

Abstract
Accurate clinical course descriptions (phenotypes) of multiple sclerosis (MS) are important for communication, prognostication, design and recruitment of clinical trials, and treatment decision-making. Standardized descriptions published in 1996 based on a survey of international MS experts provided purely clinical phenotypes based on data and consensus at that time, but imaging and biological correlates were lacking. Increased understanding of MS and its pathology, coupled with general concern that the original descriptors may not adequately reflect more recently identified clinical aspects of the disease, prompted a re-examination of MS disease phenotypes by the International Advisory Committee on Clinical Trials of MS. While imaging and biological markers that might provide objective criteria for separating clinical phenotypes are lacking, we propose refined descriptors that include consideration of disease activity (based on clinical relapse rate and imaging findings) and disease progression. Strategies for future research to better define phenotypes are also outlined.
In 1996, the US National Multiple Sclerosis Society (NMSS) Advisory Committee on Clinical Trials in Multiple Sclerosis defined the clinical subtypes of multiple sclerosis (MS).1 The definitions provided consensus on terminology to describe various clinical courses of MS and highlighted areas where there was lack of consensus, or confusion. The rationale was the perceived need for clarity and consistency in defining patient groups for natural history and demographic studies, to enhance homogeneity in clinical trials, and to clarify communications among clinicians and with individuals with MS.
The Committee provided standardized definitions for 4 MS clinical courses: relapsing-remitting (RR), secondary progressive (SP), primary progressive (PP), and progressive relapsing (PR).1 It further recommended that the term relapsing-progressive MS be dropped, as the term was believed to be vague and overlapped with other disease course subtypes. Also recommended was that the term chronic progressive be replaced with the more specific terms SP and PP. Definitions were provided for benign MS and malignant MS. These phenotype descriptions were believed to represent the spectrum of clinical subtypes of MS but it was recognized that the descriptions might change over time.
The 1996 clinical course descriptions were rapidly incorporated into clinical practice and utilized in the eligibility criteria of almost all subsequent MS clinical trials. They were also used to some extent to guide regulatory review and approval of new therapeutics. At times the course descriptions were amalgamated into relapsing (including RR, SP, and PR) and progressive (including PP, SP, and PR) forms with the major distinction being whether the subject's disease was predominantly relapsing vs predominately progressing, although the distinction was never clearly delineated.
When proposed, it was noted that these clinical course descriptors were based on subjective views of MS experts and lacked objective biological support. There was insufficient knowledge to confidently link MS clinical course with MRI findings and biological and other surrogate markers for disease course were lacking. The authors suggested that developments in imaging and biological marker research would have a future impact on modifying or complementing the purely clinical course descriptors and that the clinical course subtypes of MS should be re-addressed when such markers became available.
In 2011, the Committee (now jointly sponsored by NMSS and The European Committee for Treatment and Research in MS) and other experts (The MS Phenotype Group) re-examined MS phenotypes, exploring clinical, imaging, and biomarker advances through working groups and literature searches. In October 2012, we convened to review the 1996 clinical course descriptions and determine if sufficient progress and new insights were available to recommend changes. The specific goals of the meeting were as follows:
Re-examine the 1996 phenotype descriptions to determine whether they could be better characterized by including improved clinical descriptive terminology, MRI and other imaging techniques, analysis of fluid biomarkers, and other assays including neurophysiology.
Produce a summary of our discussions that presents what we know, what we recommend, and what we still need to know.
Recommend research strategies to move the field forward where data or consensus are lacking.
KEY CONSENSUS POINTS
Retaining the basics of the 1996 phenotype descriptions, with clarifications.
It was believed that the 1996 phenotype descriptions had become part of standard MS practice and clinical research. The Group recommended that the basic features of the original descriptions should be maintained, with modifications and clarifications, as discussed below.
We noted that the diagnosis of MS should be made on clinical grounds with input from imaging and other paraclinical studies, where needed.2 The clinical phenotype may be assessed based on current status and historical data, with the understanding that this can be a dynamic process and that the subtype on initial assessment may change over time. For example, an RR subtype may transition into an SP subtype.
New disease courses.
Clinically isolated syndrome.
Clinically isolated syndrome (CIS) was not included in the initial MS clinical descriptors. CIS is now recognized as the first clinical presentation of a disease that shows characteristics of inflammatory demyelination that could be MS, but has yet to fulfill criteria of dissemination in time.3 Natural history studies and clinical trials of MS disease-modifying therapies have shown that CIS coupled with brain MRI lesions carries a high risk for meeting diagnostic criteria for MS.4–7 Clinical trials of MS disease-modifying agents show fewer treated individuals with CIS who develop a second exacerbation (the defining event for “clinically definite MS”) and reduced MRI activity.8–11 Regulatory acceptance of agents used in CIS to delay confirmed diagnosis of MS has further established CIS as an element of the MS phenotype spectrum.12 Use of the 2010 revisions to the McDonald MS diagnostic criteria allows some patients with a single clinical episode to be diagnosed with MS based on the single scan criterion for dissemination in time and space,2 reducing the number of patients who will be categorized as CIS.
Radiologically isolated syndrome.
