

Abstract
Heparin, an anticoagulant, has been used in many forms to treat various diseases. These forms include soluble heparin and heparin immobilized to supporting matrices by physical adsorption, by covalent chemical methods and by photochemical attachment. These immobilization methods often require the use of spacers or linkers. This review examines and compares various techniques that have been used for the immobilization of heparin as well as applications of these immobilized heparins. In the applications reviewed, immobilized heparin is compared with soluble heparin for efficient and versatile use in each of the various applications.
Keywords: Heparin, Immobilization, Blood compatibility
1. INTRODUCTION
Heparin is one of the most intensively studied glycosaminoglycans (GAGs) as a result of its anticoagulant properties. Heparin is a mixture of linear anionic polysaccharides having 2-O-sulfo-α-L-iduronic acid, 2-deoxy-2-sulfamino-6-O-sulfo-α-D-glucose, β-D-glucuronic acid, 2-acetamido-2-deoxy-α-D-glucose, and α-L-iduronic acid as major saccharide units. These are joined through 1→4 glycosidic linkages. The presence and frequency of these saccharide units vary with the tissue source from which heparin is extracted. Fig. 1 compares the structure of heparin with structures of other common glycosaminoglycans, based on their major and minor repeating disaccharide units.
Fig. (1).

Structure of glycosaminoglycans (X = H or SO3−; Y = Ac or SO3−).
In addition to unfractionated pharmaceutical heparin, there are partially depolymerized forms called low molecular weight (LMW) heparins and a synthetic heparin pentasaccharide that are currently in clinical use. Systemic intravenous administration of heparin can result in serious side effects including hemorrhage, difficulty in breathing, closing of throat, swelling of lips, tongue or face and less serious ones such as mild pain, redness, warmth and hair loss. The subcutaneous injection of the newer LMW heparins for the treatment of deep venous thrombosis (DVT) has made anticoagulant therapy more effective and easier to administer, making it possible to treat DVT patients without hospitalization and at no increased risk of recurrent thromoembolism or bleeding complications [1]. Heparin immobilized to a surface, enhances various surface properties, improving blood compatibility and biocompatibility. Heparinized surfaces show reduced platelet adhesion, reduced loss of blood cells and increased plasma recalcification time and activated partial thromboplastin time, resulting in improved biocompatibility without compromising thrombo-resistant properties. Immobilized heparin, unlike soluble heparin, also inhibits the initial contact activation coagulation enzymes through an antithrombin III (AT-III) mediated pathway, and thus show better anticoagulant properties [2]. Furthermore, devices utilized in extracorporeal therapy such as dialysis machines equipped with heparin coated tubing can potentially avoid side effects associated with systemically administered heparin.
This article reviews the applications of various heparinized (or heparin immobilized) matrices and the techniques available for their preparation. Biomaterials can be defined as materials interfacing with or containing biologically derived components. Such materials have played a vital role in treating or curing diseases. Gold, a relatively inert material, is the oldest biomaterial and still is used in dentistry. Biomaterials have applications in many areas as structural materials, drug delivery matrices, supports in tissue engineering, as well as for many other biomedical applications. Biomaterials can be used as research or bioprocessing tools. Immobilized heparin, for example, is widely used for the affinity chromatographic purification of GAG binding proteins [3, 4]. With the arrival of new technologies for preparing polymers and for designing biomaterials at the molecular level, the next generation of biomaterials with potential applications in biology and medicine are being developed [5]. Polymer coated stents, for example, are being used as controlled release systems for heparin and therapeutic proteins to treat cardiovascular disease.
Depending on the application, biomaterials are required to demonstrate good biocompatibility, appropriate mechanical properties and often biodegradation. Enhanced compatibility with blood is an important property for biomaterial that will be in contact with blood during their clinical application. Heparan sulfate is specialized form of heparin that is found in proteoglycans that line the luminal surface of the endothelium making blood vessels blood compatible. Thus, it is not surprising that the most common approach in preparing synthetic matrices having blood compatibility relies on the surface immobilization of heparin.
