

Abstract
Next-gene ration sequencing is becoming the primary discovery tool in human genetics. There have been many clear successes in identifying genes that are responsible for Mendelian diseases, and sequencing approaches are now poised to identify the mutations that cause undiagnosed childhood genetic diseases and those that predispose individuals to more common complex diseases. There are, however, growing concerns that the complexity and magnitude of complete sequence data could lead to an explosion of weakly justified claims of association between genetic variants and disease. Here, we provide an overview of the basic workflow in next-generation sequencing studies and emphasize, where possible, measures and considerations that facilitate accurate inferences from human sequencing studies.
Genome-wide association studies (GWASs) have been successful in identifying genomic regions that influence the risk of human complex disease1. However, GWASs primarily make use of markers that are intended to represent causal variation indirectly, whereas, in principle, next-generation sequencing (NGS) can directly identify the causal variants. This is perhaps the central advantage of sequencing approaches over standardized genotyping panels, especially given the growing recognition that many common diseases could be influenced by fairly infrequent mutations in many different genes. At the same time, clinical geneticists are turning to next-generation sequencing approaches to overcome limitations of their traditional genetic tools. For these reasons, sequencing is rapidly becoming the primary focus of efforts to characterize the genetic bases of human diseases.
Despite the promise of NGS, our ability to generate sequence data currently outstrips our ability to interpret it accurately. It is noteworthy in this context that without the codification of a generally agreed significance threshold for GWASs2,3, many more false-positive findings would have been reported from GWASs. The central statistical guidelines that followed were strikingly simple: first, a proper account needs to be made for the number of possible independent tests; and, second, given that GWASs largely rely on indirect association to represent incompletely known variants, we lack sufficient information to make meaningful distinctions among interrogated variants in terms of their prior probabilities of truly associating with phenotypes. These two positions lead to a simple single statistical threshold to declare significance for association between a given polymorphism and phenotypes in a GWAS (by convention, P <5 × 10−8). Most of the polymorphisms that achieve P <5 × 10−8, after careful consideration of the relevant quality-control measures and confounders, share the property of having been confirmed in additional studies.
Unfortunately, the same type of solution is not applicable to sequence data, at least not immediately. The most fundamental reason is that sequence data reveal inherently different categories of variants that cannot reasonably be viewed as all having the same prior probabilities of influencing diseases. Given the sample sizes available today, treating variants anywhere in the human genome as equally likely to influence phenotypes would often constitute too great a cost in terms of power to be acceptable to most contemporary researchers. For this reason, nearly all current sequencing studies treat different classes of variants differently, either implicitly or explicitly. By design, exome-sequencing data are predicated on the idea that mutations influencing human phenotypes are more likely, base for base, to be found in coding sequence than elsewhere. Nonetheless, we currently have too little information about the full distribution of functional consequences for different kinds of variants in the human genome to allow simple quantitative priors to be applied to variants in a universal fashion. Despite these complexities, steps can still be taken to reduce the risk of false-positive claims, as outlined below. Moreover, as sample sizes increase over time, it will eventually become possible to carry out unbiased screens for variants anywhere in the entire set of the 3 billion positions that constitute the human genome.
Although many recent reviews have outlined the potential utility of NGS in studies of both complex traits4 and Mendelian diseases5, fewer reviews provide concrete overall guidelines for running an NGS study. Here, we try to fill this gap. We describe the NGS workflow in the following order: data generation, variant calling and annotation, association statistics, appropriate standards of evidence, and how to interpret functional evaluation of candidate variants alongside association evidence.
Study populations
In this article, our focus is on studies of risk factors for disease in the inherited genome as opposed to studies of tumour genomes that have been reviewed elsewhere6,7. We consider study design questions in light of three increasingly common NGS applications: Mendelian diseases involving the study of multiple affected individuals (in particular, those that are refractory to linkage, such as dominant mutations that compromise survival or reproduction); undiagnosed childhood disease; and common complex diseases, often in large case–control settings. Depending on the application and statistical tests used, different decisions are appropriate, particularly in relation to trade-offs between sensitivity and specificity. We therefore refer, where appropriate, to best practice considerations, depending on the application and study population.
Sequence data generation
Whereas the genotyping data are generally highly consistent in GWASs, making it fairly straightforward to control for spurious signals associated with experimental artefacts, the sources of variation in generating sequence data are more varied and are currently less well understood. Moreover, there are inherent trade-offs in calling properties that have no analogue in the near perfect genotype calls that emerge from GWAS data. For example, in the study of a single child with an undiagnosed genetic disease, sequencing and variant calling may be appropriately tuned to maximize sensitivity, whereas analyses of large case–control cohorts, especially when using aggregate statistics (see below), would generally seek to balance sensitivity and specificity.
For these reasons, it is currently necessary to adjust analysis routines to the precise question (or questions) being asked. Evidently, generating data for all samples that are to be analysed together using the same version of the same technology is desirable but is often difficult to achieve. Current next-generation sequencing involves numerous preparation steps using chemistry that is regularly updated and experimental procedures that can be variable (for example, fragmentation, and target enrichment in whole-exome and other preparations).
Moreover, the sequencing reactions themselves can vary across lanes within a flow cell, across flow cells and across machines owing to variations in cluster density and other features8. Gradually, many of these sources of variation are being progressively reduced owing to improvements in sequencing methodologies; however, they still remain and are only partially accounted for by variant-calling tools (discussed below). For these reasons, it is important to be aware of which kinds of analyses are and are not sensitive to such variation.
How much data?
The question of the amount of sequence to generate for a genome has been the subject of much debate. When the aim is to maximize variant-calling accuracy, without regards to cost savings, then the generation of ~125 Gb of 2 × 100 bp data per human genome has been shown to be optimal9 for good single-nucleotide variant (SNV) detection, yielding approximately 40-fold aligned depth of coverage (note that this conclusion applies to the more recent methods, which provide more uniform coverage of the genome than earlier methods). This level not only maximizes sensitivity but also minimizes false positives, which are often expensive to follow up.
In designs in which the study population size is large (such as large case–control designs studying complex diseases) and the variants of interest are expected to be present in multiple samples, lower coverage might be acceptable. For example, the 1000 Genomes Consortium estimated an optimal coverage of three- to fivefold for detecting variants in a population, given a fixed sequencing cost10,11. For many complex diseases, high locus heterogeneity and allelic heterogeneity12–14 mean that the variants of most interest may still be rare even in very large cohorts of cases. Connected to the challenge of allele heterogeneity, statistical approaches that seek to combine association evidence across multiple variants within the same gene will benefit from identifying as many of the variants as possible in each sample, and this mandates a high coverage. Low-coverage genomes further sacrifice substantial information about structural variation that can be inferred in high-coverage genomes, with most read-depth-based methods requiring a minimum coverage of tenfold for accuracy15.