A more complicated situation is the radiologically isolated syndrome (RIS), where incidental imaging findings suggest inflammatory demyelination in the absence of clinical signs or symptoms.13–15 RIS was not considered an MS subtype per se since clinical evidence of demyelinating disease (a current criterion for MS diagnosis) is lacking and MRI findings alone may be nonspecific. However, RIS may raise the suspicion of MS, depending on the morphology and location of detected MRI lesions. Changes on brain imaging that are highly suggestive of demyelinating pathology carry the greatest risk of future MS clinical symptoms.15 Asymptomatic spinal cord lesions, gadolinium-enhancing lesions, or positive CSF findings enhance the likelihood of an eventual MS diagnosis.16,17 An RIS patient with no obvious clinical signs or symptoms suggestive of MS should be followed prospectively. Until more information is available from prospective RIS cohorts, RIS should not be considered a distinct MS phenotype.
Defining SPMS.
In most clinical contexts, SPMS is diagnosed retrospectively by a history of gradual worsening after an initial relapsing disease course, with or without acute exacerbations during the progressive course. To date, there are no clear clinical, imaging, immunologic, or pathologic criteria to determine the transition point when RRMS converts to SPMS; the transition is usually gradual. This has limited our ability to study the imaging and biomarker characteristics that may distinguish this course. We suggest that modeling of existing clinical trial and natural history datasets might provide answers to these questions.
PPMS.
While some evidence suggests that PPMS represents a distinct, noninflammatory or at least less inflammatory pathologic form of MS,18 abundant clinical, imaging, and genetic data suggest that PPMS is a part of the spectrum of progressive MS phenotypes and that any differences are relative rather than absolute.16,19 Analyses of natural history cohorts demonstrate that worsening proceeds at a similar rate in SPMS and PPMS.20,21 PPMS should remain a separate clinical course because of the absence of exacerbations prior to clinical progression, but it likely does not have pathophysiologically distinct features from relapsing forms of MS that have entered a progressive course (SPMS).
Modifiers of basic MS phenotypes: Incorporating disease activity and disease progression.
MS phenotypes can be categorized as relapsing or progressive in the context of current medical status and history, but these categories do not provide temporal information about the ongoing disease process. The MS Phenotype Group believes that disease activity detected by clinical relapses or imaging (gadolinium-enhancing lesions or new or unequivocally enlarging T2 lesions) as well as progression of disability can be meaningful additional descriptors in either relapsing or progressive disease. Evidence of disease activity and clinical progression, which by current understanding reflects ongoing inflammatory or neurodegenerative processes,18 may impact prognosis, therapeutic decisions, and clinical trial designs and outcomes.
Assessment of activity.
The Group recommended at least annual assessment of disease activity by clinical and brain imaging criteria for relapsing MS. For progressive MS, annual clinical assessment is recommended, but there was no consensus on the optimal frequency of imaging that would be useful for progressive forms of MS. Because there is a strong association of brain and spinal cord MRI activity,22 and because information from spinal cord imaging findings in the absence of brain imaging findings is limited, annual cord imaging is not recommended unless there are spinal clinical findings.23 We chose an annual time frame as a minimum, which is a practical period for assessments. Shorter or longer assessment periods may be appropriate for certain situations. In any case, the assessment period for both clinical and imaging outcomes should be specified. As an example, a patient with RRMS who had a new gadolinium-enhancing lesion on a current MRI would be considered to be RR–active (figure 1). Conversely, “not active” as a phenotype modifier could be used in the same way, to indicate a patient with a relapsing course but no relapses, gadolinium-enhancing activity, or new or unequivocally enlarging T2 lesions during the assessment period. Patients not assessed over a designated time frame would be considered “activity indeterminate.” As with the MS diagnostic criteria, careful attention to technical aspects of serial scanning procedures and interpretation are critical. This is particularly important in assessing new or enlarging T2 lesions.
Figure 1. The 1996 vs 2013 multiple sclerosis phenotype descriptions for relapsing disease.
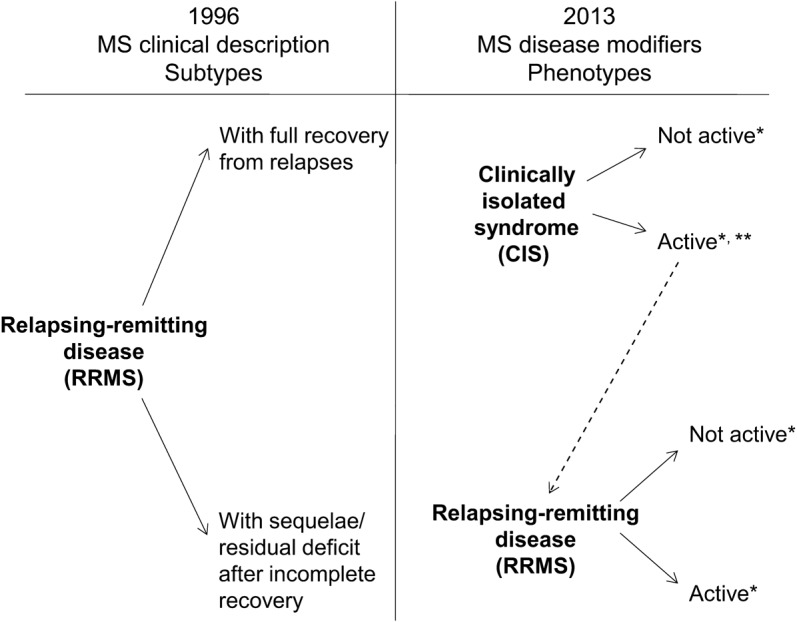
*Activity determined by clinical relapses and/or MRI activity (contrast-enhancing lesions; new or unequivocally enlarging T2 lesions assessed at least annually); if assessments are not available, activity is “indeterminate.” **CIS, if subsequently clinically active and fulfilling current multiple sclerosis (MS) diagnostic criteria, becomes relapsing-remitting MS (RRMS).
