1999). The OGN protein has been detected in cartilage (De Ceuninck et al., 2005), and it is involved in the regulation of collagen fibrillogenesis and linkages to the muscles (Ge et al., 2004; Tanaka et al., 2012). Indeed, OGN−/− mice showed an increased diameter of collagen fibrils (Tasheva et al., 2002). FGF-18, which signals through FGF receptor 3, promotes chondrogenesis (Ellman et al., 2013). A recombinant version of FGF-18 (sprifermin) may enter clinical trials as a potential treatment for OA (Ellman et al., 2013). VEGF couples hypertrophic cartilage remodeling, ossification, and angiogenesis during endochondral bone formation, proliferation, differentiation, and/or survival of osteoclasts, osteoblasts, and chondrocytes (Hans-Peter et al., 1999; Carlevaro et al., 2000; Murata et al., 2008). The limiting availability of VEGF may impact the healing and remodeling of human cartilage in OA.
Western blot analysis of human cartilage extracts confirmed the presence of HMGB1 in normal and OA-affected cartilage (unpublished data), similar to those observed in monkeys (Loeser et al., 2005). In view of the expression and secretion of HMGB1 in normal and OA-affected cartilage by relatively healthy cells, lack of apoptotic cells in normal cartilage, presence of HMGB1 in normal cartilage, and the effects of IL-1β on HMGB1 accumulation in OA-affected cartilage support the assertion that the major amount of HMGB1 was most likely released by active secretion and not apoptotic or dead cells. The increased level of HMGB1-positive cells in the deep zone of human OA-affected cartilage was similar to up-regulated HMGB1 and S100 (at week 20) in cartilage of animal models of OA (Kyostio-Moore et al., 2011). In mice, HMGB1-positive cells (hypertrophic chondrocytes) in the deep zone also secreted HMGB1 in the cartilage milieu in mice (Taniguchi et al., 2007). Likewise, distinct expression of HMGB1 has been reported in other pathophysiological conditions with sterile injury such as colitis, ischemia, sepsis, endotoxemia, and systemic lupus erythematous (Sparvero et al., 2009; Kyostio-Moore et al., 2011; Harris et al., 2012; Kang et al., 2013).
There was a similarity in HMGB1 expression in human (this study), mice, and bovine cartilage (Heinola et al., 2010): (1) Nucleosomal staining was predominant in chondrocytes from normal cartilage as compared with OA-affected cartilage; (2) OA-affected cartilage showed increased intensity of extra-/pericellular deposition of HMGB1; (3) There was also an increased staining of HMGB1 in the subchondral bone plate and osteophyte formation region of bovine cartilage similar to human cartilage (Heinola et al., 2010); (4) Similar to OA-affected bovine and human cartilage, arthritis models in mice showed increased expression of HMGB1 in the cartilage; (5) cytosolic HMGB1 was observed in hypertrophic chondrocytes, growth plate region in mice (Taniguchi et al., 2007), and human OA-affected cartilage; (6) activated murine (Johnson et al., 2003; Liu-Bryan and Terkeltaub, 2010) and human chondrocytes released a 29 kDa HMGB1; and (7) HMGB1 from murine chondrocyte (Kyostio-Moore et al., 2011) have been reported to function as a chemoattractant for osteophytes, osteoblasts, and endochondral ossification (Taniguchi et al., 2007). HMGB1 in the extracellular milieu in the deep region of human OA-affected cartilage may promote chemotaxis by forming a complex with CXCL12 and signaling via CXCR4, similar to chemokine-like function of HMGB1 in an inflammatory bone marrow microenvironment of patients with chronic idiopathic neutropenia (Velegraki et al., 2012). (8) HMGB1 induces MMP-3 and -13 in murine (Liu-Bryan and Terkeltaub, 2010) and human chondrocytes (Unpublished data); (9) HMGB1 or HMGB2 gene deletion (or decreased expression) showed decreased chondrogenesis, reduced superficial zone cellularity, and early onset of OA similar to human OA-affected cartilage (Taniguchi et al., 2009, 2011). (10) Activated human and mice synovial cells show increased levels of HMGB1 (García-Arnandis et al., 2001, Palmblad et al., 2007).
