

Abstract
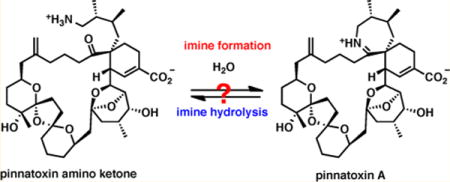
Pinnatoxins belong to the cyclic imine (CI) group of marine toxins with a unique toxicological profile. The need for structural integrity of the aliphatic 7-membered cyclic imine for the potent bioactivity of pinnatoxins has been experimentally demonstrated. In this study, we probe interconversion of the natural cyclic imine and its open form, pinnatoxin A amino ketone (PnTX AK), under physiologically relevant aqueous conditions. Our studies demonstrate the high stability of PnTX A. The unusual stability of the imine ring in PnTX A has implications for its oral toxicity and detoxification. These studies, as well the access to PnTX amino ketone, were enabled by the total synthesis of (+)-pinnatoxin A completed previously in our laboratory.
Pinnatoxin A (PnTX A) is the first structurally characterized member of a growing subfamily of marine toxins containing a spiroimine fragment as a hallmark of their molecular architecture.1 To date, the cyclic imine (CI) group of marine toxins comprises more than 30 members including the spirolides,2 gymnodimines,3 spiro-prorocentrolides,4 and other natural products sharing a macrocyclic framework similar to PnTX A (Figure 1). Pinnatoxins G and E isolated from a range of shellfish and environmental samples in Australia and New Zealand have been hypothesized to be the biosynthetic origin of all other pinnatoxins and pteriatoxins, which are believed to be produced by metabolic modification of pinnatoxins G and E after shellfish contamination by the dinoflagellate Vulcanodinium rugosum. Recent studies point out that Vulcanodinium rugosum is responsible for the production of pinnatoxins E–G in New Zealand, Australia, Japan, and France.5
Figure 1.
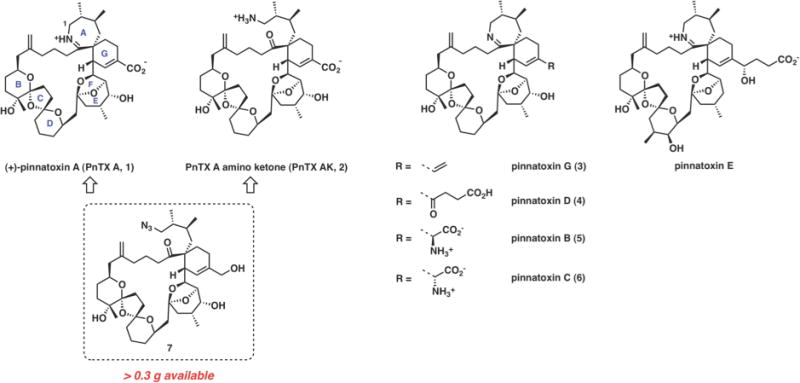
Naturally occurring pinnatoxins and non-natural pinnatoxin A amino ketone with an open imine ring.
The characteristic spiroimine system of pinnatoxins (AG-spiroimine) has been experimentally demonstrated to be a pharmacophore required for biological activity.6 Its structural integrity is necessary to maintain potent toxicity. During the course of our studies, we have found that the seven-membered cyclic aliphatic imine of PnTX A is unusually stable to hydrolysis, which has implications for its oral toxicity and contrasts the properties of gymnodimine and certain spirolides, especially spirolides E and F.7 In order to probe experimentally the interconversion of pinnatoxin A and its imine ring open form, we required access to the amino ketone form PnTX AK (2). Access to this material is hampered by an extreme scarcity of the natural toxin and unusual stability of the cyclic aliphatic imine. These factors render the total synthesis as the only realistic alternative for our intended studies. Previously, we developed a robust and scalable synthetic path to PnTX A that utilizes azido triol 7 as an advanced intermediate (Figure 1).6 We envisioned that azide 7, which we prepared in substantial quantity (>0.30 g), could be readily modified to access pinnatoxin amino ketone (PnTX AK), which in fact was considered as one of potential intermediates en route to pinnatoxin A. An examination of interconversion between pinnatoxin A and its open form (PnTX AK), would provide information on chemical stability of the marine toxin. Herein, we describe an improved synthesis8 of PnTX AK (2) from triol 7 and disclose a detailed study on the interconversion of PnTX AK and its cyclic imine form, pinnatoxin A, under aqueous conditions. Additionally, we have performed extensive computational studies of the PnTX A hydrolysis reaction that point to specific structural factors that contribute to the unusual stability of the cyclic imine.