Inclusion of activity as a modifier of a basic clinical course phenotype allows elimination of the PRMS category. A patient with PPMS who has an acute attack (thus fulfilling prior criteria for PRMS) would be considered to be PP–active. On the other hand, a patient with PPMS with no acute attacks and no MRI activity would be considered to be PP–not active.
Assessment of progression.
An additional modifier of disease course is whether or not there is clinical evidence of disease progression, independent of relapses, over a given period of time in patients who have a progressive disease course (PPMS or SPMS). Progressive disease does not progress in a uniform fashion and may remain relatively stable over periods of time.24,25 We suggest that progression be determined annually by history or objective measure of change. Thus, a patient with PPMS who has not progressed over the past year would be classified as PPMS–not progressing. A patient with SPMS who has gradually worsened and has gadolinium-enhancing lesions on MRI would be classified as SPMS–active and progressing (figure 2).
Figure 2. The 1996 vs 2013 multiple sclerosis phenotype descriptions for progressive disease.

*Activity determined by clinical relapses assessed at least annually and/or MRI activity (contrast-enhancing lesions; new and unequivocally enlarging T2 lesions). **Progression measured by clinical evaluation, assessed at least annually. If assessments are not available, activity and progression are “indeterminate.” MS = multiple sclerosis; PP = primary progressive; PR = progressive relapsing; SP = secondary progressive.
We recognize that either relapsing or progressive disease may be characterized by severity of signs and symptoms, frequency of relapses, rate of worsening, residual disability, and impairment. However, there are insufficient data to further characterize an active disease course in this way. Degree of recovery from an acute relapse was considered to be, in itself, not useful for determining or modifying MS phenotypes, but is instead a contributor to disease worsening over time. These areas represent fertile topics for future research.
Sustained or confirmed worsening: Clarifying terminology.
Many studies have used the term sustained worsening as a clinical trials outcome, referring to a worsening of the Expanded Disability Status Scale (EDSS) score that persists for a specified period of time (usually 3 or 6 months).26 This has been interpreted as a measure of worsening disability. We suggest that sustained implies a permanence that is sometimes not a characteristic of disease change in MS and is therefore a potentially misleading concept. Further, it is possible for the EDSS to worsen in different functional systems within the designated time frame and still appear to be sustained, whereas it may be improving in one or more functional systems while worsening in others. We suggest that the term confirmed be used rather than sustained to guide evaluation of worsening disability. Thus, confirmed accumulation of disability would be defined by a worsening of EDSS that persists over x months, agnostic to functional system, as has frequently been used. A more rigorous definition would require that worsening be confirmed in the same functional system.
In this context, there is a need for better clarity in the use of the terms disease or disability progression, which have been used to describe worsening from multiple attacks, poor recovery from a severe attack, or onset of a progressive phase of the illness. We suggest using the term worsening in place of progressing especially for patients with relapsing forms of disease, reserving the term progression only for those in the progressive phase of MS, independent of relapse activity. This would also hold true for the characterization of confirmed change in EDSS, discussed above.
Detecting activity and progression: Much to be discovered.
Some clinical manifestations may be too subtle to easily detect no matter how frequently assessed. Following patients closely for cognitive, visual, and other clinical changes could provide clinical evidence for disease activity. Consensus is lacking about how to use patient-reported outcomes (PRO) and their utility as indicators of disease status. Tools for the remote assessment of patient performance outside of clinical settings may prove useful in better understanding PRO and more correlative research in this area would be useful.
While T2 and gadolinium-enhancing lesions are measures of disease activity, sufficient consensus has not been achieved about other measures of tissue damage to allow their inclusion in phenotypic descriptions, although there were some initial steps to use MRI measures of inflammation and tissue loss to categorize patients into clinical subgroups.27 Assessment and interpretation of brain volume loss and black hole evolution28 lack standardization, which limits their practical application outside of research settings and may not adequately discriminate among the clinical phenotypes at the level of the individual patient. Newer imaging modalities such as diffusion tensor imaging and magnetization transfer imaging are not yet optimized for clinical use. Optical coherence tomography (OCT) may show correlations between retinal nerve fiber layer thickness and visual acuity,29 but there is currently insufficient information to suggest that OCT can be used as an indirect measure of whole brain tissue loss. Further information about these imaging assessments and their role as potential markers of disease type or course is a high research priority.
Fluid-borne biological markers and electrophysiology.
While it was hoped that the original MS phenotype descriptors would be supported and better defined by biological markers, to date no blood or CSF biological marker reliably and reproducibly differentiates between MS disease phenotypes. There is a pressing need, using large datasets of clinically and radiologically well-characterized patient pools, to explore in detail the potential that biological markers may have in supporting (and in the future, further refining) MS phenotype descriptions.
We recognized the potential value of electrophysiology studies to help define MS disease subtypes. However, we noted that there is considerable interlaboratory variability in such measures. Standardization of procedures and assessment will be essential if evoked potentials are to be of additional value in assessing MS phenotype.
Benign and malignant MS.
The terms benign and malignant are not MS phenotype descriptors per se, but rather were intended to provide an indication of disease severity over time and were described “by consensus.”1 These terms can, in theory, apply to any MS phenotype, depending on degree of activity over time or impairment/disability at any given point in time. These terms, especially the term benign, which should always be a retrospective determination, are often misunderstood and misused. In a long-term disease like MS, the severity and activity of the disease can change significantly and unpredictably. We recommend that these terms be used with caution.