The mechanism of action of HMGB1 was compared with murine models of arthritis and bovine cartilage. Briefly, a single injection (5 μg) of intra-articular HMGB1 in the knee joint of rodents induced HMGB1, which preceded the clinical onset of the diseases, resulted in moderate synovitis that lingered for about 28 days, with increased TNFα and IL-1β at this stage of the diseases (Pullerits et al., 2003; Andersson and Harris, 2004). Furthermore, an injection of HMGB1 in joints of TNFα−/− mice induced arthritis similar to TNFα+/+ mice (Pullerits et al., 2008). It should be noted that the lack of HMGB1 expression in mice had detrimental effects on homeostasis of bone and cartilage (Taniguchi et al., 2007). Seol et al. (2012) showed that injury to bovine cartilage and chondrocyte death stimulated the emergence of chondrogenic progenitor cells via HMGB1 release and RAGE-mediated chemotaxis. This process could be inhibited by Glycrrhizin, a chelator of HMGB1 and anti-HMGB1 antibodies. Thus, inflammatory cell recruitment (by all thio-HMGB1) and activation of inflammatory mediators (by disulfide HMGB1) may depend on mutually exclusive (and interchangeable) redox forms of HMGB1: These different redox forms of HMGB1 may function via different mechanisms, time lines of secretions by the same cell, binding to different receptors, and interact with different matrix proteins (such as DNA, lipids, IL-1β, CXC chemokines, and stromal cell-derived factors) in the presence of other inflammatory mediators and redox state of the tissue (Tang et al., 2012, Janko et al., 2014). It is quite possible that the release of dynamic redox-regulated HMGB1 may contribute to an orderly orchestrated response to early inflammatory response in cartilage, recruitment of progenitors of chondrocytes, and subsequent resolution of inflammation (by the oxidized HMGB1). The potential role of different redox form(s) of HMGB1 operating in the complex interplay of chondrogenesis in normal and OA-affected cartilage remains elusive. However, the recombinant HMGB1 previously reported to induce inflammatory mediators in macrophages (Wang et al., 1999) also induced NFκB, chemokines, and nitric oxide in cartilage and chondrocytes in both in vitro and ex vivo conditions.
DAMPs (including S100A4, S100A10, S100A11, LEF1, and SRY) signal via the RAGE receptors and/or Wnt pathway. S100A4 is up-regulated in synovial tissue and fluids of OA and RA with increased secretion of MMP-13 (Yammani, 2012). In mice, S100A4−/− animals showed reduced joint inflammation, impaired bone resorption and regulation of osteoclast, increased cartilage and bone destruction, and decreased expression of MMPs (Bian, 2011; Erlandsson et al., 2013). S100A10 expression decreased the production of TNFα, IL-1β, and IL-10 in chondrocytes (O'Connell et al., 2010; Song et al., 2012). The S100A11 induces chondrocyte hypertrophy and matrix catabolism (Yammani, 2012). Thus, similar to the HMGs, different species of S100 proteins have a critical role in cartilage inflammation and resolution.
Our observations suggest that there are relatively more similarities in expression and possibly common functions shared by DAMPs in man and mouse cartilage and joint tissues. For example, (1) increased levels of the S100A family of proteins stimulate chondrocyte hypertrophy and catabolic activity (Cecil and Terkeltaub, 2008; Van Lent et al., 2008; Bian, 2011; Yammani, 2012); (2) Age-related loss of HMGB2 is associated with reduced cellularity and onset of OA. HMGB2 and LEF-1 enhance Wnt signaling and promote superficial zone chondrocyte survival in mice. Loss of the HMGB2-Wnt signaling interaction is one of the mechanisms in aging-related cartilage (Taniguchi et al., 2007, 2009, 2011); (3) SRY-related high-mobility-group (HMG) box transcription factors (SOXs) exhibit a distinct role in different stages of chondrogenic differentiation and survival (Ikeda et al., 2005); (4) Similar to HMGB1, Spondin 1 was increased in OA-affected cartilage. It regulates the differentiation of chondrocytes and endochondral bone formation in mice (Palmer et al., 2010).
Several chemokines that were up-regulated (>by 1000%) by HMGB1 in human chondrocytes not only bind to more than receptor, but also influence cartilage homeostasis (Borzi et al., 2004; Haringman et al., 2004). Briefly, (1) CCL20 is required for osteoclast proliferation, differentiation, osteolytic bone lesion, and subchondral bone formation of arthritis-affected cartilage (Lisignoli et al., 2007, 2009); (2) Chondrocyte hypertrophy can be induced by GRO1 (Pulsatelli et al., 1999; Borzi et al., 2002; Cristino et al., 2008); (3) CXCL10 is noted to promote osteoclast formation (Grassi et al., 2003) and recruits subchondral bone mesenchymal progenitor cells (Endres et al., 2010); (4) CXCL1 is known to induce hypertrophic differentiation, apoptosis, and calcification in chondrocytes (Merz et al., 2003); (5) CCL5 up-regulated NO, IL-6, MMP-1, −3, and -13 production in cartilage (Alaaeddine et al., 2001; Borzi et al., 2004); and (6) CXCL12 can form a complex with HMGB1 and signal via the CXCR4 receptor to recruit cells to damaged tissue (Schiraldi et al., 2012). IL-8 can induce S100A11, which, in turn, can induce chondrocyte hypertrophy (Yammani, 2012). These observations suggest that some of the homeostatic functions in cartilage and subchondral bone by HMGB1 may be mediated via a complex interaction of chemokines.
The paracrine effects of HMGB1 may further activate macrophages and neutrophils in arthritis-affected joints (Yang and Tracey, 2005; Yang et al., 2005). Although low and variable levels of nitric oxide are essential during endochondral ossification, catabolic and homeostatic activity in human cartilage (Amin et al., 1999; Wang et al., 2011), high levels of NO have detrimental effects in cartilage (Amin and Abramson, 1998). Granted that HMGB1 can bind to a multitude of known receptors in chondrocytes, the functional receptor for HMGB1 in human OA-affected cartilage remains elusive.