Our initial goal was to develop a robust, scalable preparation of 2 by reduction of the azido group in 7 followed by oxidation of the allylic alcohol to carboxylic acid. After extensive experimentation, we determined that the optimal procedure for the synthesis of PnTX AK (2) involved a five-step approach (Scheme 1). Azido triol 7 was first subjected to TEMPO-catalyzed oxidation with PhI(OAc)2 followed by another oxidation of the intermediate to carboxylic acid 8 with buffered sodium chlorite. Methyl ester 9 was readily formed upon treatment of the carboxylic acid with (trimethylsilyl)-diazomethane, affording 9 in three steps and 86% overall yield. Under carefully optimized conditions,9 the azido group in 9 was reduced to the primary amine by hydrogenolysis in the presence of 5% Pd-BaSO4 and pyridine. The following saponification with lithium hydroxide in aqueous THF and purification by reversed-phase HPLC completed the sequence of reactions, affording 5.1 mg of pure amino acid 2 from 6.3 mg of 9 (85% isolated yield).
Scheme 1.
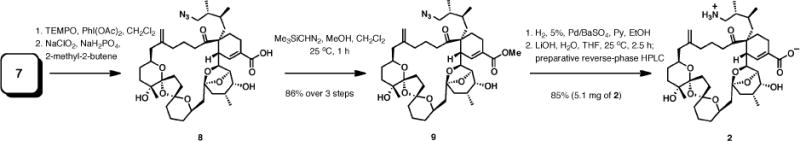
Optimized, Scalable Preparation of PnTX AK (2)
Having developed a reliable procedure for the synthesis of PnTX AK (2), we were able to undertake a detailed investigation of the interconversion of pinnatoxin A and its amino ketone form under aqueous conditions (Scheme 2). Initial studies probed cyclization of 2 to PnTX A under neutral or mildly acidic conditions. Reactivity of the amino ketone in KH2PO4 buffer (D2O, 0.01 M, pH 7.3) was monitored by NMR and mass spectroscopy (ESI). No cyclization to PnTX A was observed after 48 h at 20 °C, 19 h at 50 °C, and 72 h at 100 °C; remarkably, virtually no decomposition was observed even after prolonged heating at 100 °C. Similarly, at pH 4.1, no traces of pinnatoxin A were detected after an identical treatment (48 h at 20 °C, 19 h at 50 °C, and 72 h at 100 °C), although after 24 h at 100 °C some degree of decomposition was observed by NMR. Decomposition was complete after 72 h.
Scheme 2.
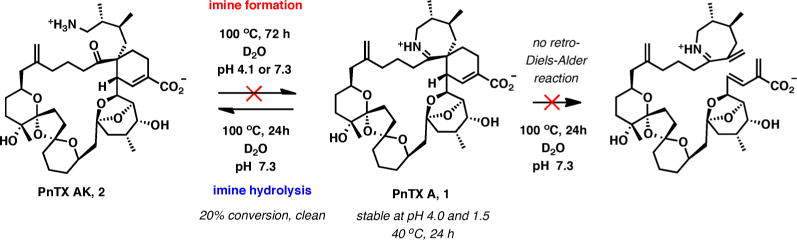
Imine Formation/Hydrolysis
In the reverse direction, a solution of pinnatoxin A in deuterium oxide was heated at 40 °C for 48 h at pH 4.0 and then acidified to pH 1.5 with concentrated aqueous hydrochloric acid followed by heating at 40 °C for an additional 24 h, which resulted in no observable decomposition. When PnTX A was heated in a KH2PO4 buffer (D2O, 0.01 M, pH 7.3) at 100 °C for 12 h, slow but clean hydrolysis was observed, giving 12% of the imine hydrolysis product (PnTX AK). After 24 h of heating, the fraction of the imine hydrolysis product increased to 20%. Notably, while we do observe a very clean hydrolysis of PnTX A to PnTX AK under these conditions, no retro-Diels-Alder reaction was noted in these experiments. This observation provides an intriguing initial insight into the proposed biosynthesis of PnTX A based on this Diels-Alder reaction.