Further refinements to MS phenotypes: The need for more research.
The original definitions of MS clinical phenotypes from 1996 cited the need for objective imaging and fluid-borne biological markers. Limited progress has been made in the intervening years, and much prospective research is needed to understand whether biological markers can enhance our understanding of MS disease subtypes.
Data still do not provide compelling imaging distinctions among the original 1996 clinical descriptors nor do they yet provide grounds for significantly altering the core disease subtypes of relapsing and progressive MS. Some of the data originally supporting imaging differences between primary and secondary progressive MS30 have been qualified by more recent data,31 suggesting that differences in pathology seen by contrast-enhancing brain lesions between the 2 forms of the disease are less clear than originally thought. Patients with PPMS based on clinical patterns may frequently show contrast-enhancing brain lesions particularly in the earlier stages,31 thus making the pathologic boundary between the 2 forms of progressive disease less clear.
The need to follow cohorts of clinically well-defined patients with a spectrum of clinical and imaging assessments, laboratory markers, and tools such as OCT is a priority. Such studies will be essential to determine whether these objective indicators of a patient's biological status can contribute to our understanding of MS disease subtypes and in particular to better understand and predict the transition between different disease subtypes.
DISCUSSION
The MS Phenotype Group has reconsidered prior MS disease course descriptors, some 16 years after their original publication. We recommend the following:
The core MS phenotype descriptions of relapsing and progressive disease should be retained with some modifications (table 1).
An important modifier of these core phenotypes is an assessment of disease activity, as defined by clinical assessment of relapse occurrence or lesion activity detected by CNS imaging.
The second important modifier of these phenotypes is a determination of whether progression of disability has occurred over a given time period.
The prior category of PRMS can be eliminated since subjects so categorized would now be classified as PP patients with disease activity.
PPMS is a part of the spectrum of progressive disease and differences from other forms are relative rather than absolute.
CIS should be included in the spectrum of MS phenotypes. Prospective follow-up of most such patients should determine their subsequent disease phenotype.
RIS should not be considered a separate MS phenotype, since such patients lack clinical signs and symptoms of the disease. Prospective follow-up is recommended.
Use of the term worsening is preferable and less confusing than the term progressing to describe a patient in the relapsing phase of disease whose disease is advancing due to frequent relapses and/or incomplete relapse recovery.
In considering clinical trial or natural history assessment of worsening disease by EDSS or other metrics, use the term confirmed rather than sustained over a defined period of time, either within (more rigorously) the functional system or without considering the specific functional systems in which worsening is detected.
The terms “benign” and “malignant” disease are often misused and should be used with caution.
Further research is needed to better define the value of imaging and biological markers in assessing, confirming, or revising MS phenotype descriptions (table 2).
Table 1.
Definitions related to multiple sclerosis phenotypes used in this article
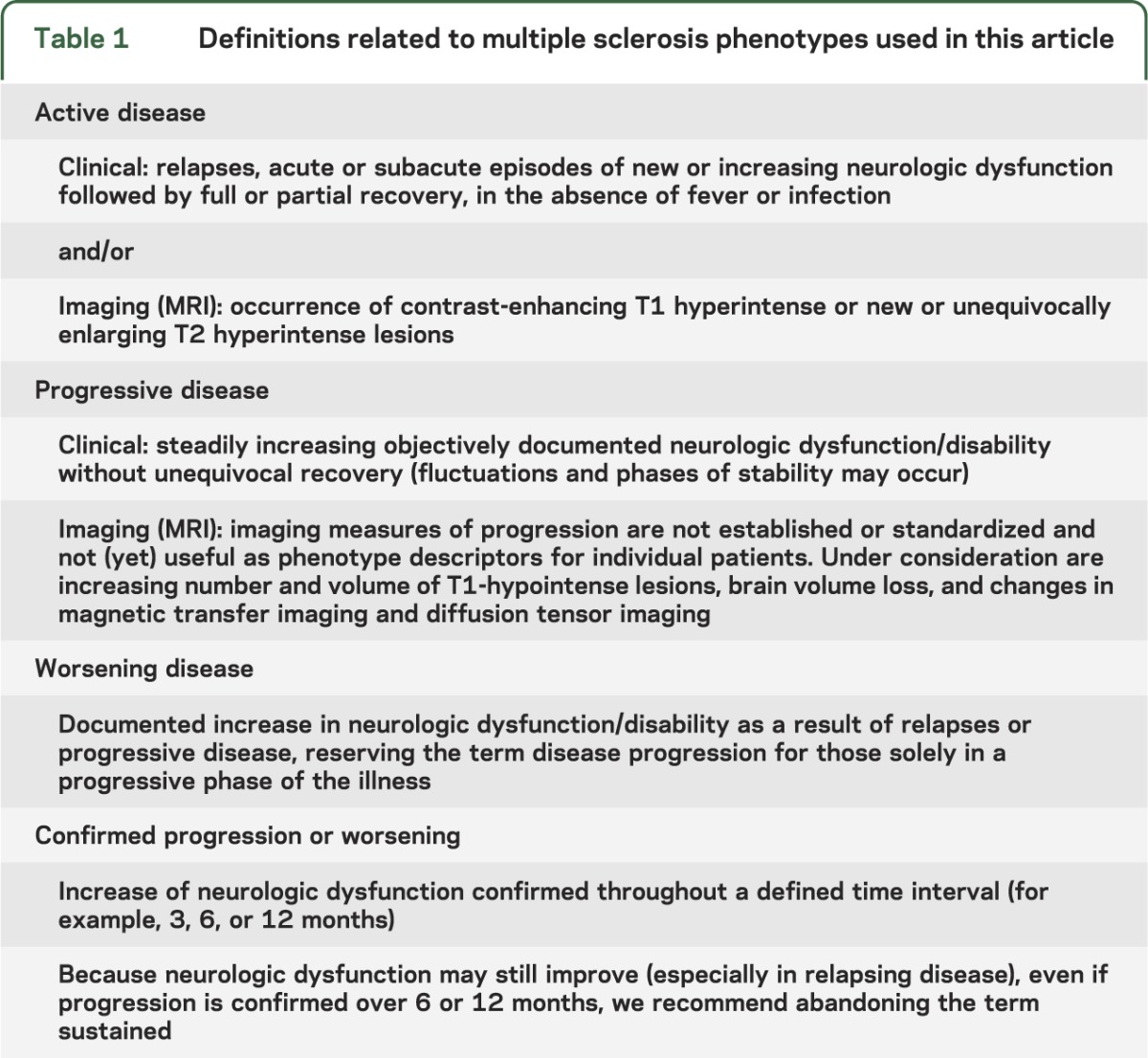
Table 2.