In summary, our observations bolster the complex role of these alarmins, DAMPs, and HMGs as innate and endogenous regulators of injury-induced sterile inflammation in the cartilage and joints, their collaborative role in chondrogenesis, hypertrophy, and maintenance of the homeostasis in mice and human cartilage as shown in Figure 9. There is significant focus on the therapeutic intervention of HMGB1 (Pulsatelli et al., 2013). A drug such as Atrovastatin (Lipitor) reduces increased levels of hyperlipidemia and HMGB1 in the serum (Jin et al., 2012). Therapeutic benefits through HMGB1-directed mechanisms involve HMGB1 inhibitory ligands such as Toll-like receptor antagonists, RAGE antagonists, and alpha7 nicotinic acetylcholine receptor agonists (Lamore et al., 2010). Antibodies that neutralize HMGB1 are known to confer protection against tissue damage and injury in joints (Yang and Tracey, 2005; Sparvero et al., 2009). Anti-HMGB1 therapy was comparable to TNF-blocking regiments in animal studies of arthritis (Andersson and Harris, 2004) and, thus, shows significant promise.
FIG. 9.

The doppelganger life of HMGB1 in OA-affected cartilage: architectural factor, extracellular signaling, sterile inflammation, and cartilage homeostasis. The up-regulation of mRNA for HMGB1, HMGN1, and HMGN3 and a decrease in expression of HMGB2 in human OA-affected cartilage exhibited the interaction among HMG. HMGB1 is observed within the nucleus (N-HMGB1) of all nucleated cells. It was also observed in chondrocytes. There are typically ∼106 HMGB1 molecules per cell. The HMGB1 protein was observed in the cytosol (C-HMGB1) of chondrocytes and extracellular milieu (E-HMGB1), where it can adopt its role as a cytokine (Yang and Tracey, 2005; Yang et al., 2005). The extracellular HMGB1 (1) may bind to its receptor (red) and induce signal transduction (Wahamaa et al., 2007; Sparvero et al., 2009). HMGB1 exhibited superinduction of the CC and CXC family of chemokines, IL-8 and iNOS gene expression (by ≥1000%), as shown throughout this study. (2) may interact with matrix and or chemokines (CXCL12) and exhibit other functions (Diener et al., 2013); (3) be released outside the cartilage (ECA-HMGB1), where it can enter the synovial fluid in the joints (Kokkola et al., 2002). ECA-HMGB1 may (1) activate other inflammatory cells and/ or (2) might also penetrate back into the cartilage and activate the HMGB1 receptors on chondrocytes (Klune et al., 2008). Increased levels of HMGN1 and HMGN3 facilitate gene transcription (Bustin, 2010) and superinduction of chemokines, iNOS and IL-8, in OA-affected cartilage. NO and IL-8 can enter the synovial fluid. IL-8 can attract inflammatory cells. Most of the HMGB1-induced genes are involved in pro- or anti-inflammatory activity, chemotaxis and cell/matrix adhesion, matrix metabolism, and cartilage homeostasis as referenced in Table 1. Increased levels of nitric acid and oxidative stress have long been recognized to alter catabolism in OA-affected cartilage, degradation, and cartilage repair (Amin and Abramson, 1998; Abramson et al., 2001a). This oxidative environment in OA-affected cartilage may create an environment for post-translational modification of HMGB1 depending on its oxidation state within the cartilage (Janko et al., 2014) as described in some in vitro experimental settings. Oxidation of di-sulfides of HMGB1 has been reported to favor an inflammatory response, and terminal oxidation of HMGB1 may promote inflammation resolution (Yang et al., 2012; Diener et al., 2013; Janko et al., 2014). The decreasing level of HMGB2 is well documented in arthritis-affected cartilage and is shown throughout this study. HMGB2 plays a critical role in chondrocyte survival and cartilage homeostasis (Taniguchi et al., 2007; Taniguchi et al., 2009). The human OA-affected cartilage also showed increased expression of other DAMPs such as S100A4, S100A10, and S100A11. They have been reported to have catabolic and proinflammatory activity in chondrocytes (Yammani, 2012). Color images available online at www.liebertpub.com/dna
Supplementary Material
Acknowledgments
This project was supported in part by NIH grants AR 47206-03 and Yamanuchi Pharmaceuticals. The authors would like to thank Drs. Kevin Tracy and M. Wisniewski for the gifts of recombinant HMGB1 and HMGI/Y, respectively. Anti-HMGB1 antibodies were provided by Dr. K. Tracy. We would like to thank the YPCL scientists for their input.
Disclosure Statement
No competing financial interests exist.
