

Abstract
Objective:
To identify factors influencing age at symptom onset and disease course in autosomal dominant Alzheimer disease (ADAD), and develop evidence-based criteria for predicting symptom onset in ADAD.
Methods:
We have collected individual-level data on ages at symptom onset and death from 387 ADAD pedigrees, compiled from 137 peer-reviewed publications, the Dominantly Inherited Alzheimer Network (DIAN) database, and 2 large kindreds of Colombian (PSEN1 E280A) and Volga German (PSEN2 N141I) ancestry. Our combined dataset includes 3,275 individuals, of whom 1,307 were affected by ADAD with known age at symptom onset. We assessed the relative contributions of several factors in influencing age at onset, including parental age at onset, age at onset by mutation type and family, and APOE genotype and sex. We additionally performed survival analysis using data on symptom onset collected from 183 ADAD mutation carriers followed longitudinally in the DIAN Study.
Results:
We report summary statistics on age at onset and disease course for 174 ADAD mutations, and discover strong and highly significant (p < 10−16, r2 > 0.38) correlations between individual age at symptom onset and predicted values based on parental age at onset and mean ages at onset by mutation type and family, which persist after controlling for APOE genotype and sex.
Conclusions:
Significant proportions of the observed variance in age at symptom onset in ADAD can be explained by family history and mutation type, providing empirical support for use of these data to estimate onset in clinical research.
Researchers have identified more than 230 different autosomal dominant Alzheimer disease (ADAD) mutations located in the genes for amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), including the canonical case discovered by Alois Alzheimer.1 There are significant differences between mutation types in age at symptom onset, and many result in onset as early as the third or fourth decade of life.2,3 Some families carrying an identical ADAD mutation can have significantly different ages at onset, suggesting the presence of other genetic or environmental modifiers of the disease process.4,5 APOE genotype was found to slightly modify age at onset in 2 ADAD kindreds,5,6 although it is not yet clear whether this is the case for ADAD in general. The factors influencing symptom onset and progression in ADAD are not fully understood, and the degree to which symptom onset may be predictable from family history has not yet been established.
Many investigators increasingly believe that treating Alzheimer disease (AD) in its early presymptomatic stages, before the accumulation of irreversible damage, may be necessary to develop effective treatments for this devastating illness.7,8 Several clinical trials are investigating treatments for AD prevention in presymptomatic ADAD mutation carriers.2,9,10 Evidence-based criteria for estimating symptom onset will significantly enhance the power of clinical research, by enabling enrollment of cohorts at well-defined time points in presymptomatic disease. In this study, we perform a meta-analysis of a large set of ADAD kindreds to investigate potential methods for estimating symptom onset in ADAD.
METHODS
Data collection.
We reviewed publications cited in the AD/Frontotemporal Dementia Mutation Database11 and the Alzheimer Research Forum Database,12 and searched PubMed with the terms “dominant Alzheimer,” “dominant AD,” “ADAD,” “presenilin,” “PSEN1,” “PSEN2,” and “APP,” identifying 137 peer-reviewed journal articles that reported age at symptom onset of affected individuals and included pedigrees recording relevant parent-offspring relationships. As further described in the discussion section, in this report “age at onset” refers to the age at onset of progressive cognitive symptoms as determined by investigators during collection of family history, rather than the age at which an individual received a clinical diagnosis of dementia or mild cognitive impairment.
Data on parent-offspring relationships and age at symptom onset were used to construct a relational database of ADAD pedigrees, represented as a data frame in the statistical computer language R.13 When available, data on sex, age at death, and APOE genotype were also included. Additional pedigree data were collected from 3 international studies: the Dominantly Inherited Alzheimer Network (DIAN) Study,9 a large Colombian kindred carrying the PSEN1 E280A mutation,14 and a set of 11 extended families of Volga German ancestry carrying an identical PSEN2 N141I mutation due to a genetic founder effect.5,15 Updated findings from a previously published multigenerational PSEN1 L286V kindred were provided by the investigator.16 To avoid potential double reporting, pedigrees for each mutation type were manually examined for possible duplicates, and these were removed where identified. The combined dataset contains 387 pedigrees including 3,275 individuals, of whom 1,307 were affected by ADAD with known age at symptom onset.