Future research needed to better understand and define multiple sclerosis phenotypes

Assessing clinical and MRI status can provide a means to determine activity for future research studies and for consideration in clinical practice. At present, there are no evidence-based guidelines for using activity assessment for management decisions in clinical practice.
There was consensus among the group that clinical assessments for activity and progression should be dictated by individual disease evolution, but should occur at least annually. Annual brain MRI scanning for activity in relapsing forms of MS was believed to be useful. There was no consensus on how frequently to scan progressive patients. However, subtyping progressive patients by MRI activity may be particularly valuable for clinical and translational research studies. As has been the case with the diagnostic criteria revisions, careful interpretation of MRI is required especially for determination of new and enlarging T2 lesions.
The addition of markers of activity (clinical exacerbations or MRI-detected lesions) and measures of disease progression should make communicating with patients and among physicians clearer and should also enhance design, recruitment, and conduct of clinical trials. In such studies, attention should be paid to stratifying enrollment and analysis by disease subtype. These markers may also prove useful in studies to determine when therapy should be discontinued.
We recognize that there may be other markers of disease activity, but there is insufficient evidence for including them at this time. We also recognize that one could consider progression as a sign of activity, but we recommend treating it in a conceptually different context so as to distinguish it from more acute changes. While some have proposed underlying pathologic differences for these clinical or MRI events, we have intentionally avoided drawing pathologic conclusions until more data become available. As we proposed to do in 1996 with the original MS phenotype descriptions, we hope that these modifications will serve to better characterize patients with MS and provide a framework for both clinical research and ongoing clinical care.
Supplementary Material
ACKNOWLEDGMENT
The activities and meeting of the MS Phenotype Group were undertaken and organized by the International Advisory Committee on Clinical Trials in Multiple Sclerosis.
GLOSSARY
- CIS
clinically isolated syndrome
- EDSS
Expanded Disability Status Scale
- MS
multiple sclerosis
- NMSS
National Multiple Sclerosis Society
- OCT
optical coherence tomography
- PP
primary progressive
- PR
progressive relapsing
- PRO
patient-reported outcomes
- RIS
radiologically isolated syndrome
- RR
relapsing-remitting
- SP
secondary progressive
Footnotes
Editorial, page 206
AUTHOR CONTRIBUTIONS
Dr. Lublin participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Reingold participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Cohen participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Cutter participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Sorensen participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Thompson participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Wolinsky participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Balcer participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Banwell participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Barkhof participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Bebo participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Calabresi participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Clanet participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Comi participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Fox participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Freedman participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Goodman participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Inglese participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Kappos participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Kieseier participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Lincoln participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Lubetzki participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Miller participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Montalban participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. O'Connor participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Petkau participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Pozzilli participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Rudick participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Sormani participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Stuve participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Waubant participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. Polman participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content.
STUDY FUNDING
The International Advisory Committee on Clinical Trials in Multiple Sclerosis is supported by the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) and by the US National Multiple Sclerosis Society (NMSS). The 2012 conference that resulted in this article was supported by these organizations and by the Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS), the Multiple Sclerosis International Federation (MSIF), and the Multiple Sclerosis Society of Canada (MSSC).