References
- Abramson S.B., Amin A.R., Clancy R.M., and Attur M. (2001a). The role of nitric oxide in tissue destruction. Best practice & research. Clin Rheumatol 15,831–845 [DOI] [PubMed] [Google Scholar]
- Abramson S.B., Attur M., Amin A.R., and Clancy R. (2001b). Nitric oxide and inflammatory mediators in the perpetuation of osteoarthritis. Curr Rheumatol Rep 3,535–541 [DOI] [PubMed] [Google Scholar]
- Adjaye J., Huntriss J., Herwig R., BenKahla A., Brink T.C., Wierling C., Hultschig C., Groth D., Yaspo M.L., Picton H.M., Gosden R.G., and Lehrach H. (2005). Primary differentiation in the human blastocyst: comparative molecular portraits of inner cell mass and trophectoderm cells. Stem Cells 23,1514–1525 [DOI] [PubMed] [Google Scholar]
- Alaaeddine N., Olee T., Hashimoto S., Creighton-Achermann L., and Lotz M. (2001). Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis Rheum 44,1633–1643 [DOI] [PubMed] [Google Scholar]
- Allen S.J., Crown S.E., and Handel T.M. (2007). Chemokine: receptor structure, interactions, and antagonism. Ann Rev Immunol 25,787–820 [DOI] [PubMed] [Google Scholar]
- Amin A.R., and Abramson S.B. (1998). The role of nitric oxide in articular cartilage breakdown in osteoarthritis. Curr Opin Rheumatol 10,263–268 [DOI] [PubMed] [Google Scholar]
- Amin A.R., Attur M., and Abramson S.B. (1999). Nitric oxide synthase and cyclooxygenases: distribution, regulation, and intervention in arthritis. Curr Opin Rheumatol 11,202–209 [DOI] [PubMed] [Google Scholar]
- Amin A.R., Attur M., Patel R.N., Thakker G.D., Marshall P.J., Rediske J., Stuchin S.A., Patel I.R., and Abramson S.B. (1997). Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J Clin Invest 99,1231–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin A.R., Attur M.G., Thakker G.D., Patel P.D., Vyas P.R., Patel R.N., Patel I.R., and Abramson S.B. (1996). A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci U S A 93,14014–14019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin A.R., Thompson S., and Amin S. (2007). Future of Genomics in Diagnosis of Human Arthritis: the hype, hope and metamorphosis for tomorrow. Future Med 2,4–7 [Google Scholar]
- Andersson U., and Harris E. (2004). HMGB1 is a potent trigger of arthritis. J Intern Med 255,344–350 [DOI] [PubMed] [Google Scholar]
- Attur M.G., Dave M., Akamatsu M., Katoh M., and Amin A.R. (2002a). Osteoarthritis or osteoarthrosis: the definition of inflammation becomes a semantic issue in the genomic era of molecular medicine. Osteoarthritis Cartilage 10,1–4 [DOI] [PubMed] [Google Scholar]
- Attur M.G., Dave M., Cipolletta C., Kang P., Goldring M.B., Patel I.R., Abramson S.B., and Amin A.R. (2000). Reversal of autocrine and paracrine effects of interleukin 1 (IL-1) in human arthritis by type II IL-1 decoy receptor. Potential for pharmacological intervention. J Biol Chem 275,40307–40315 [DOI] [PubMed] [Google Scholar]
- Attur M.G., Dave M.N., Leung M.Y., Cipolletta C., Meseck M., Woo S.L., and Amin A.R. (2002b). Functional genomic analysis of type II IL-1beta decoy receptor: potential for gene therapy in human arthritis and inflammation. J Immunol 168,2001–2010 [DOI] [PubMed] [Google Scholar]
- Attur M.G., Dave M.N., Stuchin S., Kowalski A.J., Steiner G., Abramson S.B., Denhardt D.T., and Amin A.R. (2001). Osteopontin: an intrinsic inhibitor of inflammation in cartilage. Arthritis Rheum 44,578–584 [DOI] [PubMed] [Google Scholar]
- Attur M.G., Dave M.N., Tsunoyama K., Akamatsu M., Kobori M., Miki J., Abramson S.B., Katoh M., and Amin A.R. (2002c). “A system biology” approach to bioinformatics and functional genomics in complex human diseases: arthritis. Curr Issues Mol Biol 4,129–146 [PubMed] [Google Scholar]
- Attur M.G., Palmer G.D., Al-Mussawir H.E., Dave M., Teixeira C.C., Rifkin D.B., Appleton C.T., Beier F., and Abramson S.B. (2009). F-spondin, a neuroregulatory protein, is up-regulated in osteoarthritis and regulates cartilage metabolism via TGF-beta activation. FASEB J 23,79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attur M.G., Patel I.R., Patel R.N., Abramson S.B., and Amin A.R. (1998). Autocrine production of IL-1 beta by human osteoarthritis-affected cartilage and differential regulation of endogenous nitric oxide, IL-6, prostaglandin E2, and IL-8. Proc Assoc Am Phys 110,65–72 [PubMed] [Google Scholar]
- Bian L. (2011). The role of S100A4 protein as a regulator of inflammation and bone metabolism in experimental arthritis. PhD thesis. Dept of Rheumatology and Inflammtion Research. The Sahlgtrenska Academy. University of Gothenburg; Gothenburg. Sweden [Google Scholar]
- Borzi R.M., Mazzetti I., Magagnoli G., Paoletti S., Uguccioni M., Gatti R., Orlandini G., Cattini L., and Facchini A. (2002). Growth-related oncogene alpha induction of apoptosis in osteoarthritis chondrocytes. Arthritis Rheum 46,3201–3211 [DOI] [PubMed] [Google Scholar]