Standard protocol approvals, registrations, and patient consents.
The DIAN Study received approval from institutional review boards of participating sites, and written informed consent was obtained from all participants (or designated guardians of participants).
Associations between individual age at onset and family history measures.
We assessed 3 potential measures of family history (parental age at onset, mean age at onset of all other affected family members, and mean age at onset of all other individuals with the same mutation type) and evaluated associations between each of these measures and the actual age at onset for each affected individual.
For each affected individual, a script in the computer language R was used to calculate the mean age at onset of all other affected family members, after excluding the value for that individual to avoid ascertainment bias. A similar process was used to calculate the mean age at onset of all others affected by the same mutation type, again excluding the value for that individual to avoid bias. Correlations between individual age at onset and each of these family history measures were assessed by linear regression. We replicated these in confirmatory analyses using stratified sampling of one individual per family, and in subgroup analyses after excluding the Volga German and Colombian kindreds. We additionally assessed intragroup agreement in onset by calculating intraclass correlation coefficients (ICCs) for mutation type and family using the multilevel package in R.17
Effects of APOE genotype and sex.
Three hundred eighty-seven affected individuals (29.6%) additionally had information about APOE genotype. APOE genotypes were represented as a categorical ordered factor in R, with values ranked by order of increasing relative risk (ε2ε2 < ε2ε3 < ε3ε3 < ε2ε4 < ε3ε4 < ε4ε4) based on prior studies.18,19 Multiple regression was used to evaluate associations between individual age at onset and each of the family history measures described above, controlling for individual sex and APOE genotype as fixed effects.
Analysis of DIAN longitudinal data.
We also performed a survival analysis using data on clinician-determined ages at symptom onset for 183 ADAD mutation carriers followed longitudinally in the DIAN observational study. For mutation carriers who are currently presymptomatic, data were censored as of their age at last assessment. We compared Kaplan-Meier estimates of median onset with the mean and median ages at onset calculated from the meta-analysis dataset.
Comparison of age at onset and disease course.
For 600 affected individuals (45.6%) with known age at symptom onset and known age at death, the difference between these values was used to calculate the disease course in years from symptom onset to death. The relationship between age at onset and disease course was investigated by linear regression and polynomial regression.
RESULTS
Table e-1 on the Neurology® Web site at Neurology.org shows summary statistics for each of the 176 mutations in our dataset. Although ADAD is generally described as having early onset and rapid progression,20 our findings emphasize that this is not necessarily the case for all ADAD mutations or for all patients. Mean onset was 46.2 years for all affected patients in the combined dataset, clearly younger than the mean symptom onset of 68 years reported in late-onset AD.21 However, 57 mutations in our dataset had mean ages at onset as late as the sixth or seventh decade of life. Figure 1 displays ages at onset for all individuals grouped by the mutated gene. A mixed model adjusting for family membership as a random effect showed that mutations in PSEN2 had significantly later onset than mutations in PSEN1 and APP, and mutations in PSEN1 had significantly earlier onset than all other groups (all values significant at p < 0.0001).
Figure 1. Age at symptom onset by mutated gene.

Age at symptom onset for all affected individuals are shown grouped by the gene affected by their autosomal dominant Alzheimer disease mutation, with APP mutations having an additional subclass for gene duplications. APP = amyloid precursor protein; PS1 = presenilin 1; PS2 = presenilin 2.
Linear regression revealed strong and highly significant associations between individual age at symptom onset and each of the 3 measures of family history we assessed. Scatterplots in figure 2 show the actual age at symptom onset for each individual on the y-axis, plotted against that individual's estimated value using parental onset, mean onset of all other family members, and mean onset for all others with the same mutation type along the x-axes. Correlations between individual age at onset and each of these measures of family history were highly significant at p < 10−16. The r2 values were 0.3838 for parental onset, 0.5021 for mean onset of all other family members, and 0.5303 for mean onset by mutation type, indicating that these variables account for a substantial proportion of the observed variance in age at symptom onset.
Figure 2. Individual age at symptom onset vs measures of family onset history.