MOLECULAR MODELING
In order to gain a deeper mechanistic understanding for the atypical hydrolytic stability for the aliphatic 7-membered cyclic imine of pinnatoxins, we carried out a computational analysis of the hydrolysis reaction by comparing the energy along the reaction coordinate for two systems: the complete structure of PnTX A (1) and a simplified derivative (13), containing only the spiroimine fragment (Figure 2).
Figure 2.
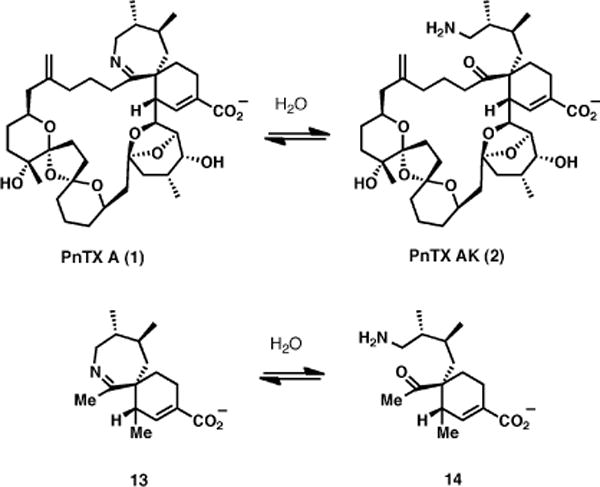
Hydrolysis of PnTX A and simplified spiroimine fragment 13 for computational studies.
The bioactive conformation of PnTX A presents a globular form, stabilized by several intramolecular hydrophobic interactions, and consequently, the spiroimine group is accessible for reaction with a water molecule only from one side (Figure 3). This stereochemistry was considered throughout in the molecular modeling part of our study.
Figure 3.
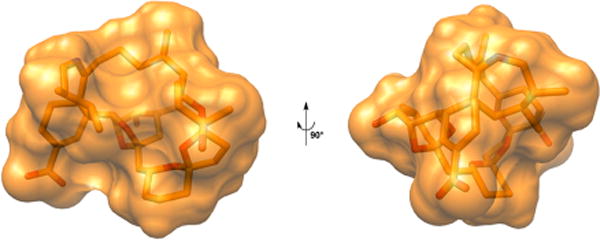
Three-dimensional structure of pinnatoxin A.
Hall and Smith have investigated, using ab initio quantum calculations, the Schiff base formation from methylamine and formaldehyde and showed that this process is strongly influenced by the number of water molecules facilitating the proton transfer.10 Therefore, a similar approach was used in the present study, which is focused on the imine formation from the neutral reactants in the presence of zero or one bridging water molecules.
In the absence of bridging water molecules, the energy barriers for both systems 1 and 13 considered in this study are similar, of about 45–48 kcal/mol, in both directions (Figure 4). Although very high, these values are in good agreement with the results previously reported by Hall and Smith for a smaller system.11
Figure 4.
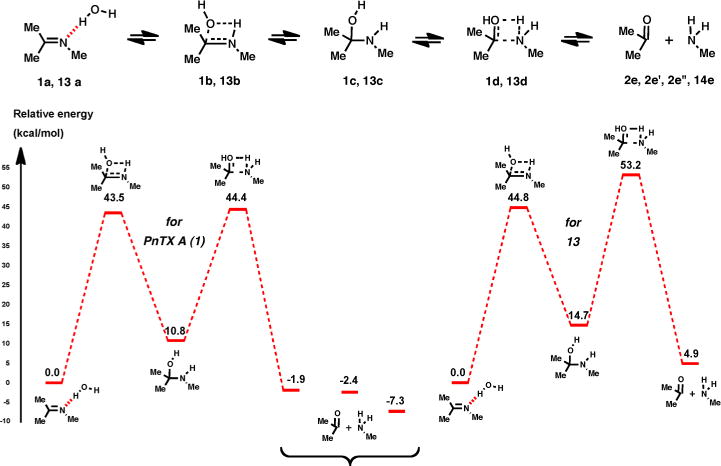
Energy profile for imine hydrolysis of 1 and 13 with no bridging water molecules.