- Borzi R.M., Mazzetti I., Marcu K.B., and Facchini A. (2004). Chemokines in cartilage degradation. Clin Orthop Relat Res, 427,S53–S61 [DOI] [PubMed] [Google Scholar]
- Bustin M. (2010). High mobility group proteins. Biochim Biophys Acta 1799,1–2 [DOI] [PubMed] [Google Scholar]
- Carlevaro M.F., Cermelli S., Cancedda R., and Cancedda F. (2000). Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci 113,59–69 [DOI] [PubMed] [Google Scholar]
- Cecil D.L., Johnson K., Rediske J., Lotz M., Schmidt A.M., and Terkeltaub R. (2005). Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol 175,8296–8302 [DOI] [PubMed] [Google Scholar]
- Cecil D.L, and Terkeltaub R. (2008). Transamidation by transglutaminase 2 transforms S100A calgranulin into a procatabolic cytokine for chondrocytes J Immunol 180, 8378–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.Y., and Nuñez G. (2010). Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10,826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristino S., Piacentini A., Manferdini C., Codeluppi K., Grassi F., Facchini A., and Lisignoli G. (2008). Expression of CXC chemokines and their receptors is modulated during chondrogenic differentiation of human mesenchymal stem cells grown in three-dimensional scaffold: evidence in native cartilage. Tissue Eng Part A 14,97–105 [DOI] [PubMed] [Google Scholar]
- Cuddapah S., Schones D.E., Cui K., Roh T.Y., Barski A., Wei G., Rochman M., Bustin M., and Zhao K. (2011). Genomic profiling of HMGN1 reveals an association with chromatin at regulatory regions. Mol Cell Biol 31,700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave M., and Amin A.R. (2013). Yin-Yang regulation of prostaglandins and nitric oxide by PGD2 in human arthritis: reversal by celecoxib. Immunol Lett 152,47–54 [DOI] [PubMed] [Google Scholar]
- De Ceuninck F., Marcheteau E., Berger S., Caliez A., Dumont V., Raes M., Anract P., Leclerc G., Boutin J.A., and Ferry G. (2005). Assessment of some tools for the characterization of the human osteoarthritic cartilage proteome. J Biomol Tech 16,256–265 [PMC free article] [PubMed] [Google Scholar]
- De Jonge H.J., Fehrmann R.S., de Bont E.S., Hofstra R.M., Gerbens F., Kamps W.A., de Vries E.G., van der Zee A.G., te Meerman G.J., and ter Elst A., (2007). Evidence based selection of housekeeping genes. PloS One 2, e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener K.R., Dasooqi N.A., Lousberg E.L., Hayball J.D. (2013). Immunology and Cell Biology 91, 443–450 [DOI] [PubMed] [Google Scholar]
- Ellman M.B., Yan D., Ahmadinia K., Chen D., An H.S, and Im H.J., (2013). Fibroblast growth factor control of cartilage homeostasis. J Cell Biochem 114,735–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M., Andreas K., Kalwitz G., Freymann U., Neumann K., Ringe J., Sittinger M., Haupl T., and Kaps C., (2010). Chemokine profile of synovial fluid from normal, osteoarthritis and rheumatoid arthritis patients: CCL25, CXCL10 and XCL1 recruit human subchondral mesenchymal progenitor cells. Osteoarthritis Cartilage 18,1458–1466 [DOI] [PubMed] [Google Scholar]
- Erlandsson M.C., Svensson M.D., Jonsson I.M., Bian L., Ambartsumian N., Andersson S., Peng Z., Vaaraniemi J., Ohlsson C., Andersson K.M., and Bokarewa M.I. (2013). Expression of metastasin S100A4 is essential for bone resorption and regulates osteoclast function. Biochim Biophys Acta 1833,2653–2663 [DOI] [PubMed] [Google Scholar]
- García-Arnandis I., Guillén M.I., Gomar F., Pelletier J.P., Martel-Pelletier J.M., and Alcaraz M.J. (2001). High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1β in osteoarthritic synoviocytes. Arthritis Rheum 44,1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge G., Seo N.S., Liang X., Hopkins D.R., Höök M., and Greenspan D.S. (2004). Bone morphogenetic protein-1/tolloid-related metalloproteinases process osteoglycin and enhance its ability to regulate collagen fibrillogenesis. J Biol Chem 279,41626–41633 [DOI] [PubMed] [Google Scholar]
- Goldstein R.S., Bruchfeld A., Yang L., Qureshi A.R., Gallowitsch-Puerta M., Patel N.B., Huston B.J., Chavan S., Rosas-Ballina M., Gregersen P.K., Czura C.J., Sloan R.P., Sama A.E., and Tracey K.J. (2007). Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med 13,210–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi F., Piacentini A., Cristino S., Toneguzzi S., Cavallo C., Facchini A., and Lisignoli G. (2003). Human osteoclasts express different CXC chemokines depending on cell culture substrate: molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem Cell Biol 120,391–400 [DOI] [PubMed] [Google Scholar]
- Hans-Peter G., Thiennu H., Ryan A.M., Kowalski J., Werb Z., and Ferrara N. (1999). EGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 5,623–628 [DOI] [PubMed] [Google Scholar]
- Haringman J.J., Ludikhuize J., and Tak P.P. (2004). Chemokines in joint disease: the key to inflammation? Ann Rheum Dis 63,1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H.E., Andersson U., and Pisetsky D.S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 8,195–202 [DOI] [PubMed] [Google Scholar]