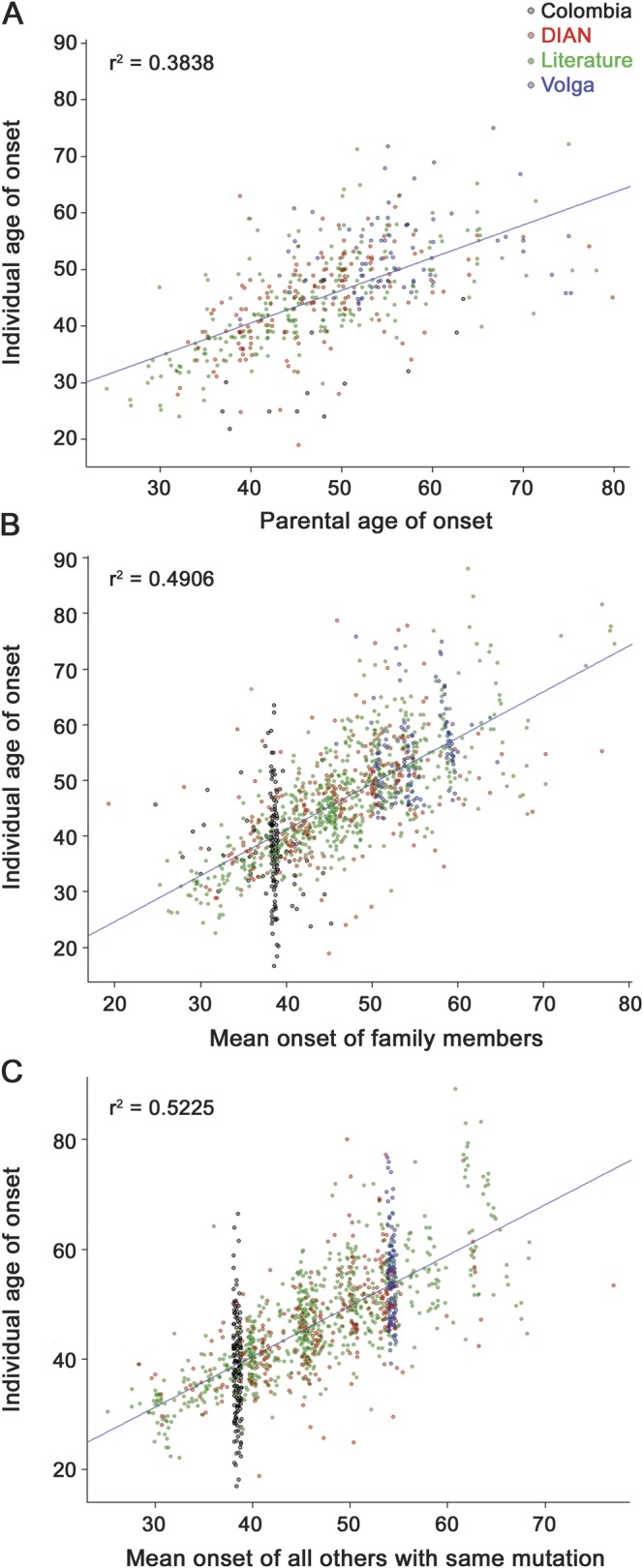
The actual age at symptom onset for each affected individual is shown on the y-axis, plotted against values predicted from family history on the x-axis. (A) Each individual's age at onset vs the reported age at onset of their affected parent. (B) Each individual's age at onset vs the mean age at onset for all other individuals in their extended family. (C) Each individual's age at onset vs the mean age at onset for all other individuals sharing the same autosomal dominant Alzheimer disease mutation type. Plot points for each individual are colored according to their study of origin, as shown in the legend at the upper right. Jitter was added to the x- and y-axes for easier visualization. Regression lines and adjusted r2 values showing correlations between individual age at onset and each of these family history measures are displayed on the respective figures. DIAN = Dominantly Inherited Alzheimer Network.
Each of these correlations remained highly significant (p < 10−12) after adjusting for APOE genotype and sex as fixed effects. Neither APOE (p > 0.4) nor sex (p > 0.5) reached significance or added to the variance explained in any of these models. Overall mean age at onset was slightly but not significantly later in males (46.58 vs 45.20 years). Figure 3 shows the difference between each individual's actual age at onset and the mean onset for all others with the same ADAD mutation type. Differences in onset were in the same direction as those previously identified for late-onset AD risk,18,19 with ε4 allele carriers having earlier onset and ε2 carriers having later onset; however, this did not reach significance or add to the variance explained.