When one water molecule is present to facilitate the proton transfer process, the intermediate carbinolamine:water complex can be represented in two forms, with the oxygen and nitrogen atoms as either hydrogen bond donors or acceptors (Figure 5).
Figure 5.
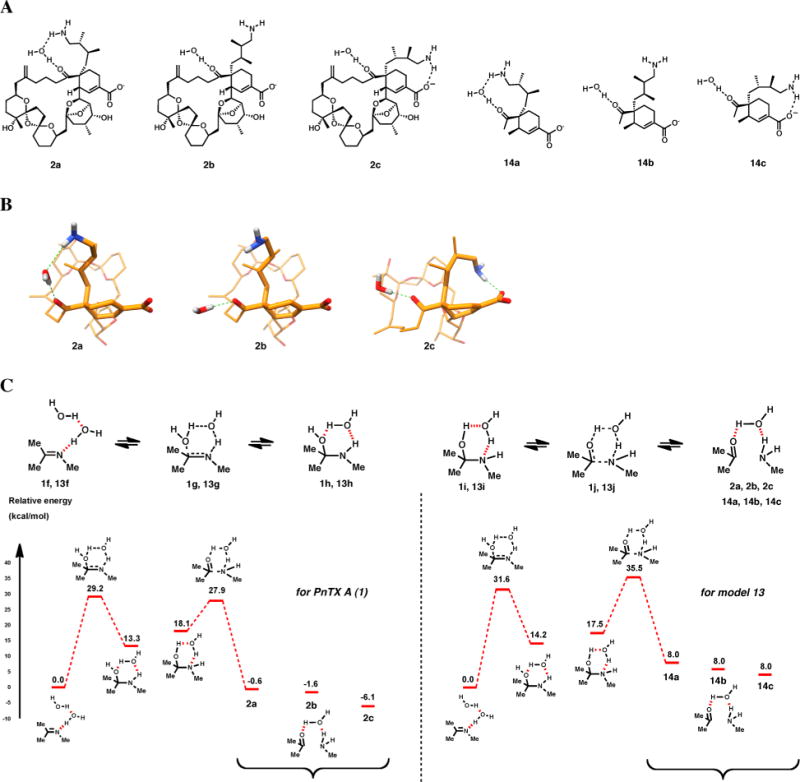
Structures (A) and conformations (B) of intermediates 2a, 2b, 2c, 14a, 14b, and 14c and a schematic energy profile for the hydrolysis of 1 and 13 with one bridging water molecule.
Under these conditions, the computed energy barriers are much lower (Figure 5C), as it was also noted previously.11 Relatively similar values are obtained for the water addition on imine (29.2 kcal/mol for 1 and 31.6 kcal/mol for 13), whereas important differences are observed for the reaction between carbonyl and amine groups (34.0 and 27.5 kcal/mol for 1 and 13, respectively), the amino-ketone form of PnTX A being more stable thermodynamically. These differences, which might explain the difficulties witnessed for the direct preparation of PnTX A from the amino-ketone form (see above), are mainly due to the conformational flexibility of the aminoalkyl chain in PnTX A, for which three representative conformations were considered, with the amino group in the proximity of carbonyl (1f) and carboxylate (1f″) moieties, or in an intermediate position (1f′). As the aminoalkyl chain moves progressively from the carbonyl toward the carboxylate moiety, a decrease in the total energy is observed, leading to the most stable conformer, 1f″. In our previous study,6a this conformation was also used to explain the lack of biological activity of the amino ketone form of PnTX A against nAChRs.
CONCLUSIONS
Collectively, our experiments demonstrate the remarkable chemical stability of pinnatoxin A under neutral and strongly acidic aqueous, which most likely is an important factor contributing to its potent oral toxicity compared to other related toxins within the cyclic imine group. For PnTX A itself, when a transformation was observed, it was a clean hydrolysis of the cyclic imine (~20% after 24 h at 100 °C) rather than the retro-Diels–Alder reaction which, in the reverse direction, was postulated as a step in the biosynthesis of pinnatoxin A.11 In addition, we provided evidence for the higher thermodynamic stability of the amino ketone form of PnTX A, compared with the cyclic imine form, which supports the notion that the stability of PnTX A is due to kinetic, rather than thermodynamic factors. Future computational work will be focused on the study of other factors influencing the interconversion of aminoketone and imine forms of PnTX A, e.g., the presence of two bridging water molecules to facilitate the proton transfer and the protonation state of the nitrogen atom (imine vs iminium), as well as the potential of the biomimetic Diels–Alder reaction to assemble the macrocyclic framework of pinnatoxins.12
The observed high stability of pinnatoxins in aqueous solution has important implications for detoxification of shellfish contaminated with cyclic imine toxins and regulatory issues concerning this class of toxins.