In studies focused on identifying highly penetrant mutations, accurately determining the exact genotype in all samples is essential. For example, in studies of undiagnosed conditions12 and of multiple patients affected with a Mendelian disease16, it has often been essential to identify de novo mutations in the proband by comparing the affected genome to those of the parents. Here, high coverage of all samples is essential (for example, 60× or greater average coverage17) to facilitate improved sensitivity particularly in the parents to ensure that one of the parental alleles is not probabilistically missed, which would lead to suggestions of a putative de novo mutation in the child. On balance, our opinion is that many of the key goals of human genetics depend on determining individual genotypes with high confidence, and this requires high coverage.
Alignment and variant detection
Various algorithms are available for short-read alignment and variant detection18,19. Although this process is sometimes considered to involve two separate tasks, in reality it comprises several interdependent steps. As variant callers make assumptions about how the alignment process works, it is also necessary to harmonize variant callers and the alignment process18,20,21.
Detecting SNVs is often the most straightforward variant detection process. Many algorithms converge on a Bayesian approach that differs slightly in the prior assumption20–23 (TABLE 1). Several programs now enhance the initial SNV and small-indel variant detection approach in various ways. These approaches include a more sensitive read re-alignment for orphan read pairs and initially aligned reads with other anomalies, looking for clusters of read pairs with aberrant insert size distributions, de novo assembly of anomalously aligned reads24, and looking at copy number changes from differences in read depth.
Table 1.
Examples of implemented algorithms for single-nucleotide variant detection
| Program and URL (if available) | Notes | Refs |
|---|---|---|
| CASAVA (http://support.illumina.com/sequencing/sequencing_software/casava.ilmn) | Illumina platform software that is compatible with raw data produced by the Genome Analyzer and HiSeq sequencers | |
| GATK UnifiedGenotyper | Implements a Bayesian genotype likelihood model to estimate simultaneously the most likely genotypes and allele frequency in one or multiple samples | 20,24 |
| Platypus (www.well.ox.ac.uk/platypus) | Uses local realignment and assembly to detect sensitively and specifically SNPs and short indels in low- and high- coverage data; most efficiently used with alignments produced by Stampy | 21 |
| Polybayes/PbShort (http://bioinformatics.bc.edu/marthlab/PbShort) | A Bayesian approach designed from various short-read technologies; packaged with computer programs for converting text format sequence files and assembly formats | 88 |
| SAMtools | Provides various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing, generating alignments and creating a consensus sequence. Also includes a Bayesian single-nucleotide variant (SNV) or short indel caller | 22,89 |
| SOAPsnp | A Bayesian algorithm used to call consensus genotype incorporating the data quality, alignment and recurring experimental errors; supports alignments produced by SOAPaligner | 23 |
For a more complete listing, see SNP discovery website.
Another way to gain sensitivity and consistency in variant detection is to make the variant calls among multiple samples together. The GATK suite of tools has successfully adopted this approach24. A consideration for this approach is that multi-sample variant calling can introduce batch effects, depending on which genomes are considered together; this could in turn create subtle patterns of signal in some types of case–control analyses. For this reason, some large case–control studies may be best analysed using samples that have all been individually called or by carrying out multi-sample variant calling on all samples (both cases and controls) to be analysed together, as a set. Obvious challenges for the latter strategy include computational restrictions on how many samples can be called together and the fact that adding new samples to a data set would necessitate the recalling of samples in this framework. However, some studies clearly benefit from multi-sample calling. For example, in screens for de novo mutations, there is a clear advantage to calling variants within each trio as a set25. In this way, variants that are securely called in the child but that show some evidence for the presence of the variant in the parents will be ‘caught’, reducing the number of candidate de novo mutations for follow-up evaluation. Recently, methods that are explicitly aware of the expected inheritance pattern have been proposed and can formally evaluate the likelihood of a violation of those patterns (for example, de novo mutations)17,26,27.
It is also worth noting that all stages of the process can, in principle, be tailored to calling specific kinds of variants. For example, adding sequence data from a library with larger insert sizes will facilitate detection of structural variants. There are aligners that have been specifically developed for structural variant calling, such as MR and MRS FAST28,29. There is, however, an advantage in cost and consistency to use not only a single data type but a single alignment routine for all inferences about variation. It has recently been shown that it is possible to infer copy number status accurately in both unique regions of the genome and in segmental duplication regions using the same alignment data that are relied on to call SNVs and small indels15.
At present, SNVs seen across multiple samples are often sufficiently reliable in NGS data so that external validation is not required. However, screens for de novo mutations amount to a combined screen for both real de novo variants and sequencing anomalies. For this reason, putative de novo mutations should be confirmed by Sanger sequencing25,30–34 or some other independent technology. Similarly, indel and structural variant calling remains less reliable than SNV calling, and in most applications such variants of interest should also be separately confirmed: for example, by Sanger sequencing or by real-time quantitative PCR, respectively.
Measuring sequence completeness and quality
Computationally, it is simple to run analyses on variant calls that are represented through variant call format files (VCF files)20,35, which is a standardized output format from variant-calling algorithms. Many types of analyses, however, require data for sites in individual samples in which no variant was called. For example, in studies of undiagnosed genetic disease in children12,36, it is necessary to identify all genotypes that are clearly present in children, yet clearly absent in parents, to suggest de novo mutations. Here, it is important to know whether it is possible to have confidence that the parents do not have the genotype in question by checking the genotype quality and sequencing depth at the relevant site in the parents. A similar situation is found in case–control studies, in which it is desirable to know whether a putative causal variant is truly not present in a set of controls or merely not called owing to missing or poor-quality data and in studying patients with a known Mendelian disease when it is desirable to establish how well known genes are ‘covered’.
Although the tertiary analyses and the common exchange of secondary results are made possible by the standardization of the file formats, there is currently no standardized set of criteria for assessing the quality of variant calls versus no calls nor is there a standard framework for structuring the data for larger study designs across hundreds or thousands of samples. Modern alignment and variant-calling algorithms produce an abundance of quality-control metrics for each base call (for example, reads supporting an alternative allele, total read depth, read mapping quality, haplotype score, genotype likelihoods and combinations of metrics reflected by the variant quality score log odds ratio (VQSLOD score)20). Deciphering which metric or combination of metrics to include in downstream analysis is not always a simple task. It is desirable to maintain the flexibility of leveraging many of the available metrics across variants called in individual samples or arbitrary groups of samples, depending on the primary aims of particular analyses.