DISCLOSURE
F. Lublin: sources of funding for research: Acorda Therapeutics, Inc., Biogen Idec, Novartis Pharmaceuticals Corp., Teva Neuroscience, Inc., Genzyme, Sanofi, Celgene, NIH, NMSS; consulting agreements/advisory boards/DSMB: Bayer HealthCare Pharmaceuticals, Biogen Idec, EMD Serono, Inc., Novartis, Teva Neuroscience, Actelion, Sanofi-Aventis, Acorda, Questcor, Roche, Genentech, Celgene, Johnson & Johnson, Revalesio, Coronado Bioscience, Genzyme, MedImmune, Bristol-Myers Squibb, Xenoport, Receptos, Forward Pharma; co-chief editor: Multiple Sclerosis and Related Diseases; stock ownership: Cognition Pharmaceuticals, Inc. S. Reingold has served as a consultant to the National Multiple Sclerosis Society (USA) and the European Committee for Treatment and Research in MS and has received honoraria and travel reimbursement for service on data safety monitoring boards or advisory boards for Bayer HealthCare, Coronado Biosciences Inc., Eli Lilly & Company, EMD Merck Serono, Genentech, F. Hoffmann-LaRoche, Ironwood Pharmaceuticals Inc., ISIS Pharmaceuticals Inc., MedImmune Inc., Novartis Pharmaceuticals Corporation, Observatoire Français de la Sclérosis en Plaques, Opexa Therapeutics, Sanofi-Aventis, SK Biopharmaceuticals, Synthon Pharmaceuticals Inc., and Teva Pharmaceutical Industries; and served as an editorial board member of the Multiple Sclerosis Journal. J. Cohen reports personal compensation for consulting from Novartis and Teva and research support paid to his institution from Biogen Idec, Genzyme, Novartis, Receptos, Synthon, Teva, and Vaccinex. G. Cutter: participation on data and safety monitoring committees: Apotek, Biogen-Idec, Cleveland Clinic, GlaxoSmithKline Pharmaceuticals, Gilead Pharmaceuticals, Modigenetech/Prolor, Merck/Ono Pharmaceuticals, Merck, Neuren, PCT Bio, Revalesio, Sanofi-Aventis, Teva, Vivus, NHLBI (Protocol Review Committee), NINDS, NMSS, NICHD (OPRU oversight committee). Consulting, speaking fees, and advisory boards: Alexion, Allozyne, Bayer, Consortium of MS Centers (grant), Klein-Buendel Incorporated, Genzyme, Medimmune, Novartis, Nuron Biotech, Receptos, Revalesio, Sanofi-Aventis, Spiniflex Pharmaceuticals, Somahlution, Teva Pharmaceuticals, Xenoport. Dr. Cutter is employed by the University of Alabama at Birmingham and President of Pythagoras, Inc., a private consulting company located in Birmingham. P. Sorensen received personal compensation from Biogen Idec, Merck Serono, Novartis, Genmab, Teva, Elan, GSK, Bayer Schering, Sanofi-Aventis, and Genzyme as member of scientific advisory boards, steering committees, or independent data monitoring boards in clinical trials, or as speaker at meetings. His research unit has received research support from Biogen Idec, Bayer Schering, Merck Serono, Sanofi-Aventis, and Novartis. A. Thompson is chair of the Eisai-UCL Joint Steering Committee for Neuroscience (honorarium to UCL); member, Imanova Advisory Board (no remuneration); chair, MSIF International Medical and Scientific Board (support for travel); trustee, Development Foundation Brain Appeal, National Hospital for Neurology and Neurosurgery, Queen Square (no remuneration); received grants from NIHR as a Senior Investigator; is co-recipient of grants from the Wolfson Foundation, MRC, Wellcome Trust, MS Society of GB, SRH Holding, and Eisai Inc.; received an honorarium and support for travel for consultancy from Genzyme; honoraria and support for travel for invited lectures from Novartis, Serono Symposia International Foundation, Teva, and Remedica; receives an honorarium as Editor-in-Chief for Multiple Sclerosis Journal, a free subscription as member of Lancet Neurology Editorial Board, and publishing royalties from Cambridge University Press. J. Wolinsky has received compensation for service on steering committees or data monitoring boards for Novartis Pharmaceuticals, Sanofi, and Teva Pharmaceuticals; reimbursement for services as consultant to Athersys, Inc., Bayer HealthCare, Celgene, Genentech, Genzyme, Novartis, F. Hoffmann-La Roche, Ltd., Jansen RND, Teva and Teva Neurosciences, and XenoPort. Honoraria were provided from ACTRIMS, the Consortium of Multiple Sclerosis Centers, Medscape CME, Prime, Serono Symposia International Foundation, Teva Pharmaceuticals, and Teva Neuroscience. He has received or receives research support from Genzyme, Sanofi, NIH, the Clayton Foundation for Research, and the National Multiple Sclerosis Society through the University of Texas Health Science Center at Houston (UTHSCH) and royalties for monoclonal antibodies out-licensed to Chemicon International through UTHSCH. L. Balcer has received consulting and/or advisory board service fees from Biogen-Idec, Genzyme, and Questcor. B. Banwell serves as a senior editor for Multiple Sclerosis and Related Disorders and on the editorial board of Neurology®. Dr Banwell serves on a steering committee for Biogen Idec, Novartis, and Sanofi. F. Barkhof serves as a consultant for Bayer-Schering Pharma, Sanofi-Aventis, Biogen Idec, Teva, Merck Serono, Novartis, Roche, Synthon, and Jansen Research. B. Bebo reports no disclosures relevant to the manuscript. P. Calabresi has received personal compensation for consulting and serving on scientific advisory boards from Vertex, Vaccinex, Medimmune, Prothena, and Abbott, and has received research funding from Biogen-Idec, Abbott, Vertex, Novartis, and Bayer. M. Clanet has received consulting fees for advisory board membership from Novartis, Genzyme, and Teva and for participation in a symposium from Merck Serono; and has received a grant in support of a scientific workshop from Teva and a grant for research