EXPERIMENTAL SECTION
Carboxylic Acid 86a
Iodobenzene diacetate (0.120 g, 0.37 mmol) was added to a solution of triol 7 (35 mg, 47.2 mmol) in dichloromethane (3.0 mL) and 2,2,6,6-tetramethylpiperidin-1-oxyl (1.0 mg, 6.4 mmol) in dichloromethane (2.0 mL) at rt. After 2 h 15 min, the solution was directly subjected to column chromatography (silica, 50% → 60% ethyl acetate/hexanes) to afford the expected aldehyde (S1)6a,13 (30.7 mg, 41.5 mmol, 88%). [α]23 D = +17.7 (c 0.94, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ (ppm): 9.44 (s, 1H); 6.27 (s, 1H); 4.83 (s, 1H); 4.78 (s, 1H); 4.44 (d, J = 2.5 Hz, 1H); 4.10–4.01 (m, 3H); 3.58 (s, 1H); 3.17 (d, J = 7.5 Hz, 2H); 3.10 (bd, J = 11.0 Hz, 1H); 2.57–2.42 (m, 3H); 2.27 (dd, J1=9.0 Hz, J2=3.5 Hz, 1H); 2.22 (m, 23H); 1.43–1.16 (m, 8H); 1.27 (s, 3H); 1.01 (d, J = 7.0 Hz, 3H); 0.85 (d, J = 7.0 Hz, 3H); 0.79 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 212.6, 192.8, 146.0, 145.2, 143.0, 112.7, 111.4, 109.1, 108.0, 80.2, 77.4, 76.7, 70.5, 69.0, 68.9, 67.5, 56.1, 52.3, 46.2, 44.2, 43.4, 39.6, 39.4, 38.4, 37.2, 36.2, 34.7, 34.0, 32.2, 31.4, 31.2, 30.5, 29.9, 29.6, 26.6, 21.7, 21.6, 19.8, 16.4, 15.9, 12.5. HRMS (ESI): calcd for C41H61O9N3Na [M + Na] 762.4306, found 762.4281.
Sodium chlorite (24.4 mg, 0.270 mmol) was added to a solution of the aldehyde (S1)13 (20 mg, 27.0 mmol), sodium dihydrogen phosphate (41 mg, 0.30 mmol), and 2-methyl-2-butene (1.20 mL, 11.3 mmol), in tert-butyl alcohol (3.2 mL) and water (0.8 mL) at 0 °C and stirred for 10 min. The reaction was warmed to rt and stirred for an additional 50 min. After dilution with water and ethyl acetate, the aqueous layer was extracted with ethyl acetate (4 × 20 mL). The combined organic layers were dried with sodium sulfate and concentrated, and the residue was submitted to the next reaction without further purification. 1H NMR (500 MHz, CDCl3) δ (ppm): 6.66 (s, 1H); 4.83 (s, 1H); 4.78 (s, 1H); 4.50 (s, 1H), 4.11–4.03 (m, 2H), 3.98 (dd, J1=13.8 Hz, J2=4.0 Hz, 1H); 3.65 (s, 1H); 2.99 (d, J = 11.5 Hz, 1H); 2.52 (m, 3H); 2.32–1.57 (m, 28H); 1.41 – 1.21 (m, 8H); 1.27 (s, 3H); 0.99 (d, J = 7.0 Hz, 3H); 0.87 (d, J = 7.0 Hz, 3H); 0.80 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 213.4, 145.9, 113.0, 111.9, 109.2, 108.5, 80.7, 77.8, 77.5, 77.2, 70.9, 69.5, 69.4, 67.9, 56.6, 51.9, 46.2, 44.6, 44.0, 40.2, 40.0, 38.8, 37.5, 36.7, 35.3, 34.4, 32.6, 31.8, 31.6, 30.9, 30.3, 30.1, 30.0, 27.7, 23.0, 22.3, 22.1, 20.3, 16.9, 16.3, 13.0. HRMS (ESI): calcd for C41H61O10N3Na [M + Na] 778.42546, found 778.4252.