The challenge is to structure variant calls, variant annotations, coverage data, quality metrics and sample relationships in such a way that maximizes the available computational resources while facilitating flexible data querying by downstream software. Ideally, software tools that allow interaction with such data should be usable by investigators who have little computational background. Some example approaches include batch VCF searches with VCFtools35, PLINK/SEQ and SVA37. The most general and flexible solution currently is to house the relevant data in a relational database (BOX 1). Finally, a direction that could gain popularity for addressing these types of analyses is the development of a specialized use case for the VCF by incorporating quality and depth information at homozygous reference positions, allowing verification of the absence of a variant. An example of this is the genome VCF (gVCF).
Box 1. Frameworks for large-scale next-generation sequencing variant analyses.
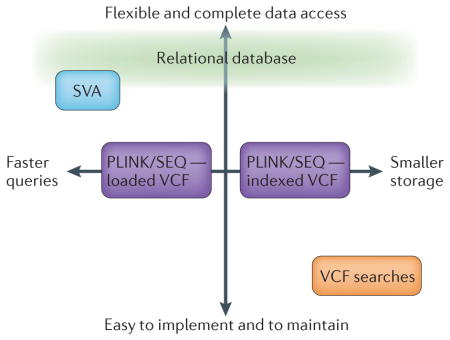
Any adopted framework will make various trade-offs that should be carefully considered. The diagram shows the approximate relative positions of four possible approaches within the context of two typical trade-offs: query speed versus required data storage (horizontal axis); and flexible and complete data access versus ease of implementation and maintenance (vertical axis).
The most flexible and complete approach is to implement a relational database using an industry platform (for example, MySQL) to incorporate all coverage data, quality metrics, variant calls and variant annotations of interest. A relational database presents information in relations (tables) that are themselves collections of non-redundant objects (rows) that have the same attributes (columns). Each table has an attribute or a set of attributes (primary key), the values of which uniquely define each row, and every table shares at least one attribute with another table in a one-to-one, one-to-many or many-to-many relationship. The ability to retrieve related data through these relationships forms the basis of the term ‘relational database’. The database can be coupled with an abstracting front-end application that permits complex data queries without requiring detailed knowledge of the underlying data structure. This framework can be optimally configured to incorporate both fast queries and efficient disk usage but is adopted at a high cost of initial implementation difficulty and downstream maintenance. However, a relational database can be updated with additional genomes and novel annotations and enables a host of industry-tested third-party tools to be used for database maintenance and optimization. Finally, individual database instances can be optimally configured for certain tasks or specific studies by leveraging the replication capabilities of a master database to multiple slaves.
SVA is a suite of tools for annotating and visualizing sequence data80 that organizes all relevant data into an easily searchable format that is optimized for efficient data access. SVA is more tightly bound at the speedy side of the query-time–storage-space spectrum and shares many flexibility and completeness attributes with a relational database approach. However, SVA is not easily updated with additional samples or new annotations. The PLINK/SEQ approach maintains a library of VCF files, creates its own relational database behind the scenes and indexes the contained information for efficient querying without requiring much more disk space than storing the original VCF. PLINK/SEQ allows filtering on arbitrary quality metrics but provides no comprehensive solution for searches based on precise coverage data for samples without a genotype of interest, as it is not currently compatible with gVCF files. PLINK/SEQ can be operated either by fully loading VCFs, permitting faster searches or by indexing the VCF files on disk, sacrificing query time to use less disk space. The VCF search approach amounts to storing libraries of compressed VCF/gVCF files and using the available toolsets (VCFtools/gVCFtools) for querying the libraries. This approach is easier to implement than the others as the tools are available for download but sacrifices arbitrarily customizable queries and flexibility in how the samples are grouped among the individual or multi-called VCFs. This approach may also be substantially slower with analyses of many samples as the indexing has not been developed to the same degree as database indexes.
Prioritizing and analysing variants
Currently, the fundamental challenge in interpreting NGS data is how appropriately to distinguish and to prioritize among types of genetic variants in interpreting NGS. In a GWAS, it is clearly appropriate to treat all interrogated variants as having an equal prior probability of real association with disease because the variants studied are generally markers for unknown causal variants. In the case of sequencing studies, however, we do have more information available for many of the interrogated sites, and this allows more meaningful distinctions to be made in terms of the a priori probabilities. Here we discuss data types that are commonly used to make distinctions among variants and the analysis routines that are currently available for incorporating them.
Information from population genetics
Ultimately, interpreting population sequencing data and designing association studies depends on how many human alleles have any effect on molecular function and phenotype. Here, population genetics provides a useful complement to association evidence by characterizing and quantifying natural selection against deleterious alleles (that is, alleles that negatively affect fitness). However, we do not imply that all damaging and even pathogenic alleles are necessarily deleterious in an evolutionary sense or that all deleterious alleles are pathogenic.
Population genetics approaches are based, in principle, on two related effects. First, deleterious alleles are much less likely to reach fixation in populations than neutral alleles. Second, deleterious alleles are less likely to be observed as common variants that segregate in the population. Proportionally more deleterious than neutral polymorphic variants are expected to be rare38. Thus, the inference of selection against deleterious alleles is usually based on a comparison of polymorphism-to-divergence ratios and site frequency spectra between functional categories of variants. An excess of rare alleles and a higher polymorphism-to-divergence ratio for variants in a more important functional category, such as nonsense and frameshift mutations, is often interpreted as presence of deleterious alleles.
The appropriate quantitative use of both of these signals can help to establish meaningful priors on variants in interpreting sequence data. For example, the power of various statistical methods for detecting association with multiple rare alleles at a locus depends on the fraction of alleles in a unit of analysis (for example, genes) that are functionally important. Selecting the appropriate tests should therefore be informed by the expected proportion of functionally significant alleles.
Computational tools for predicting functional effects
A more fine-grained functional stratification can be achieved by applying computational methods for predicting the functional effect of amino acid changes, such as SIFT39, PolyPhen2 (REF. 40), MAPP41, SNAP42, MutationTaster43 and others44. These methods can discriminate between damaging and benign amino acid changes with an accuracy of 75–80%, and their utility in prioritizing variants has been reviewed in depth elsewhere44,45. Variants predicted to have damaging effects on protein function have, on average, markedly lower allele frequencies and higher polymorphism-to-divergence ratios than do benign variants46,47. Despite the clear utility of these approaches in distinguishing classes of variants, the community must remain vigilant always to distinguish the assessments of whether a variant is or is not likely to be deleterious in an evolutionary context, from the question of whether a variant is or is not pathogenic for a given condition or set of conditions under study.