from Biogen-Idec. G. Comi has received compensation for consulting services and/or speaking activities from Novartis, Teva, Sanofi, Genzyme, Merck Serono, Biogen, Bayer, Serono Symposia International Foundation, Almirall, and Actelion. R. Fox has received consultant fees from Allozyne, Avanir, Biogen Idec, EMD Serono, Novartis, Questcor, Teva, and XenoPort; has received research support from Biogen Idec and Novartis; and serves on the editorial boards of Neurology and Multiple Sclerosis Journal. M. Freedman: receipt of research or educational grants: Bayer Healthcare, Genzyme; receipt of honoraria or consultation fees: Bayer Healthcare, Biogen Idec, EMD Canada, Genzyme, Novartis, Sanofi-Aventis, Teva Canada Innovation; member of a company advisory board, board of directors, or other similar group: Actelion, Bayer Healthcare, Biogen Idec, Celgene, Hoffman LA-Roche, Merck Serono, Novartis, Opexa, Sanofi-Aventis; participation in a company-sponsored speaker's bureau: Genzyme. A. Goodman has received consulting fees from Acorda Therapeutics, Avanir, Biogen-Idec, EMD Serono, Genzyme-Sanofi, Novartis, Pfizer, and Teva; and has received grant support from Acorda Therapeutics, Avanir, Biogen-Idec, EMD Serono, Genzyme, Novartis, Ono, Sun Pharma, Takeda, and Teva. M. Inglese has received research grants from Novartis Pharmaceuticals and is a consultant for Vaccinex Inc. L. Kappos reports fees paid to his institution for his services related to advisory boards, steering committees, or data safety monitoring committees from Actelion, Advancell, Allozyne, Bayer HealthCare, Biogen-Idec, Genmab, Merck Serono, MediciNova, Novartis, Santhera, Roche, Sanofi-Aventis, UCB, and Wyeth; fees paid to his institution for his consulting services provided to NovoNordisk, Peptimmune, Glenmark, BioMarin, GeNeuro SA, Eli Lilly, SCL Behring, and Bayhill; payments to his institution for his lectures or speakers bureau services from Bayer HealthCare, Biogen-Idec, Merck Serono, Novartis, Sanofi-Aventis, Roche, and Teva; payments to his institution for development of educational presentations from Bayer HealthCare, Merck Serono, Novartis, Sanofi-Aventis, Roche, Teva, and UCB; and grants or pending grants to his institution from Bayer HealthCare, Biogen-Idec, Merck Serono, Novartis, Sanofi-Aventis, Teva Roche, the Swiss Multiple Sclerosis Society, the Swiss National Research Foundation, the European Union, the Gianni Rubatto Foundation, the Roche Research Foundation, and the Novartis Research Foundation. B. Kieseier has received honoraria for lecturing, travel expenses for attending meetings, and financial support for research from Bayer Health Care, Biogen Idec, Genzyme/Sanofi Aventis, Grifols, Merck Serono, Mitsubishi Europe, Novartis, Roche, Talecris, and Teva. J. Lincoln has received honoraria from serving as a speaker for Acorda Pharmaceuticals, Biogen Idec, Novartis, Pfizer, Sanofi-Aventis, and Teva Pharmaceuticals and served on the advisory board for Questcor, Sanofi-Aventis, and Teva Pharmaceuticals. C. Lubetzki received personal compensation for activities (participation in advisory boards, lectures) with Roche, Genzyme, Novartis, Biogen, Serono Symposia International Foundation, and Teva-Aventis. A. Miller has served as a consultant and/or participant in advisory board meetings for Genzyme/Sanofi-Aventis, Biogen Idec, Glaxo Smith Kline, EMD Serono (Merck Serono), Novartis, ONO, Acorda, Nuron Biotech, Teva, Questcor, and Accordant Health Services. He has received research support from Acorda, Novartis, Genentech, Genzyme/Sanofi-Aventis, Biogen Idec, Roche, and Questcor. He has served as Editor of Continuum, a continuing medical education publication of the AAN, and currently serves as Editor of Continuum audio. He is a member of the editorial board of Multiple Sclerosis and Related Disorders. He occasionally performs expert reviews of medical records or serves as an expert witness in medical malpractice cases. X. Montalban has received consulting fees, speaker fees, and/or travel support from Almiral, Bayer HealthCare, Biogen-Idec, EMD Serono, Merck, Genentech, GeNeuro, Genzyme, Neurotec, Novarits, Sanofi-Genzyme, and Teva. P. O'Connor is a consultant for Sanofi Genzyme, Biogen Idec, Roche, Teva, EMD Serono, and Novartis. He served on DMCs for Teva. J. Petkau holds research funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes for Health Research (CIHR), and over the last 2 years has received research funds from Bayer Pharma and travel support, honoraria, consulting fees, or fees for service on data safety monitoring boards or steering committees from Bayer Canada, Bayer Pharma, Bayhill Therapeutics, EMD Serono, Merck-Serono, Novartis, the Canadian Study Group on CCSVI, and the Myelin Repair Foundation. C. Pozzilli received honoraria from serving on the scientific advisory board of Biogen Idec, Teva Pharmaceutical, Merck Serono, Novartis, Actelion, Almirall, Genzyme, Sanofi Aventis, and GW Pharma, and received research support from Novartis, Merck Serono, Biogen, and institutional support from the Sapienza University of Rome. R. Rudick reports consulting income from Biogen Idec, Genzyme, and Novartis, and research funding to his institution from Genzyme, Novartis, the National MS Society, and NIH. M. Sormani reports personal consulting fees from Merck Serono, Biogen Idec, Teva, Actelion, Synthon, and Novartis, outside the submitted work. She serves as an editorial board member of the Multiple Sclerosis Journal. O. Stuve serves on the editorial boards of JAMA Neurology, Multiple Sclerosis Journal, and Therapeutic Advances in Neurological Disorders. He has received grant support from Teva Pharmaceuticals. E. Waubant has received honorarium from Teva, Sanofi-Aventis, and Genentech for 3 educational lectures, and is on the advisory board for a trial of Novartis. Dr. Waubant has received free medication from Biogen Idec and Sanofi-Aventis for a trial. C. Polman has received compensation for activities from Actelion, Biogen Idec, Glaxo Smith Kline, Merck Serono, MorphoSys AG, Novartis, Receptos, and Teva as consultant/committee member/speaker. He has received research support from Biogen Idec, Bayer Schering, Glaxo Smith Kline, Merck Serono, Novartis, Teva, UCB, and Roche. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology 1996;46:907–911 [DOI] [PubMed] [Google Scholar]