2. THE PHYSIOCHEMICAL PROPERTIES AND THE CHEMICAL REACTIVITY OF HEPARIN
Heparin is a relatively large polysaccharide of average molecular weight ~12,000 (corresponding to 20 disaccharide units) and having a polydispersity of ~ 1.2 – 1.4 and a molecular weight range of 5,000 to 40,000 [6]. The heparin polymer is hydrophilic holding ~ 2-10% water even after extensive drying and is a polyelectrolyte having an average net charge of −75/chain. This charge is maintained through a wide range of pH values as the pKa of its anionic groups are ~3.3 (carboxyl) and ~ 1.0-1.5 (O-sulfo and N-sulfo). While metallic (i.e., sodium, potassium, etc.) salts are infinitely soluble in water, and insoluble in virtually all organic solvents, quarternary alkyl (aryl) ammonium salts are extremely hydrophobic and water insoluble but are soluble in organic solvents [7]. Heparin sodium salt in water exists as an extended helix. The length of a heparin dodecasaccharide is 5 nm, suggesting that heparin is ~9 nm in length [8].
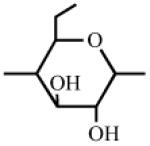
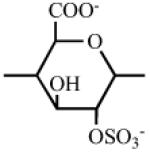
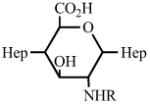
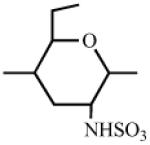
Heparin has a number of chemically reactive functional groups. Each disaccharide repeating unit contains a carboxyl group. All repeating units contain one or more 1° or 2° hydroxyl groups and an average of 2-2.5 sulfo groups. N-sulfo groups are found in 75-85% of all repeating units. Approximately 15-25% of these repeating units contain a vicinal diol [9]. Each chain contains a reducing end hemiacetal, which is a masked aldehyde group. Heparin contains ~0.3 unsubstituted amino groups/chain. These functional groups can be utilized directly are activated to moieties capable of either directly attaching to a supporting matrix or accepting the introduction of a linker or spacer for attachment to a matrix. Table 1 shows the chemistries that have been employed to prepare immobilized heparins.
Table 1.
List of Chemistries Used in the Immobilization of Heparin
| Functional Group | # of Sites/ Heparin Chain |
Activation with, to | Reaction with | Linkage |
|---|---|---|---|---|
|
|
1 | None 
|
R-NH2 or RNH-NH2, NaBH3CN R-NH2, RNH-NH2 |
RNHCH2Hep RNHNHCH2Hep RNHCOHep RNHNHCOHep |
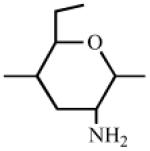
|
~ 0.3 | none | RCO2X RCOH, NaBH3CN |
RCONHHep RCH2NHHep |
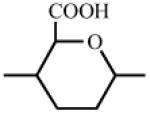
|
~ 20 |
|
RNH2 | RNHCOHep, RNHNHCOHep |

|
~ 60 | CNBr, Hep-O-CN (or Triazine) |
RNH2
|
RNHCOO-Hep 
|
|
~ 5 |

|
RNH2, NaBH3CN RNHNH2, NaBH3CN |

|
|
~ 15 |

|
RNH2 |
|
|
~ 15 |

|
RNH2, NaBH3CN |

|
3. APPLICATIONS
Many methods have been used to make biomaterials with enhanced blood compatible surface properties. These properties include increased biocompatibility, decreased platelet adhesion and decreased blood cell loss. Enhanced blood compatibility is often estimated based on increased plasma recalcification time and increased activated partial thromboplastin time.
When a biomaterial is implanted inside a living tissue, or when blood comes in contact with any foreign body, the blood coagulation cascade is triggered ultimately resulting in the formation of a clot. Such clots can stop a biomaterial from properly functioning, endangering the patient’s health. All of the factors influencing the compatibility of biomaterials are not currently understood [10]. Nevertheless, there are various techniques available that are known to improve the biocompatibility of biomaterials.
3.1. Immobilization of Heparin to Vinyl Copolymer by Covalent Bonding
A commercial ethylene-vinyl alcohol (EVAL) copolymer (3:7 molar ethylene –vinylalcohol ratio) has been used as a biocompatible polymer matrix. Using such a matrix, Marconi and coworkers prepared biocompatible vinyl type polymer surfaces using two bifunctional reagents - adipoyl chloride (APC) and hexamethylene diisocyanate (HMDI) [11]. To increase the surface-volume ratio, the polymer was supported by casting from dimethylformamide onto glass microspheres having an average diameter of 165 μm. Fig. 2 shows two schemes used for the preparation of these biocompatible vinyl based polymers. Heparin was incorporated onto the surface of the polymer matrices using 0.1 and 1% heparin concentrations in these reactions.
Fig. (2).