Azido Ester 96a
(Trimethylsilyl)diazomethane (1.16 M in ether, 0.120 mL, 0.135 mmol) was added dropwise to a solution of the crude residue in a mixture of dichloromethane (1.6 mL) and methanol (0.4 mL) at rt. After 10 min, the mixture was concentrated on a rotary evaporator and the residue was purified by column chromatography (silica, 25% ethyl acetate/hexanes) to afford azido ester 9 (20.3 mg, 26.4 mmol, 98%). [α]23D +14.9 (c 0.5, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ (ppm): 6.44 (s, 1H); 4.82 (s, 1H); 4.78 (s, 1H); 4.43 (dd, J1=6.8 Hz, J2=2.5 Hz, 1H); 4.13–4.08 (m, 1H); 4.06–4.02 (m, 1H); 3.96 (dd, J1=17.3 Hz, J2=5.5 Hz, 1H); 3.75 (s, 3H); 3.58 (bd, J = 10.0 Hz, 1H); 3.17 (d, J = 9.0 Hz, 2H); 2.99 (bd, J = 15.0 Hz, 1H); 2.54–2.48 (m, 3H); 2.28 (dd, J1 = 13.0 Hz, J2 = 3.5 Hz, 1H); 2.24–2.17 (m, 2H); 2.11 – 1.86 (m, 6H); 1.86–1.64 (m, 16H); 1.60–1.56 (m, 1H); 1.39 (dd, J1 = 13.5 Hz, J2 = 12.0 Hz, 1H); 1.33–1.21 (m, 3H); 1.26 (s, 3H); 0.99 (d, J = 8.5 Hz, 3H); 0.87 (d, J = 6.5 Hz, 3H); 0.80 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 213.2, 166.7, 145.5, 135.4, 132.8, 112.6, 111.5, 108.8, 108.1, 80.3, 77.4, 70.5, 69.1, 69.0, 67.5, 56.2, 52.2, 51.6, 45.7, 44.2, 43.8, 39.9, 39.5, 38.3, 37.2, 36.3, 34.9, 34.0, 32.2, 31.4, 31.2, 30.5, 29.8, 29.6, 27.3, 22.7, 21.9, 21.7, 19.9, 16.5, 15.9, 12.6. HRMS (ESI): calcd for C42H63N3O10Na [M + Na] 792.44111, found 792.44155.
Pinnatoxin Amino Ketone (PnTX AK, 2)6a
Hydrogen gas was bubbled through a solution containing azido acid 8 (3.0 mg, 3.97 μmol) and Lindlar catalyst (5.1 mg, 2.4 μmol) in EtOH (0.5 mL). After 5 min, the hydrogen needle was placed above the solution and the reaction was allowed to stir for 36 h. The solution was filtered through Celite with MeOH and then concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (YMC ODS-A; 250 × 10 mm; 210 nm; 70% MeOH-H2O containing 0.1% CF3CO2H, 3.0 mL/min; t1 = 15.5 min) to afford PnTXAK, 2 (1.0 mg, 1.39 μmol, 35%). [α]23D + 10.8 (c 0.33, MeOH). 1H NMR (800 MHz, CD3OD) δ (ppm): 6.58 (s, 1H); 4.81 (s, 1H); 4.79 (s, 1H); 4.35 (d, J = 2.5 Hz, 1H); 4.30–4.22 (m, 1H); 4.11–4.06 (m, 1H); 3.94 (dd, J1 = 12.0 Hz, J2 = 4.5 Hz, 1H); 3.62 (s, 1H); 3.17 (d, J = 12.0 Hz, 1H); 2.94 (dd, J1 = 12.8 Hz, J2 = 5.5 Hz, 1H); 2.77–2.71 (m, 2H); 2.52 (bd, J = 16.8 Hz, 1H); 2.38–2.34 (m, 2H); 2.23–1.17 (m, 32H); 1.27 (s, 3H); 0.97 (m, 6H); 0.83 (d, J = 7.2 Hz, 2H). C NMR (150 MHz, CD3OD) δ (ppm): 213.2, 147.3, 137.4, 113.5, 112.8, 109.9, 109.2, 82.0, 78.3, 72.2, 70.3, 70.0, 67.7, 52.8, 49.5, 46.3, 45.9, 45.2, 44.4, 40.8, 39.7, 39.6, 39.5, 37.1, 35.9, 35.1, 35.0, 33.4, 32.8, 32.4, 31.8, 31.0, 30.6, 28.0, 23.9, 23.4, 23.2, 21.4, 16.7, 16.4, 12.5. HRMS (SI): calcd for C41H63NO10Na [M + Na] 752.4350, found 730.4338.