- Haseeb A., and Haqqi T.M. (2013). Immunopathogenesis of osteoarthritis. Clin Immunol 146,185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.S, and Ghosh S. (2012). NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26,203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinola T., Kouri V.P., Clarijs P., Ciferska H., Sukura A, Salo J., and Konttinen YT.2010. High mobility group box-1 (HMGB-1) in osteoarthritic cartilage. Clin Exp Rheumatol 28, 511–518 [PubMed] [Google Scholar]
- Ikeda T., Kawaguchi H., Kamekura S., Ogata N., Mori Y., Nakamura K., Ikegawa S., and Chung U. (2005). Distinct roles of Sox5, Sox6, and Sox9 in different stages of chondrogenic differentiation. Bone Miner Metab 23,237–240 [DOI] [PubMed] [Google Scholar]
- Janko C., Filipović M., Munoz L.E., Schorn C., Schett G., Ivanović-Burmazović I., and Herrmann M. (2014). Redox Modulation of HMGB1-Related Signaling. Antioxid Redox Signal 20,1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D., Wu Y., Zhao L., Guo J., Zhang K., and Chen Z. (2012). Atorvastatin reduces serum HMGB1 levels in patients with hyperlipidemia. Exp Ther Med 4,1124–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.A., van Etten D., Nanda N., Graham R.M., and Terkeltaub R.A. (2003). Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J Biol Chem 278,18824–18832 [DOI] [PubMed] [Google Scholar]
- Kang R., Tang D., Schapiro N.E., Loux T., Livesey K.M., Billiar T.R., Wang H., Van Houten B., Lotze M.T., and Zeh H.J. (2013). The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 33,567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew R.R., Penzo M., Habiel D.M., and Marcu K.B. (2012). The IKKα-dependent NF-κB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol 188,2380–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.A., Lee Y.J., Seong S.C., Choe K.W., and Song W. (2000). Apoptotic chondrocyte death in human osteoarthritis. J Rheumatol 27,455–462 [PubMed] [Google Scholar]
- Klune J.R., Dhupar R., Cardinal J., Billiar T.R., and Tsung A. (2008). HMGB1: Endogenous danger signaling. Mol Med 14,476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola R., Sundberg E., Ulfgren A.K., Palmblad K., Li J., Wang H., Ulloa L., Yang H., Yan X.J., Furie R., Chiorazzi N., Tracey K.J., Andersson U., and Harris H.E. (2002). High mobility group box chromosomal protein 1: A novel proinflammatory mediator in synovitis. Arthritis Rheum 46,2598–2603 [DOI] [PubMed] [Google Scholar]
- Kyostio-Moore S., Nambiar B., Hutto E., Ewing P.J., Piraino S., Berthelette P., Sookdeo C., Matthews G., and Armentano D. (2011). STR/ort mice, a model for spontaneous osteoarthritis, exhibit elevated levels of both local and systemic inflammatory markers. Comp Med 61,346–355 [PMC free article] [PubMed] [Google Scholar]
- Kyriakides T.R., Jessica W.Y., and Bornstein P. (1999). Wound Healing in Mice with a disruption of the Thrombospondin 2 Gene. J Invest Dermatol 113,782–787 [DOI] [PubMed] [Google Scholar]
- Lamore S.D., Cabello C.M., and Wondrak G.T. (2010). HMGB1-directed drug discovery targeting cutaneous inflammatory dysregulation. Curr Drug Metab 11,250–265 [DOI] [PubMed] [Google Scholar]
- Li G., Liang X., and Lotze M.T. (2013). HMGB1: the central cytokine for all lymphoid cells. Front Immunol 4,68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Wang H., Mason J.M., Levine J., Yu M., Ulloa L., Czura C.J., Tracey K.J., and Yang H. (2004). Recombinant HMGB1 with cytokine-stimulating activity. J Immunol Methods 289,211–223 [DOI] [PubMed] [Google Scholar]
- Lisignoli G., Manferdini C., Codeluppi K., Piacentini A., Grassi F., Cattini L., Filardo G., and Facchini A. (2009). CCL20/CCR6 chemokine/receptor expression in bone tissue from osteoarthritis and rheumatoid arthritis patients: different response of osteoblasts in the two groups. J Cell Physiol 221,154–160 [DOI] [PubMed] [Google Scholar]
- Lisignoli G., Piacentini A., Cristino S., Grassi F., Cavallo C., Cattini L., Tonnarelli B., Manferdini C., and Facchini A. (2007). CCL20 chemokine induces both osteoblast proliferation and osteoclast differentiation: Increased levels of CCL20 are expressed in subchondral bone tissue of rheumatoid arthritis patients. J Cell Physiol 210,798–806 [DOI] [PubMed] [Google Scholar]
- Liu-Bryan R., and Terkeltaub R. (2010). Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum 62,2004–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser R.F., Yammani R.R., Carlson C.S., Chen H., Cole A., Im H.J., Bursch L.S., and Yan S.D. (2005). Arthritis Rheum 52, 2376–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz M., Terkeltaub R., and Villiger P.M. (1992). Cartilage and joint inflammation. Regulation of IL-8 expression by human articular chondrocytes. J Immunol 148,466–473 [PubMed] [Google Scholar]