Information from Mendelian disease associations
Another possible resource for understanding the impact of variants is leveraging what is already known about the phenotypes associated with mutation in genes that are responsible for Mendelian diseases48,49. Resources housing this information, such as Online Mendelian Inheritance in Man (OMIM), can be readily interrogated for additional support regarding whether the variant (or gene) identified in a given study has been linked to the same or a similar phenotype in a previous study. It should be noted, however, that it is sometimes difficult to assess, in a formal way, whether a particular phenotypic presentation is or is not similar enough to a reported Mendelian condition then to prioritize the relevant genes. Coupling this with the reality that all human genomes carry many deleterious mutations, in many different genes, it is clear that all human genomes have a high level of ‘narrative potential’ to provide compelling but statistically poorly justified connections between mutations and phenotypes (BOX 2). Moreover, it must always be kept in mind that the strength of evidence supporting the pathogenicity of the variants presented in these databases dramatically varies. For example, in one resequencing study, it was determined that up to one-third of the mutations listed in the Human Gene Mutation Database (HGMD) as pathogenic were in fact apparently benign owing to being common polymorphisms in the population, sequencing errors or owing to a lack of appropriate evidence required for pathogenicity37.
Box 2. Illustrating ‘narrative potential’ in a control genome.
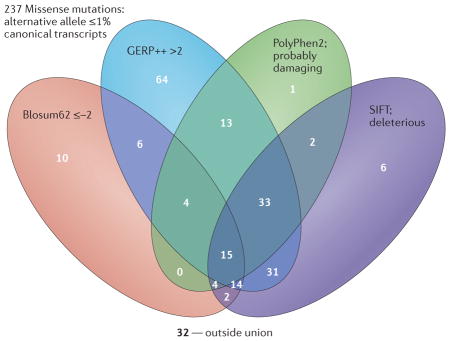
To illustrate the ‘narrative potential’ of a control genome, we analysed the sequence data from a female control12. We choose a filtering strategy favouring a reduction in false positives (Supplementary information S1 (box)). We restrict our analysis to protein-coding variants within CCDS transcripts81. We set ‘qualifying’ variants to be those with an alternative allele frequency ≤1% in the internal control population of sequences generated using the same sequencing machines, alignment, quality-control and variant-calling pipelines. Moreover, we compare the allele frequencies obtained here with allele frequencies from the ESP6500 European–American exome-sequenced representatives (allele frequencies are taken from the Exome Variant Server). We also adopt four algorithms to assign further ‘qualifying’ criteria in putative missense mutations: SIFT39, PolyPhen2 (REF. 40), GERP++82 and Blosum62 (REF. 83; Supplementary information S2 (box)). These represent examples of the algorithms that are available to assess ‘qualifying’ variant status and have been reviewed in detail elsewhere45. Here we select qualitative thresholds in these tools for illustrative purposes, although selection can differ on the basis of expected genetic model and ideally should be quantitatively assessed through a weighting system. Two resources for connecting mutations and genes to specific phenotypes were used in this illustration (namely, Online Mendelian Inheritance in Man (OMIM) and the Human Gene Mutation Database (HGMD)48).
We find that this control genome contains nine variants that are likely to be gene disrupting (that is, nonsense, splice acceptors or donor sites) that are rare in controls (allele frequency ≤1%), five of which are in genes that can be connected to specific phenotypes through OMIM or HGMD (Supplementary information S3 (table)). Moreover, we find 237 rare missense mutations (allele frequency ≤1%) in this genome, of which 86.5% are judged to be damaging by at least one of the four algorithms used. Moreover, 32.5% of these variants are in genes that can be connected to specific phenotypes through consideration of OMIM (Supplementary information S4 (table)) or HGMD. Thus, the potential to assign a disease-related narrative to these mutations is high in a single control sample.
Information from multiple variants
We currently do not know the precise weights that should be given in the prior probabilities discussed above to incorporate them into appropriate formal statistical models, and there is a substantial risk that post hoc deployment of plausibility arguments based on genes or variants could lead to misinterpretation of association evidence. In addition, it is plausible that at least in some genes or pathways, there are multiple different mutations that each changes the risk of disease to a similar degree. For example, if a gene increases disease risk when its expression is reduced, all possible complete knockout mutations for the gene would be expected to have the same impact on risk. As such, deleterious variants are often rare, and it is difficult to detect association using single-variant analyses. In this setting, a better alternative is to collapse or to aggregate statistical information across qualifying mutations within a functional unit (for example, a gene or pathway), resulting in a single gene-based or pathway-based test. The simplest such approach is the burden test50,51. For a binary trait, the burden test compares frequencies of individuals carrying a damaging variant in a gene between cases and controls. For quantitative traits, most variations of the burden test can be regarded as a regression of phenotypic value on presence of any damaging mutation in a gene. The approaches can be generalized to pathways rather than to genes and to multiple types of genetic variants.
Several existing burden tests vary in the way that they take into account allele frequencies of individual variants and whether they take weighted combinations of variants based on external information52,53. One limitation of these approaches is that they assume that all variants act in the same direction with respect to disease risk. This assumption may not hold well for genes that carry both loss-of-function and gain-of-function mutations or that carry different kinds of gain-of-function mutations; the assumption is further complicated if the unit of analysis is a pathway. Numerous tests have therefore been developed that relax the assumption that mutations all act in the same direction, including the C-alpha test54, the sequence kernel association test (SKAT)55 and the estimated regression coefficient test (EREC)56. Multiple reviews have summarized much of the work done on rare variant methodologies, including the effect of genetic architecture57–61.
Currently, however, the impact of incorporating different types of prior information into different types of tests has not been systematically evaluated either through simulation or by empirical means.
One attractive feature of these collapse or aggregate approaches is that the number of tests is clarified before statistical assessment. Clearly, significance should account for all regions that are considered. For example, if genes are the unit of analysis — and for demonstrative purposes we define the number of assessable genes as 20,000 — then the Bonferroni significance threshold would be set at a value no higher than 0.05/20,000. However, as there are many different ways to define qualifying variants and weight by variant characteristics, it will also be important to ensure that alternatives are not explored in order to maximize significance, as the threshold of 0.05/20,000 would apply to only a single set of rules. It should also be recognized that smaller studies may suffer from deflation of P values because there are insufficient counts to generate low P values. In this case, more accurate correction for multiple testing can be achieved using permutation60.