- 2.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis, part I: natural history, pathogenesis, diagnosis, and prognosis. Lancet Neurol 2005;4:281–288 [DOI] [PubMed] [Google Scholar]
- 4.Tintore M, Rovira A, Martinez MJ, et al. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. AJNR Am J Neuroradiol 2000;21:702–706 [PMC free article] [PubMed] [Google Scholar]
- 5.O'Riordan JI, Thompson AJ, Kingsley DP, et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS: a 10-year follow-up. Brain 1998;121:495–503 [DOI] [PubMed] [Google Scholar]
- 6.Filippi M, Horsfield MA, Morrissey SP, et al. Quantitative brain MRI lesion load predicts the course of clinically isolated syndromes suggestive of multiple sclerosis. Neurology 1994;44:635–641 [DOI] [PubMed] [Google Scholar]
- 7.Morrissey SP, Miller DH, Kendall BE, et al. The significance of brain magnetic resonance imaging abnormalities at presentation with clinically isolated syndromes suggestive of multiple sclerosis: a 5-year follow-up study. Brain 1993;116:135–146 [DOI] [PubMed] [Google Scholar]
- 8.Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis: CHAMPS Study Group. N Engl J Med 2000;343:898–904 [DOI] [PubMed] [Google Scholar]
- 9.Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet 2001;357:1576–1582 [DOI] [PubMed] [Google Scholar]
- 10.Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006;67:1242–1249 [DOI] [PubMed] [Google Scholar]
- 11.Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet 2009;374:1503–1511 [DOI] [PubMed] [Google Scholar]
- 12.Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler 2008;14:1157–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lebrun C, Bensa C, Debouverie M, et al. Association between clinical conversion to multiple sclerosis in radiologically isolated syndrome and magnetic resonance imaging, cerebrospinal fluid, and visual evoked potential: follow-up of 70 patients. Arch Neurol 2009;66:841–846 [DOI] [PubMed] [Google Scholar]
- 14.Siva A, Saip S, Altintas A, Jacob A, Keegan BM, Kantarci OH. Multiple sclerosis risk in radiologically uncovered asymptomatic possible inflammatory-demyelinating disease. Mult Scler 2009;15:918–927 [DOI] [PubMed] [Google Scholar]
- 15.Okuda DT, Mowry EM, Beheshtian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurology 2009;72:800–805 [DOI] [PubMed] [Google Scholar]
- 16.Okuda DT, Mowry EM, Cree BA, et al. Asymptomatic spinal cord lesions predict disease progression in radiologically isolated syndrome. Neurology 2011;76:686–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giorgio A, Stromillo ML, Rossi F, et al. Cortical lesions in radiologically isolated syndrome. Neurology 2011;77:1896–1899 [DOI] [PubMed] [Google Scholar]
- 18.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8:647–656 [DOI] [PubMed] [Google Scholar]
- 19.Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol 2007;17:210–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebers GC. Natural history of primary progressive multiple sclerosis. Mult Scler 2004;10(suppl 1):S8–S13 [DOI] [PubMed] [Google Scholar]
- 21.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain 2006;129:606–616 [DOI] [PubMed] [Google Scholar]
- 22.Thorpe JW, Kidd D, Moseley IF, et al. Serial gadolinium-enhanced MRI of the brain and spinal cord in early relapsing-remitting multiple sclerosis. Neurology 1996;46:373–378 [DOI] [PubMed] [Google Scholar]
- 23.Bot JC, Barkhof F. Spinal-cord MRI in multiple sclerosis: conventional and nonconventional MR techniques. Neuroimaging Clin N Am 2009;19:81–99 [DOI] [PubMed] [Google Scholar]
- 24.Wolinsky JS. The PROMiSe trial: baseline data review and progress report. Mult Scler 2004;10(suppl 1):S65–S71 [DOI] [PubMed] [Google Scholar]
- 25.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688 [DOI] [PubMed] [Google Scholar]
- 26.Rudick RA, Kappos L. Measuring disability in relapsing-remitting MS. Neurology 2010;75:296–297 [DOI] [PubMed] [Google Scholar]
- 27.Bielekova B, Kadom N, Fisher E, et al. MRI as a marker for disease heterogeneity in multiple sclerosis. Neurology 2005;65:1071–1076 [DOI] [PubMed] [Google Scholar]
- 28.van den Elskamp IJ, Boden B, Dattola V, et al. Cerebral atrophy as outcome measure in short-term phase 2 clinical trials in multiple sclerosis. Neuroradiology 2010;52:875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galetta KM, Calabresi PA, Frohman EM, Balcer LJ. Optical coherence tomography (OCT): imaging the visual pathway as a model for neurodegeneration. Neurotherapeutics 2011;8:117–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson AJ, Kermode AG, Wicks D, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann Neurol 1991;29:53–62 [DOI] [PubMed] [Google Scholar]
- 31.Ingle GT, Sastre-Garriga J, Miller DH, Thompson AJ. Is inflammation important in early PPMS? A longitudinal MRI study. J Neurol Neurosurg Psychiatry 2005;76:1255–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.