- Merz D., Liu R., Johnson K., and Terkeltaub R. (2003). IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol 171,4406–4415 [DOI] [PubMed] [Google Scholar]
- Muller S., Scaffidi P., Degryse B., Bonaldi T., Ronfani L., Agresti A., Beltrame M., and Bianchi M.E. (2001). New EMBO members' review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J 20,4337–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M., Yudoh K., and Masuko K. (2008). The potential role of vascular endothelial growth factor (VEGF) in cartilage: how the angiogenic factor could be involved in the pathogenesis of osteoarthritis? Osteoarthritis Cartilage 16,279–286 [DOI] [PubMed] [Google Scholar]
- O'Connell P.A., Surette A.P., Liwski R.S., Svenningsson P., and Waisman D.M. (2010). S100A10 regulates plasminogen-dependent macrophage invasion. Blood 116,1136–1146 [DOI] [PubMed] [Google Scholar]
- Palmblad K., Sundberg E., Diez M., Soderling R., Aveberger A.C., Andersson U., and Harris H.E. (2007). Morphological characterization of intra-articular HMGB1 expression during the course of collagen-induced arthritis. Arthritis Res Ther 9, R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer G.D., Piton A.H., Thant L.M., Oliveira S.M., D'Angelo M., Attur M.G., Abramson S.B., and Teixeira C.C. (2010). F-spondin (Spondin 1) regulates chondrocyte terminal differentiation and endochondral bone formation. J Orthop Res 28,1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel I.R., Attur M.G., Patel R.N., Stuchin S.A., Abagyan R.A., Abramson S.B., and Amin A.R. (1998). TNF-alpha convertase enzyme from human arthritis-affected cartilage: isolation of cDNA by differential display, expression of the active enzyme, and regulation of TNF-alpha. J Immunol 160,4570–4579 [PubMed] [Google Scholar]
- Pelletier J.P., Martel-Pelletier J., and Abramson S.B. (2001). Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum 44,1237–1247 [DOI] [PubMed] [Google Scholar]
- Perez-Llamas C., and Lopez-Bigas N. (2011). Gitools: analysis and visualisation of genomic data using interactive heat-maps. PloS One 6, e19541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritzker K.P., Gay S., Jimenez S.A., Ostergaard K., Pelletier J.P., Revell P.A., Salter D., and van den Berg W.B. (2006). Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14,13–29 [DOI] [PubMed] [Google Scholar]
- Pullerits R., Jonsson I.M., Kollias G., and Tarkowski A. (2008). Induction of arthritis by high mobility group box chromosomal protein 1 is independent of tumor necrosis factor signaling. Arthritis Res Ther 10, R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullerits R., Jonsson I.M., Verdrengh M., Bokarewa M., Andersson U., Erlandsson-Harris H., and Tarkowski A. (2003). High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum 48,1693–1700 [DOI] [PubMed] [Google Scholar]
- Pulsatelli L., Addimanda O., Brusi V., Pavloska B., and Meliconi R. (2013). New findings in osteoarthritis pathogenesis: therapeutic implications. Ther Adv Chronic Dis 4,23–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsatelli L., Dolzani P., Piacentini A., Silvestri T., Ruggeri R., Gualtieri G., Meliconi R., and Facchini A. (1999). Chemokine production by human chondrocytes. J Rheumatol 26,1992–2001 [PubMed] [Google Scholar]
- Rao J., Elliott M.R., Leitinger N., Jensen R.V., Goldberg J.B., and Amin A.R. (2011). RahU: an inducible and functionally pleiotropic protein in Pseudomonas aeruginosa modulates innate immunity and inflammation in host cells. Cell Immunol 270,103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiraldi M., Raucci A., Munoz L.M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A., Thelen M., Varani L., Mellado M., Proudfoot A., Bianchi M.E., and Uguccioni M. (2012). HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 209,551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol D., McCabe D.J., Choe H., Zheng H., Yu Y., Jang K., Walter M.W., Lehman A.D., Ding L., Buckwalter J.A., and Martin J.A. (2012). Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum 64,3626–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims G.P., Rowe D.C., Rietdijk S.T., Herbst R., and Coyle A.J. (2010). HMGB1 and RAGE in inflammation and cancer. Ann Rev Immunol 28,367–388 [DOI] [PubMed] [Google Scholar]
- Smyth G.K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3. [DOI] [PubMed] [Google Scholar]
- Song C., Zhou X., Dong Q., Fan R., Wu G., Ji B., Meng Q., and Zheng M. (2012). Regulation of inflammatory response in human chondrocytes by lentiviral mediated RNA interference against S100A10. Inflamm Res 61,1219–1227 [DOI] [PubMed] [Google Scholar]