Many methodological challenges, however, remain in implementing these prioritization approaches. For example, aggregate methods often focus on rare variants as those that are most likely to influence disease62,63. Because control populations used to determine allele frequencies (for example, the Exome Variant Server) are far from comprehensive in their ethnic background, population groups that are not well represented may be systemically more likely to show signals of spurious association. In addition, aggregate methods are particularly prone to sequencing artefacts, and we have little current guidance on how variations in sequence quality will influence how well aggregate signals will be replicated across studies.
Functional evaluation
Although certain classes of mutations and certain genes are more likely to cause particular phenotypes, the basic challenge remains that all genomes carry ‘narrative potential’ (BOX 2) in the sense that even genomes of ‘control’ individuals with no known diagnosis carry functional mutations in many genes of known or suspected relevance to a broad range of different phenotypes12 (BOX 2). We find that there is considerable potential to link variants found in a randomly selected ‘control’ individual to different diseases (BOX 2) and phenotypes with plausible sounding arguments. Thus, such plausibility arguments are clearly never sufficient to link a variant to a phenotype, and other criteria are required. Delineation of the full range of functional studies that might follow the identification of a disease-causing mutation would span all of human biology and is evidently beyond the scope of this or any single review. Nonetheless, there are several general principles that functional programs dedicated to elucidating the mechanism of disease causing mutations should consider (BOX 3).
Box 3. Suggested workflows for functional evaluation of novel disease variants.
Quality management and collaboration
Before initiating functional experiments, we recommend that standardization of laboratory procedures and informatics approaches be agreed on by participant groups. This would extend to standard informatics issues, such as selection of metadata and database design, data storage, access and security. These databases will link functional data on each variant with disease phenotype and provide a context in which novel variants with similar functional profiles can be interpreted.
Overall management and procedural issues must be finalized to maximize data quality and to reduce time and costs. For example, this may involve incorporating quality systems into the procedure — including standardization of protocols, replication policies, statistical analysis, performance management and reporting — as well as developing policies for publication, presentation, attribution and protection of intellectual property. An important consideration is the assignment of particular tasks and genes to qualified laboratories.
Provision of reagents and functional platforms
Core services that provide reagents for functional assays include DNA-cloning services for expression vectors of exonic variants or mini-genes for intronic variants and probes for transcript analysis, as well as services for the provision of antibodies, cell lines, statistics and data curation, and viral vectors for expression and knockdown. For assessment of gene agnostic properties, several technology platforms are available, such as: Biacore (GE Healthcare) and LabChip GXII (PerkinElmer) for protein analysis; droplet-based assays for studying protein–protein interactions84; and high-content screening platforms such as Operetta (PerkinElmer) and ImageXpress (Molecular Devices) for detecting morphology and compartmental expression in cells. Systems-biology workflows and platforms85 incorporate many computational tools to facilitate modelling of complex disorders that cross multiple temporal and spatial scales.
Functional assays
General
First-pass functional assays address questions at the molecular cellular scale that are relevant to broad range of protein classes, such as:
Are there any changes in protein synthesis, folding and degradation?
Is the protein trafficked to appropriate cellular compartments?
Are protein–protein interactions changed by amino acid alterations, expression levels or trafficking changes?
Fortunately, these assessments can be viewed as almost entirely generic and in principle can be applied to any mutation in any gene. These assays are therefore, in principle, amenable to deployment in high-throughput platforms that would allow characterization of the hundreds to thousands of mutations that will be identified in every human gene in the years to come.
Specialized
At higher levels of organization, protein function can diverge, and highly specialized assays are needed. Ion channels, for example, constitute ~1% of the total human genome, yet they have been unequivocally implicated in more than 50 human genetic disorders86 and are grossly over-represented as drug targets, being the primary therapeutic mode of action for 13.4% of the some 1,200 currently known small-molecule drugs78. It is also sobering that of the potential more than 20,000 proteins in the human genome, fewer than 300 are validated drug targets78. An important consideration in devising workflows and advancing genes through functional programs is the ultimate ‘druggability’ of that target: some estimates suggest that only 5% of the genome will be druggable and disease-relevant87. Within this framework, ion channels are likely to remain an important protein family, and existing platforms that have been devised for drug discovery can be exploited for automated high-throughput analysis of variants in diseases such as epilepsy, in which the path to translation is clear.
The role of functional data
There are two very distinct ways in which functional work has been used in relation to human genetic studies. The first and most problematic is an attempt to ‘validate’ potential associations between identified variants and diseases. The second is to understand the biological basis of disease caused by variants that have been securely implicated in diseases on the basis of the genetic work alone. In reality, these two applications of functional work in human genetic studies are too often blurred together, making it difficult to assess the overall strength of pathogenicity claims. Although recognizing that there are specific settings in which functional evaluations may help to make the case for pathogenicity, in general our view is that the case for being pathogenic itself will depend on the genetic analyses, whereas functional work will help us to understand how the variant influences pathogenicity.
This perspective somewhat contradicts our earlier argument about making distinctions among variants in genetic association studies. Thus, we have argued that it is appropriate to make distinctions among, for example, substitutions that result in frameshift mutations and those that do not, among variants that are rare in the population versus common, and so on. This seems justifiable, as we generally have much better data to distinguish types of variants (for example, nonsense mutations compared with those at least 5 kb from any exon) than we have for assessing the probability that a gene will influence phenotypes of interest. For example, before GWASs, there was a burgeoning literature in neuropsychiatric genetics that primarily related common polymorphisms in ‘obvious’ candidate genes to both disease endophenotypes and cognitive traits65,66. However, the evidence supporting the suggested polymorphisms evaporated when they were interrogated in statistically more careful study designs67 and in GWASs68.
As sample sizes grow, we believe that entirely unbiased discovery of mutations of large effect influencing any base in the human genome will become feasible. In such an entirely unbiased approach, no prior knowledge of the genome would need to be used, and moreover it would not be necessary to assume that different mutations in the same gene would affect the same phenotype (or phenotypes) (BOX 4). Given that this approach would need to account for testing any of the 3 billion different sites in the human genome that could carry a causal mutation, sample sizes would have to considerable. Interestingly, we find that under a range of genetic architectures, the total sample size of controls is the most important factor.
Box 4 . Detecting association in the human genome without prior information.