Figure 3. Age at onset differences by APOE genotype.

For affected individuals with known APOE genotype, the difference between each individual's actual age at onset and the mean age at onset of all others sharing the same autosomal dominant Alzheimer disease mutation type is plotted on the y-axis.
To investigate the robustness of these results and control for potential biases resulting from large families, we performed a subgroup analysis excluding data from the Volga German and Colombian kindreds, which did not substantially alter our results, giving r2 values of 0.402 for parental onset, 0.502 for family, and 0.538 for mutation type, with all correlations significant at p < 10−16. We also performed 10 iterations of a confirmatory analysis using stratified sampling of one subject per family. For each of these analyses, all correlations remained highly significant at p < 10−16. The r2 values ranged between 0.356 and 0.457 for parental onset, 0.454 and 0.503 for mean onset of all other family members, and 0.522 and 0.589 for mean onset by mutation type. We additionally calculated ICCs17 by mutation type and family. ICC1 and ICC2 values were 0.5619 and 0.9069 for mutation type, and 0.5837 and 0.8290 for family, which is again supportive of a relatively strong association of these variables with age at symptom onset.
A multivariate model including all 3 family history variables together gave an adjusted r2 value of 0.527, only slightly higher than the value of 0.5225 for mutation type alone. However, mean onset by mutation type and family each continued to show statistically significant (p < 0.01) independent associations with individual onset. This indicates that much of the variance in individual onset in the overall ADAD population can be accounted for by ADAD mutation type itself, but that in certain families, other genetic or environmental factors may have independent effects in modifying age at onset.
Overall mean onset in the DIAN and literature datasets was very similar at 46.11 and 46.61 years, respectively, although SD was greater in the literature than in DIAN (10.1 and 8.9 years, respectively). Remarkably, these values are each closely comparable to the mean onset of 48.8 years reported in early case series of familial AD.22 For 3 mutations reported with n > 10 prevalence in both DIAN and other studies, mean onset did not differ significantly (APP V717I: 49.4 vs 51.3 years, p = 0.4; PSEN1 G206A: 53.3 vs 53.8 years, p = 0.8; and PSEN1 H163R: 45.2 vs 47.2 years, p = 0.2).
To further validate these data using longitudinal observations of presymptomatic and symptomatic mutation carriers, we performed a survival analysis of 183 mutation carriers followed in the DIAN observational study. Kaplan-Meier curves representing survival to symptom onset are shown in figure e-1. For all mutation carriers, median survival to symptom onset was 47 years (95% confidence interval 45–48 years), compared with a median 46 years and mean 46.3 years in the meta-analysis. For PSEN1 mutation carriers, median survival to onset was 45 years (95% confidence interval 42–48 years; meta-analysis median = 43 and mean = 43.8). For APP mutation carriers, median survival to onset was 47 years (meta-analysis median = 49 and mean = 49.7). Repeating the above analyses using one individual per family gave similar results (45 years for all mutations, 43 years for PSEN1, 46 years for APP). We additionally performed direct comparisons of 5 individual ADAD mutation types with the most longitudinal data. These data are not shown directly to avoid unblinding the mutation status of DIAN participants, but median survival to symptom onset was within 0 to 4 years of the mean and median onset from meta-analysis data in each case.
We also replicated each of our analyses in a subset of 131 mutations identified as meeting strict criteria for definitely pathogenic ADAD mutations by the DIAN Genetics Core (indicated with asterisks in table e-1).23 This did not substantially alter any of our results, likely because among the 1,307 affected individuals in our dataset, 1,113 (85%) have mutations meeting strict criteria for definite pathogenicity.