- Sparvero L.J., Asafu-Adjei D., Kang R., Tang D., Amin N., Im J., Rutledge R., Lin B., Amoscato A.A., Zeh H.J., and Lotze M.T. (2009). RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Matsumoto E., Higashimaki Y., Katagiri T., Sugimoto T., Seino S., and Kaji H. (2012). Role of osteoglycin in the linkage between muscle and bone. J Biol Chem 287,11616–11628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Billiar T.R., and Lotze M.T. (2012). A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med 18,1360–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N., Carames B., Hsu E., Cherqui S., Kawakami Y., and Lotz M. (2011). Expression patterns and function of chromatin protein HMGB2 during mesenchymal stem cell differentiation. J Biol Chem 286,41489–41498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N., Carames B., Ronfani L., Ulmer U., Komiya S., Bianchi M.E., and Lotz M. (2009). Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc Natl Acad Sci U S A 106,1181–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N., Yoshida K., Ito T., Tsuda M., Mishima Y., Furumatsu T., Ronfani L., Abeyama K., Kawahara K., Komiya S., Maruyama I., Lotz M., Bianchi M.E., and Asahara H. (2007). Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol 27,5650–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasheva E.S., Koester A., Paulsen A.Q., Garrett A.S., Boyle D.L., Davidson H.J., Song M., Fox N., and Conrad G.W. (2002). Mimecan/osteoglycin-deficient mice have collagen fibril abnormalities. Mol Vis 31,407–415 [PubMed] [Google Scholar]
- Taylor D.K., Meganck J.A., Terkhorn S., Rajani R., Naik A., O'Keefe R.J., Goldstein S.A., and Hankenson K.D. (2009). Thrombospondin-2 influences the proportion of cartilage and bone during fracture healing. J Bone Miner Res 24,1043–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonaviciute V., Meister S., Fürnrohr B.G., Frey B., Gückel E., Schett G., Herrmann M., and Voll R.E. (2009). Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity 42,305–307 [DOI] [PubMed] [Google Scholar]
- Van Lent P.L., Grevers L.C., Blom A.B., Arntz O.J., van de Loo F.A., van der Kraan P., Abdollahi-Roodsaz S., Srikrishna G., Freeze H., Sloetjes A., Nacken W., Vogl T., Roth J, and van den Berg WB. (2008). Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum 58,3776–3787 [DOI] [PubMed] [Google Scholar]
- Velegraki M., Koutala H., Tsatsanis C., and Papadaki H.A. (2012). Increased levels of the high mobility group box 1 protein sustain the inflammatory bone marrow microenvironment in patients with chronic idiopathic neutropenia via activation of toll-like receptor 4. J Clin Immunol 32,312–322 [DOI] [PubMed] [Google Scholar]
- Venereau E., Casalgrandi M., Schiraldi M., Antoine D.J., Cattaneo A., De Marchis F., Liu J., Antonelli A., Preti A., Raeli L., Shams S.S., Yang H., Varani L., Andersson U., Tracey K.J., Bachi A., Uguccioni M., and Bianchi M.E. (2012). Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209,1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahamaa H., Vallerskog T., Qin S., Lunderius C., LaRosa G., Andersson U., and Harris H.E. (2007). HMGB1-secreting capacity of multiple cell lineages revealed by a novel HMGB1 ELISPOT assay. J Leukoc Biol 81,129–136 [DOI] [PubMed] [Google Scholar]
- Wang G., Yan Q., Woods A., Aubrey L.A., Feng Q., and Beier F. (2011). Inducible nitric oxide synthase-nitric oxide signaling mediates the mitogenic activity of Rac1 during endochondral bone growth. J Cell Sci 124,3405–3413 [DOI] [PubMed] [Google Scholar]
- Wang H., Bloom O., Zhang M., Vishnubhakat J.M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L., Manogue K.R., Faist E., Abraham E., Andersson J., Andersson U., Molina P.E., Abumrad N.N., Sama A., and Tracey K.J. (1999). HMG-1 as a later mediator of endotoxin lethality in mice. Science 285,248–251 [DOI] [PubMed] [Google Scholar]
- Yammani R.R. (2012). S100 proteins in cartilage: role in arthritis. Biochim Biophys Acta 1822,600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Tewary P., de la Rosa G., Wei F., and Oppenheim J.J. (2010). The alarmin functions of high-mobility group proteins. Biochim Biophys Acta 1799,157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Lundbäck P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M.E., Al-Abed Y., Andersson U., Tracey K.J., and Antoine D.J. (2012). Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18,250–259 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang H., and Tracey K.J. (2005). High mobility group box 1 (HMGB1). Crit Care Med 33,S472–S474 [DOI] [PubMed] [Google Scholar]
- Yang H., Wang H., Czura C.J., and Tracey K.J. (2005). The cytokine activity of HMGB1. J Leukoc Biol 78,1–8 [DOI] [PubMed] [Google Scholar]
- Zhang X., Wang H., and Wang J. (2013). Expression of HMGB1 and NF-κB p65 and its significance in non-small cell lung cancer. Contemp Oncol (Pozn) 17,350–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.H., Liu S.Q., and Peng H. (2008). The effect of hyaluronic acid on IL-1beta-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res 26,1643–1648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.