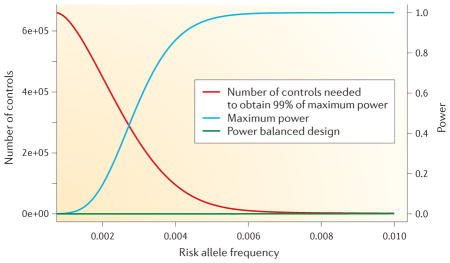
We investigated the viability of a site-based approach using standard power calculations, assuming that the α-level is given by the Bonferroni threshold (0.05/3 × 109 ≈ 2 × 10−11). Specifically, we assumed a prevalence of 1%, although we note that diseases with differing prevalence would result in similar results as long as the control sample excluded those with disease. We fixed the number of cases at 100 throughout. We assumed that a rare variant acted in a dominant fashion on disease risk. We allowed the frequency of this variant to vary and investigated the impact of the size of the control group on power. It is well known that a balanced design, in which the number of cases and controls are similar, gives the greatest power for a fixed number of samples (that is, both cases and controls). However, our analysis suggested that as the risk allele reduces in frequency, it pays to have a larger and larger control sample. We began by deriving a power formula for the hypothetical situation in which the control sample is infinite. The figure shows a given genetic model (with a relative risk of 10) for a range of rare risk allele frequencies. We computed not only the maximum power (infinite control group size) but also the size of control group necessary to achieve 99% of the maximum power available. From this figure, we can see that a control group with 100,000 samples will yield 80% power of detecting the risk variant down to an allele frequency of 4 × 10−3. The balanced design has no power throughout the presented risk allele frequency spectrum.
Regardless of the viewpoint taken concerning the use of functional data to implicate variants in disease, best practice dictates that researchers should be clear about where the evidence for pathogenicity comes from, and whether it depends on the genetic data alone or whether it requires functional data. If it requires the functional data, then the specificity of the functional assay must be carefully justified, and in many settings, this may not currently be possible.
A role for computation in functional evaluation
Computation will have an enabling role in functional evaluation. For example, variants that have been characterized on the basis of a number of functional assays will need to be quantitatively evaluated to determine whether and which characteristics are suggestive of pathogenicity. More specifically, multi-scale simulations of specific human systems have been developed, such as IBM’s Cardioid project that seeks to build a full simulation of the human heart for investigating the impacts of mutations69 and drugs on heart function. Further development of this and of similar models for other systems will provide the platforms on which genomics and functional data can be effectively integrated. Other methods, such as the dynamic clamp, which creates ‘real-time’ biological–computer hybrid models, can be used to assess mutations in cardiac genes70, to facilitate direct translation of protein dysfunction into disease-relevant phenotypes71 and to assess drug safety72. Although there are well-known reasons for caution relating to the accuracy of such models of complex biological systems, there is increasing optimism that useful models of many key processes are and will be possible73.
The role of animal models in functional assessment of variants
Models of genetic disorders from animal studies have provided unexpected insights into disease mechanisms and can give insights into mechanisms of clinical heterogeneity that are important for disease risk assessment and establishing mechanisms. In one recent example, a mouse model was used to show how a single mutation causes distinct clinical phenotypes under distinct genetic models. A comparison of the GABRG1 ion channel mutant (GABRG1(R43Q)) epilepsy mouse model with the GABRG1 ion channel knockout (GABRG1(KO)) mouse showed that although both models shared one of the disease phenotypes, a dominant-negative interaction of the mutant protein must be proposed to account for additional phenotypes74. The study also showed that only one phenotype was affected by genetic background. Similarly, a recent study in a mouse asthma model demonstrates that consideration of epistasis is vital for predicting drug action and for selecting animal models to evaluate novel therapies75.
More generally, large-scale systematic analysis of biological epistasis can be undertaken in model organisms such as yeast, worms and flies to develop interaction networks for the identification of ‘disease modifiers’, which are genetic hubs that are functionally poised to affect multiple traits76. Specific hypotheses of disease-relevant biological epistasis can then be modelled in mice to dissect these pathways further with higher-order phenotypic features that are lacking in simpler model systems. These studies clearly show that there is no ‘one size fits all’ approach in animal modelling of human genetic disease, and multiple complementary approaches are needed to bridge the gap from genetic discovery to disease therapy.
Induced pluripotent cells and functional evaluation
Induced pluripotent stem cells (iPSCs) from patients with genetic disorders have the promise to deliver ‘diseases in a dish’77. Although technical issues remain, such as creating homogeneous populations of cells with disease-relevant phenotypes, the ability to move from patient sample to model system is still enticing. Many assay platforms currently in use could be rapidly adapted for stem cells. For gene variant functional analysis and pharmacogenomics, iPSC-derived cardiomyocyte platforms already exist that can readily characterize well-validated predictors of cardiac efficacy and safety. One enticing aspect of these approaches is the fact that the effect of the mutation of interest can be assessed in both the correct genetic background (by comparing clones from the patient in which the site has and has not been ‘edited’ to match the wild-type allele) and in the standardized control background (by editing in the mutant allele in standardized and widely used control clones). Perhaps most importantly, for any diseases that have clear cellular phenotypes, high-throughput screening systems can quickly be designed and implemented.
Functional variants and the control population
It is also necessary to acknowledge the importance of understanding the spectrum of functional change to be expected in the unaffected population. For example, how do some of the most commonly characterized functional effects of mutations depend on population minor allele frequency? How does the distribution of functional effects of variants from controls compare with disease cases, especially in the case of polygenic inheritance and/or strong genetic interactions? Do functional effects in cases and controls overlap, and can the effects be separated? First forays into this area should be followed by exhaustive analysis of a single gene class to provide proof of principle. Coupled with this, it is increasingly clear that there is a need for well-curated and universally available databases of the variants that are observed in patients with precisely defined phenotypes as well as databases of the distribution of variants in patients without any clinical diagnoses.
The relevance of functional information for therapies
Despite the investment of enormous resources by both public and private researchers, the number of targets being exploited in drug discovery campaigns is fairly stagnant78. Unfortunately, not all genes implicated in disease will prove to be ‘druggable’, and accurately predicting how drugs act on a gene or pathway will crucially depend on the functional effects of disease-causing mutations. Thus, it is almost universally true that translating genomics discoveries into improved clinical outcomes will depend on a functional understanding of the effects of pathogenic variants that have been implicated in disease through genetic analysis. An important corollary is that some level of convergence of disease mechanisms across multiple individual genomes must be found, as the number of drugs that can and will be developed is limited.