To evaluate the relationship between symptom onset and progression of disease, we plotted the disease course from onset to death for all affected individuals with known age at death (figure 4). Regression showed no significant linear relationship (p > 0.5), however the “inverted-U” shape of the resulting plot suggested a nonlinear effect. Indeed, polynomial regression revealed a second-order relationship between age at onset and disease course, which was highly significant at p < 0.001, and this remained significant at p < 0.01 when replicated in each of the confirmatory subgroup analyses described above. Patients with early (younger than 35 years) or late (older than 65 years) onset each had a shorter disease course than patients with onset in midlife (35–65 years). The average course from symptom onset to death in our dataset (9.7 years ± 5.06 SD) was only modestly shorter than the average course of 11.3 years from symptom onset to death reported in a large population with sporadic AD.21
Figure 4. Age at symptom onset vs disease course in years.

(A) For all individuals with known ages of symptom onset and death, age at symptom onset is plotted against disease course in years, estimated as the total time from symptom onset to death. Jitter was added to the x- and y-axes. A nonparametric smoothing function (LOESS) is shown in blue. Dashed red lines indicate median age at symptom onset and median survival from symptom onset in sporadic Alzheimer disease based on published data.21 (B) Individuals are shown partitioned into 3 groups by age at symptom onset.
DISCUSSION
We report the largest dataset to date of individual-level data on symptom onset, disease course, and family relationships for multiple ADAD mutation types and kindreds. The relatively strong associations observed between individual age at symptom onset and each of 3 measures of family onset history provide encouraging evidence that a majority of the observed variance in onset can be accounted for by ADAD mutation type. Given these findings, the potential predictability of symptom onset in ADAD can provide powerful advantages for prevention trials in presymptomatic ADAD.
Although mutation type can account for a large part of the observed variance in onset, substantial variation remains within many ADAD families and mutation types, suggesting that other genetic, environmental, or stochastic factors may have significant roles in modifying onset in some families. To investigate a broad range of preclinical disease, DIAN treatment trials currently enroll participants up to 15 years before an estimated age at onset based on parental history, and will closely monitor AD-associated biomarkers in each participant.24,25 In the future, additional work will focus on further refining age at onset estimates in ADAD utilizing additional factors including genetics, biomarkers, and epidemiologic risk factors.
Although a widespread perception exists that ADAD always presents with much earlier onset than is seen in sporadic AD,20 our findings emphasize that this is not uniformly the case. For mutations associated with later-onset disease, many carriers may die from unrelated causes before onset, making penetrance difficult to determine conclusively. It has been hypothesized that the low numbers of reported PSEN2 mutations may result from genetic screening being performed less frequently in late-onset familial dementia.26 Because of the diverse characteristics of ADAD mutations, screening for ADAD should still be considered in cases of late-onset AD with a consistent dominantly inherited pattern.
The duration of disease caused by ADAD mutations may also be more similar to sporadic AD than has been widely believed, and our data show that not all ADAD mutations cause a more rapidly progressive course. In longitudinal studies of sporadic AD in a large multiethnic population, mean survival from symptom onset was 11.3 years for all patients, and 12.1 years for patients with onset below 60 years of age.21 Mean survival from symptom onset in our dataset was modestly lower at 9.7 years. On further analysis, we detected an inverted-U relationship between onset age and disease course, where patients with the youngest and oldest ages at onset had the shortest survival from symptom onset to death, and patients with onset in midlife had the longest survival. This is consistent with a model in which highly pathogenic mutations lead to rapid accumulation of insults and cause early-onset disease with a rapidly progressive course. A shorter disease course in older individuals is expected because of their chronologic age. Mutations causing more gradual pathogenesis may cause later onset of disease, but could also show decreased survival times resulting from frailty of the older population affected at incidence, or from a decrease in amyloid clearance with advancing age.