Pinnatoxin Amino Ketone from Ester 9 (PnTX AK, 2)
Hydrogen gas was bubbled through a solution containing ester 9 (6.3 mg, 8.18 μmol), 5% Pd-BaSO4 (44.0 mg, 20.7 μmol) and pyridine (6.5 μL, 80.7 μmol) in EtOH (0.5 mL). After 4 min, the hydrogen needle was placed above the solution and the reaction was allowed to stir for 16 min. The reaction was filtered through a syringe filter using methanol and then concentrated under reduced pressure. The crude residue was submitted to the next reaction without further purification.
Lithium hydroxide (0.63 mL, 0.2 M in H2O, 0.126 mmol) was added to a solution of the residue in THF (1.90 mL) at 0 °C. The reaction was stirred at 0 °C for 30 min, warmed to rt, and stirred for an additional 2 h 15 min. The reaction was quenched with trifluoroacetic acid (1.20 mL, 0.2 M in MeOH). The solution was concentrated under reduced pressure and the residue was purified by reversed-phase HPLC (YMC ODS-A; 250 × 10 mm; 210 nm; 70% MeOH-H2O containing 0.1% CF3CO2H, 3.0 mL/min; t1 = 15.5 min) to afford PnTX AK, 2 (5.1 mg, 6.99 μmol, 85%).
Urea 116a
1H NMR (800 MHz, CD3OD) δ (ppm): 6.53 (s, 2H); 4.82 (s, 2H); 4.79 (s, 2H); 4.37 (d, J = 2.4 Hz, 2H); 4.23–4.19 (m, 2H); 4.14–4.10 (m, 2H); 3.95 (dd, J1 = 12.0 Hz, J2 = 4.8 Hz, 2H); 3.74 (s, 6H); 3.58 (d, J = 2.4 Hz, 2H); 3.08 (d, J = 10.4 Hz, 2H); 3.01–2.94 (m, 4H); 2.67 (ddd, J1 = 17.6 Hz, J2 = J3 = 8.4 Hz, 2H); 2.50–2.44 (m, 4H); 2.34–1.14 (m, 68H); 1.26 (s, 6H); 0.96 (d, J = 6.8 Hz, 6H); 0.83 (d, J = 6.4 Hz, 6H); 0.81 (d, J = 6.8 Hz, 6H). 13C NMR (150 MHz, CD3OD) δ (ppm): 214.4, 168.3, 161.3, 147.3, 137.4, 133.6, 113.2, 112.8, 109.8, 109.3, 81.9, 78.5, 72.0, 70.2, 67.7, 53.1, 52.5, 50.0, 46.6, 45.7, 45.6, 44.8, 41.4, 41.1, 39.5, 37.9, 35.9, 35.4, 35.3, 33.5, 32.5, 32.3, 31.8, 30.9, 30.8, 30.7, 30.6, 30.4, 28.2, 23.8, 23.3, 21.2, 16.8, 16.4, 12.8. LRMS (ESI): calcd for C85H128N2O21Na [M + Na] 1535.89, found 1535.92.
Supplementary Material
Acknowledgments
We are indebted to Dr. Hongjun Zhao (University of California, Santa Barbara) for continued assistance with NMR spectroscopy. This work was supported by the NIH/NIGMS (GM 077379) and kind gifts from Amgen and Eli Lilly.