Furthermore, it would be satisfying if all genetic diseases could be reversed by rescuing the molecular phenotype, but this is not always possible as irreversible changes can occur in utero and in early development before there has been an opportunity to intervene. Even with the best prognostic genomics tools, small-molecule and biological-based therapies cannot necessarily target the temporally and spatially sensitive dysfunction caused by a pathogenic variant. In such cases, a complete understanding of the disease mechanism and the properties of that disease state garnered from functional programs will provide the clues to novel treatments that exploit therapeutic nodes to affect disease progression in a range of different individual genomes.
Conclusion and future directions
In broad summary, we now know many of the strategies that are relevant to the interpretation of sequenced genomes, including appropriate measures to control for the accuracy of sequence data, genetic and bioinformatic evidence that can help to provide prior distinctions among variants in probability of influencing disease, and functional characterization of the variants in both in vivo and in vitro models. But, although interpretation of protein- coding parts of the genome is mature, interpreting other parts of the genome is much less so. In the single-exome snapshot (BOX 2), we solely focus on the CCDS-defined protein-coding regions of the genome. Currently, our ability to interrogate the genome as a whole for variants that influence phenotypes is limited owing to the potential number of variants in the genome and current sample sizes. Eventually, this will change. As noted above, well-powered screens for mutations of major effect anywhere in the genome will become possible as the number of control individuals with comprehensive genetic data increases (BOX 4). Moreover, increasing knowledge of what parts of the genome are important in regulating gene expression and how they do so79 will further facilitate our ability to interrogate the full human genome, as will the simultaneous analysis of multiple omic layers, most immediately the genome and the transcriptome. Unfortunately, for most of these classes of evidence, it will take time to develop appropriate statistical criteria for reaching firm conclusions about pathogenicity.
Perhaps the most general lesson of the considerations reviewed here is that arguments for pathogenicity should always be made in full awareness of the opportunity for both association and narrative potential that is inherent in sequence data. If an observation of a particular kind of functional effect of a variant is used to argue for its pathogenicity, it is necessary to assess how commonly other variants in the genome produce similar functional effects. The considerations above make clear that a variant being in a class that looks suspicious (for example, rare and in silico predicted damaging) by itself can provide little evidence of pathogenicity, even if the judgment that the variant is deleterious in a population genetic sense is more secure. It is thus clear that a central challenge for the field is developing appropriate statistical criteria that incorporate disparate data types in the interpretation of sequenced genomes. An even more difficult challenge will be to develop sufficiently high-throughput assays with supporting neuronal, animal and computational models to assess the biological effects of implicated variants.
Supplementary Material
Acknowledgments
The authors thank the reviewers for their helpful comments. D.B.G. thanks L. Biesecker (NGHRI) for helpful discussions that contributed to the development of this Review. S.P. is a National Health and Medical Research Council (NHMRC) CJ Martin Fellow.
Glossary
- Priors
Used to reflect assumptions about the involvement of different classes of mutations before the evidence available from a given study is considered
- Cluster density
The density of clonal double-stranded DNA fragment clusters bound to an Illumina flow cell, typically expressed as clusters per mm2. It is used as a quality-control metric early during the sequencing reaction; low cluster densities will result in a lower sequencing yield in the resulting fastq library, whereas very high cluster densities will result in poor sequence quality
- Locus heterogeneity
Refers to the number of different genes in the genome that can carry mutations that influence risk of given disease
- Allelic heterogeneity
Refers to the number of different mutations at a single gene that can influence risk of disease
- Structural variation
Occurs in DNA regions generally greater than 1 kb in size, and includes genomic imbalances (namely, insertions and deletions (also known as copy number variants)), inversions and translocations
- De novo mutations
Non-inherited novel mutations in an individual that result from a germline mutation
- Indel
An alternative form of genetic variation to single- nucleotide variants that represents small insertion and deletion mutations
- Insert size
The length of the fragmented sequence between ligate adaptors. In paired-end sequencing, the insert size generally ranges from 200 to 500bp
- Batch effects
Differences observed for samples that are experimentally handled in different ways that are unrelated to the biological or scientific variables being studied. If batch effects are not properly accounted for in sequence studies, they can generate false signals of association between genetic variation and the traits under study
- Library
The collection of processed genome fragments that are prepared for sequencing. In a bioinformatics context, the term may also generally refer to the set of sequences found in a single fastq file
- Variant call format files (VCF files)
A flexible text file format developed within the 1000 Genomes Project that contains data specific to one or more genomic sites, including site coordinates, reference allele, observed alternative allele (or alleles) and base-call quality metrics (see Further information)
- Polymorphism-to-divergence ratios
Comparing sequence divergence across species with population polymorphism data (for example, McDonald–Kreitman test) facilitates identifying where selective forces are acting on the genomic sequence
- Site frequency spectra
Reflecting the distribution of allele frequencies. They are defined by the number of sites that has each of the possible allele frequencies. Different forms of selection perturb the site frequency spectrum in known ways
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. This influential Review compiles into one paper the basics of doing a GWAS, including best practice guidelines, such as controlling for population stratification. The Review also reinforces the universally followed guideline of 5 × 10−8 as a threshold for significance in GWAS. [DOI] [PubMed] [Google Scholar]
- 3.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 4.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 5.Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 7.Ding L, Wendl MC, Koboldt DC, Mardis ER. Analysis of next-generation genomic data in cancer: accomplishments and challenges. Hum Mol Genet. 2010;19:R188–R196. doi: 10.1093/hmg/ddq391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotech. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 9.Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011;21:1498–1505. doi: 10.1101/gr.123638.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genomes Project C. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendl MC, Wilson RK. The theory of discovering rare variants via DNA sequencing. BMC Genomics. 2009;10:485. doi: 10.1186/1471-2164-10-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Need AC, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49:353–361. doi: 10.1136/jmedgenet-2012-100819. This is the first study that estimates the ‘success rate’ of getting a genetic diagnosis through whole-exome sequencing of undiagnosed conditions in a real clinical setting considering 12 children with a broad range of severe childhood genetic conditions. The primary conclusion is that the success rate is remarkably high but depends in many cases on functional characterization of previously unidentified mutations in already known disease genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinzen EL, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. The largest epilepsy exome-sequencing study to date is reported in this paper. The results suggest high locus and allelic heterogeneity for both disorders, requiring larger sample sizes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Need AC, et al. Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. Am J Hum Genet. 