In interpreting our findings, it should be emphasized that this dataset is based on information about the age at first onset of progressive cognitive symptoms as assessed by clinicians in taking family history, rather than the age at which an individual received a clinical diagnosis of dementia or mild cognitive impairment. Because the age at which all individuals within an extended family were formally assigned to clinical diagnoses is unavailable for most participants, and may be confounded by factors such as regional/generational differences in diagnostic practices and individual differences in access to medical care, the age at symptom onset determined by clinicians when collecting family history has been preferentially used by clinicians and researchers working with ADAD families.2,4,27 In DIAN, information is obtained from the participant, a collateral source, and/or other informants who may know the parental history of disease. Clinicians then determine the year of symptom onset after careful discussion with family informants and review of all available sources of information that may be useful, such as medical records and peripheral family members. Encouragingly, prior studies have demonstrated substantial agreement (r = 0.88, p < 0.001) between unstructured estimates of symptom onset and duration reported by collateral sources and formal estimates prepared by physician raters after thorough medical record review and patient and informant interviews.28 It remains difficult to assess the extent to which differences among investigators may affect some reports, and obtaining thorough history may be particularly challenging in large multigenerational families. However, the replication of our findings in several sensitivity analyses and in longitudinal data suggests that family history obtained by many clinicians can provide valuable information about the onset and course of disease.
In this large and diverse dataset, the strong associations observed between individual age at symptom onset and several measures of family history demonstrate that ADAD mutation type can account for a substantial proportion of the observed variance in age at symptom onset. These findings can be used by researchers and clinicians to derive estimates for the timing of onset before the development of symptoms, and perhaps before many of the progressive neuropathologic insults that precede dementia in AD. In addition to ongoing prevention trials in presymptomatic ADAD, future directions in ADAD clinical research include identification of genetic and environmental modifiers of disease onset and progression, and continued investigation of the relationships between laboratory and imaging biomarkers and the clinical course of disease.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge the contributions of all participants and family members involved in this research, and also thank the many dedicated investigators involved in the prior studies included in this meta-analysis. They particularly thank Denise Levitch, Joanne Norton, Malia Rumbaugh, Angela Oliver, and M. Scot Fague for their assistance with the DIAN and Volga German datasets. The DIAN Expanded Registry (http://dianxr.org) welcomes contact from any ADAD families or treating clinicians interested in research.
GLOSSARY
- AD
Alzheimer disease
- ADAD
autosomal dominant Alzheimer disease
- APP
amyloid precursor protein
- DIAN
Dominantly Inherited Alzheimer Network
- ICC
intraclass correlation coefficient
- PSEN1
presenilin 1
- PSEN2
presenilin 2
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Ryman: writing the manuscript, study concept and design, analysis of data, acquisition of data, statistical analysis. Dr. Acosta-Baena: acquisition of data, analysis of data. Dr. Aisen: revising the manuscript, analysis of data, study coordination. Dr. Bird: acquisition of data, revising the manuscript, analysis of data, study coordination. Dr. Danek and Dr. Fox: acquisition of data, analysis of data. Dr. Goate: revising the manuscript, study concept and design, analysis of data, acquisition of data, study coordination. Dr. Frommelt, Dr. Ghetti, and Dr. Langbaum: acquisition of data, analysis of data. Dr. Lopera: acquisition of data, revising the manuscript, analysis of data, study coordination. Dr. Martins, Dr. Masters, Dr. Mayeux, Dr. McDade, Ms. Moreno, Dr. Reiman, Dr. Ringman, Dr. Salloway, Dr. Schofield, Dr. Sperling, and Dr. Tariot: acquisition of data, analysis of data. Dr. Xiong: revising the manuscript, statistical analysis, study concept and design, analysis of data. Dr. Morris and Dr. Bateman: revising the manuscript, study concept and design, analysis of data, study coordination, obtaining funding.
STUDY FUNDING
The Dominantly Inherited Alzheimer Network (DIAN) is supported by NIA grant U19 AG032438 to J.C. Morris, and by the generous support of F. Simmons and O. Mohan and an anonymous foundation, in addition to DIAN site support from the German Center for Neurodegenerative Diseases (DZNE), the NIHR Queen Square Dementia Biomedical Research Unit, the DIAN Expanded Registry (http://dianxr.org), and J.O. and J.R. Wicking Trust grants 13026 and 20821. Work with the Volga German PSEN2 N141I kindred was supported by NIH grants AG 05136, AG 11762, AG 21544, and GM 46255 to T. Bird and by the Department of Veterans Affairs. Work with the Colombian PSEN1 E280A kindred was supported by grant 1115 545 31651 from Colciencias and Universidad de Antioquia to F. Lopera and by an anonymous foundation.