Footnotes
General procedures, copies of 1H, 13C NMR spectra, and modeling data and procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Cembella A, Krock B. In: Seafood and Freshwater Toxins. 2. Botana LM, editor. CRC Press; Boca Raton: 2008. pp. 561–580. [Google Scholar]; (b) Gueret SM, Brimble MA. Nat Prod Rep. 2010;27:1350–1366. doi: 10.1039/c005400n. [DOI] [PubMed] [Google Scholar]; (c) Munday R. In: Seafood and Freshwater Toxins. 2. Botana LM, editor. CRC Press; Boca Raton: 2008. pp. 581–594. [Google Scholar]; (d) Aune T. In: Seafood and Freshwater Toxins. 2. Botana LM, editor. CRC Press; Boca Raton: 2008. pp. 3–20. [Google Scholar]
- 2.Hu T, Burton IW, Cembella AD, Curtis JM, Quilliam MA, Walter JA, Wright JLC. J Nat Prod. 2001;64:308–312. doi: 10.1021/np000416q. [DOI] [PubMed] [Google Scholar]
- 3.Munday R, Towers NR, Mackenzie L, Beuzenberg V, Holland PT, Miles CO. Toxicon. 2004;44:173–178. doi: 10.1016/j.toxicon.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 4.Lu C-K, Lee G-H, Huang R, Chou H-N. Tetrahedron Lett. 2001;42:1713–1716. [Google Scholar]
- 5.(a) Rhodes L, Smith K, Selwood AI, McNabb P, van Ginkel R, Holland PT, Munday R. Harmful Algae. 2010;9:384–389. [Google Scholar]; (b) Selwood AI, Miles CO, Wilkins AL, van Ginkel R, Munday R, Rise F, McNabb PJ. Agric Food Chem. 2010;58:6532–6542. doi: 10.1021/jf100267a. [DOI] [PubMed] [Google Scholar]; (c) Nézan EL, Chomérat N. Cryptogam: Algol. 2011;32:3–18. [Google Scholar]; (d) Rhodes L, Smith K, Selwood A, McNabb P, Molenaar S, Munday R, Wilkinson C, Hallegraeff G. New Zealand J Mar Freshwater Res. 2011;45:703–709. [Google Scholar]; (e) Rhodes L, Smith K, Selwood A, McNabb P, Munday R, Suda S, Molenaar S, Hallegraeff G. Phycologia. 2011;50:624–628. [Google Scholar]; (f) Smith KF, Rhodes LL, Suda S, Andrew I, Selwood AI. Harmful Algae. 2011;10:702–705. [Google Scholar]; (g) Hess P. 8th International Conference on Molluscan Shellfish Safety; Charlottetown, Prince Edwards Island, Canada. June 12–17 2011. [Google Scholar]
- 6.(a) Aráoz R, Servent D, Molgó J, Iorga BI, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala C, Zakarian A. J Am Chem Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stivala CE, Zakarian A. J Am Chem Soc. 2008;130:3774. doi: 10.1021/ja800435j. [DOI] [PubMed] [Google Scholar]; For additional recent synthetic studies on other CI toxins, see:; (c) Zhang YC, Furkert DP, Gueret SM, Lombard F, Brimble MA. Tetrahedron Lett. 2011;52:4896–4898. [Google Scholar]; (d) Stivala CE, Gu Z, Smith LL, Zakarian A. Org Lett. 2012;14:804–807. doi: 10.1021/ol203342e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kong K, Moussa Z, Lee C, Romo D. J Am Chem Soc. 2011;133:19844–19856. doi: 10.1021/ja207385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourne Y, Radic Z, Araoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgo J, Marchot P. Proc Natl Acad Sci USA. 2010;107:6076–6081. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.PnTX AK has been prepared previously in three steps and 31% overall yield from azido triol 7 (see ref 6).
- 9.See the Supporting Information for a detailed account of the optimization studies.
- 10.Hall NE, Smith BJ. J Phys Chem. 1998;102:4930–4938. [Google Scholar]
- 11.Uemura D, Chuo T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J Am Chem Soc. 1995;117:1155–1156. [Google Scholar]
- 12.For a recent relevant study, see: ; Marcoux D, Bindschädler P, Speed AWH, Chiu A, Pero JE, Borg GA, Evans DA. Org Lett. 2011;13:3758–3761. doi: 10.1021/ol201448h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.See the Supporting Information for the structure.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.