2012;91:303–312. doi: 10.1016/j.ajhg.2012.06.018. The largest schizophrenia exome-sequencing study to date is reported in this paper. The results suggest high locus and allelic heterogeneity for both disorders, requiring larger sample sizes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu M, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91:408–421. doi: 10.1016/j.ajhg.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinzen EL, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nature Genet. 2012;44:1030–1034. doi: 10.1038/ng.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, et al. A likelihood-based framework for variant calling and de novo mutation detection in families. PLoS Genet. 2012;8:e1002944. doi: 10.1371/journal.pgen.1002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nature Rev Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nature Methods. 2009;6:S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 20.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 2011;43:491–498. doi: 10.1038/ng.806. This paper describes what has become the most widely used variant-calling environment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011;27:1157–1158. doi: 10.1093/bioinformatics/btr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meacham LR, et al. Diabetes mellitus in long-term survivors of childhood cancer. Increased risk associated with radiation therapy: a report for the childhood cancer survivor study. Arch Intern Med. 2009;169:1381–1388. doi: 10.1001/archinternmed.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. This paper was one of the first to analyse a large number of patients with a common disease using a trio design. Importantly, the authors established a formal framework for assessing whether excess de novo mutations are observed over expectation under the null hypothesis and found that autism genomes carry only modest excess of such mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, et al. Genotype calling and haplotyping in parent-offspring trios. Genome Res. 2013;23:142–151. doi: 10.1101/gr.142455.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nature Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nature Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hach F, et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nature Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 32.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 33.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saunders CJ, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4:154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell CJ, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge Press; 1983. [Google Scholar]
- 39.Sim NL, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jordan DM, Ramensky VE, Sunyaev SR. Human allelic variation: perspective from protein function, structure, and evolution. Curr Opin Struct Biol. 2010;20:342–350. doi: 10.1016/j.sbi.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 44.Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature Rev Genet. 2011;12:628–640. doi: 10.1038/nrg3046. A comprehensive Review is presented here of the priors, such as evolutionary knowledge, in silico protein effect assessment and others, that can be used to prioritize variants on the basis of putative damaging impact scores. [DOI] [PubMed] [Google Scholar]
- 46.Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 47.Asthana S, et al. Widely distributed noncoding purifying selection in the human genome. Proc Natl Acad Sci USA. 2007;104:12410–12415. doi: 10.1073/pnas.0705140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stenson PD, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 49.Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price AL, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neale BM, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu MC, et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nature Rev Genet. 2010;11:773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011;12:227. doi: 10.1186/gb-2011-12-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiezun A, et al. Exome sequencing and the genetic basis of complex traits. Nature Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ladouceur M, Dastani Z, Aulchenko YS, Greenwood CM, Richards JB. The empirical power of rare variant association methods: results from Sanger sequencing in 1,998 individuals. PLoS Genet. 2012;8:e1002496. doi: 10.1371/journal.pgen.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Q, et al. A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am J Hum Genet. 2011;88:458–468. doi: 10.1016/j.ajhg.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 64.Prathikanti S, Weinberger DR. Psychiatric genetics—the new era: genetic research and some clinical implications. Br Med Bull. 2005;73–74:107–122. doi: 10.1093/bmb/ldh055. [DOI] [PubMed] [Google Scholar]
- 65.Mutsuddi M, et al. Analysis of high-resolution HapMap of DTNBP1 (Dysbindin) suggests no consistency between reported common variant associations and schizophrenia. Am J Hum Genet. 2006;79:903–909. doi: 10.1086/508942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Need AC, et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 2009;5:e1000373. doi: 10.1371/journal.pgen.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoefen R, et al. In silico cardiac risk assessment in patients with long QT syndrome: type 1: clinical predictability of cardiac models. J Am Coll Cardiol. 2012;60:2182–2191. doi: 10.1016/j.jacc.2012.07.053. [DOI] [PubMed] [Google Scholar]
- 68.Berecki G, Zegers JG, Wilders R, Van Ginneken AC. Cardiac channelopathies studied with the dynamic action potential-clamp technique. Methods Mol Biol. 2007;403:233–250. doi: 10.1007/978-1-59745-529-9_16. [DOI] [PubMed] [Google Scholar]
- 69.Zareba W, Moss AJ, le Cessie S. Dispersion of ventricular repolarization and arrhythmic cardiac death in coronary artery disease. Am J Cardiol. 1994;74:550–553. doi: 10.1016/0002-9149(94)90742-0. [DOI] [PubMed] [Google Scholar]
- 70.Redfern WS, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 71.Di Ventura B, Lemerle C, Michalodimitrakis K, Serrano L. From in vivo to in silico biology and back. Nature. 2006;443:527–533. doi: 10.1038/nature05127. [DOI] [PubMed] [Google Scholar]
- 72.Reid CA, et al. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology. 2013;80:1003–1008. doi: 10.1212/WNL.0b013e3182872867. This paper uses an animal model to provide remarkable resolution in dissecting how a single mutation can result in two distinct clinical manifestations with one seizure type resulting from haploinsufficiency and the other from a distinct gain of function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freimuth J, et al. Epistatic interactions between Tgfb1 and genetic loci, Tgfbm2 and Tgfbm3, determine susceptibility to an asthmatic stimulus. Proc Natl Acad Sci USA. 2012;109:18042–18047. doi: 10.1073/pnas.1205374109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nature Rev Genet. 2013;14:168–178. doi: 10.1038/nrg3404. [DOI] [PubMed] [Google Scholar]
- 75.Tiscornia G, Vivas EL, Izpisua Belmonte JC. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nature Med. 2011;17:1570–1576. doi: 10.1038/nm.2504. [DOI] [PubMed] [Google Scholar]
- 76.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 77.Consortium EP, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ge D, et al. SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27:1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pruitt KD, et al. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davydov EV, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi JW, Kang DK, Park H, deMello AJ, Chang SI. High-throughput analysis of protein-protein interactions in picoliter-volume droplets using fluorescence polarization. Anal Chem. 2012;84:3849–3854. doi: 10.1021/ac300414g. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh S, Matsuoka Y, Asai Y, Hsin KY, Kitano H. Software for systems biology: from tools to integrated platforms. Nature Rev Genet. 2011;12:821–832. doi: 10.1038/nrg3096. [DOI] [PubMed] [Google Scholar]
- 84.Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 85.Owens J. Determining druggability. Nature Rev Drug Discov. 2007;6:187. [Google Scholar]
- 86.Marth GT, et al. A general approach to single-nucleotide polymorphism discovery. Nature Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 87.Bruce HA, et al. Long tandem repeats as a form of genomic copy number variation: structure and length polymorphism of a chromosome 5p repeat in control and schizophrenia populations. Psychiatr Genet. 2009;19:64–71. doi: 10.1097/YPG.0b013e3283207ff6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.