DISCLOSURE
D. Ryman, N. Acosta-Baena, and P. Aisen report no disclosures relevant to the manuscript. T. Bird receives licensing fees from Athena Diagnostics, Inc. A. Danek, N. Fox, A. Goate, P. Frommelt, B. Ghetti, J. Langbaum, F. Lopera, R. Martins, C. Masters, R. Mayeux, E. McDade, S. Moreno, E. Reiman, J. Ringman, and S. Salloway report no disclosures relevant to the manuscript. P. Schofield has received speaking fees from Janssen Pharmaceuticals. R. Sperling, P. Tariot, C. Xiong, J. Morris, and R. Bateman report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Müller U, Winter P, Graeber MB. A presenilin 1 mutation in the first case of Alzheimer's disease. Lancet Neurol 2013;12:129–130 [DOI] [PubMed] [Google Scholar]
- 2.Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer's disease. Am J Pathol 2001;158:2165–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer's disease associated with mutations of the presenilin-1 gene. J Neurol 2006;253:139–158 [DOI] [PubMed] [Google Scholar]
- 5.Wijsman EM, Daw EW, Yu X, et al. APOE and other loci affect age-at-onset in Alzheimer's disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet 2005;132B:14–20 [DOI] [PubMed] [Google Scholar]
- 6.Pastor P, Roe CM, Villegas A, et al. Apolipoprotein epsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol 2003;54:163–169 [DOI] [PubMed] [Google Scholar]
- 7.Reiman EM, Brinton RD, Katz R, et al. Considerations in the design of clinical trials for cognitive aging. J Gerontol A Biol Sci Med Sci 2012;67:766–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selkoe DJ. Preventing Alzheimer's disease. Science 2012;337:1488–1492 [DOI] [PubMed] [Google Scholar]
- 9.Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Investig 2012;2:975–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011;26(suppl 3):321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 2012;33:1340–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertram L, McQueen M, Mullin K, Blacker DTR. The AlzGene database [online]. Available at: http://alzforum.org/mutations. Accessed January 6, 2013 [DOI] [PubMed]
- 13.R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: http://www.R-project.org. Accessed January 11, 2013 [Google Scholar]
- 14.Lopera F, Ardilla A, Martínez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 1997;277:793–799 [PubMed] [Google Scholar]
- 15.Bird TD, Lampe TH, Nemens EJ, Miner GW, Sumi SM, Schellenberg GD. Familial Alzheimer's disease in American descendants of the Volga Germans: probable genetic founder effect. Ann Neurol 1988;23:25–31 [DOI] [PubMed] [Google Scholar]
- 16.Frommelt P, Schnabel R, Kühne W, Nee LE, Polinsky RJ. Familial Alzheimer disease: a large, multigeneration German kindred. Alzheimer Dis Assoc Disord 1991;5:36–43 [DOI] [PubMed] [Google Scholar]
- 17.Bliese P. Within-group agreement, non-independence, and reliability: implications for data aggregation and analysis. In: Klein K, Kozlowski S, editors. Multilevel Theory, Research and Methods in Organizations. San Francisco: Jossey-Bass; 2000:349–381 [Google Scholar]
- 18.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356 [PubMed] [Google Scholar]
- 19.Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 2011;16:903–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyoshi K. What is “early onset dementia”? Psychogeriatrics 2009;9:67–72 [DOI] [PubMed] [Google Scholar]
- 21.Waring SC, Doody RS, Pavlik VN, Massman PJ, Chan W. Survival among patients with dementia from a large multi-ethnic population. Alzheimer Dis Assoc Disord 2005;19:178–183 [DOI] [PubMed] [Google Scholar]
- 22.Masters CL, Gajdusek DC, Gibbs CJ. The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer's disease. Brain 1981;104:535–558 [DOI] [PubMed] [Google Scholar]
- 23.Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging 2010;31:725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU Trial. Rev Neurol 2013;169:737–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moulder KL, Snider BJ, Mills SL, et al. Dominantly Inherited Alzheimer Network: facilitating research and clinical trials. Alzheimers Res Ther 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010;133:1143–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doody RS, Dunn JK, Huang E, Azher S, Kataki M. A method for estimating duration of illness in Alzheimer's disease. Dement Geriatr Cogn Disord 2004;17:1–4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.