Schemes for the immobilization of heparin onto the EVAL polymer using (a) APC and (b) HMDI as linkers.
The heparinized microspheres were filtered after the completion of the reaction and washed at room temperature first with water and then with phosphate buffered saline (PBS) for 24 h. The amount of heparin immobilized was measured by employing toluidine blue dye binding assay. The difference in the absorbance value at 631 nm of the heparin solution before and after the immobilization reaction can be used to calculate the amount of heparin immobilized. This method was modified for use in the presence of organic solvents [12]. Linkers of various lengths such as tetraethylene-pentamine (TEPA) and poly (2-hydroxyethyl) methacrylate (p(HEMA)) were evaluated to study the impact of polymer surface hydrophilicity on the biological activity of the immobilized heparin. Fig. 3 shows the protocol to improve surface biocompatibility using such linkers.
Fig. (3).

Protocols for making biocompatible vinyl surfaces with longer spacers such as TEPA and pHEMA.
Acrolyl chloride (AC) was used as a linker for TEPA reactions, and HMDI was used as a linker for the pHEMA reactions. The chemical bonding of heparin (as opposed to non-specific adsorption) in the above reactions was confirmed by attenuated total reflection/ Fourier transform infrared (ATR/FTIR) spectroscopy. Heparinized EVAL was also made ionically (Fig. 4) using N,N-diethylethylenediamine (DED) with AC and HMDI as linkers.
Fig. (4).

Immobilization of heparin ionically through DED (DEDQ = quarternized DED).
All the surfaces made were then evaluated for their anticoagulant activity by measuring activated partial thromboplastin time (APTT) following contact with plasma [12]. Better biocompatibility (elongated APTT) was observed in the cases of AC-linked surfaces when compared to HMDI-linked surfaces. The reason for the increased blood compatibility of AC-linked surface was explained by the unreacted AC giving an acidic character to the surface upon hydrolysis, thus resulting in repulsion between these carboxyl groups and the electronegative groups of heparin. This repulsion presumably increases the exposure of heparin molecule for binding to its cofactor AT-III, enhancing its bioactivity. Table 2 gives the amount of immobilized heparin and measured APTT of AC-linked or HMDI-linked heparinized surfaces with TEPA as spacer.
Table 2.
APTT of Heparinized Surfaces Through AC and HMDI Linkers (Made from 0.1% or 1% Heparin Soln)
| Sequence | Immobilized Heparin (μg/cm2) |
APTT (s) |
|---|---|---|
| AC-TEPA-HMDI-hep (0.1%) | 1.6 ± 0.7 | 96 ± 17 |
| AC-TEPA-HMDI-hep (1%) | 27 ± 7 | no clot |
| HMDI-TEPA-HMDI-hep (0.1%) | 1.9 ± 0.4 | 230 ± 100 |
| HMDI-TEPA-HMDI-hep (1%) | 23 ± 8 | no clot |
| HMDI-pHEMA-HMDI-hep (1%) | 50 ± 1 | no clot |
Fig. 5 evaluates the efficiency difference between the covalent and ionic immobilization of heparin on the EVAL surface.
Fig. (5).

A comparison of the bioefficiency of covalently (dotted line) and ionically formed surfaces (solid line).
The enhanced efficiency of the ionic immobilization was postulated to result from the slow release of heparin during biological evaluation. This view was supported by the remarkable elongation of APTT at high concentrations of heparin. A correlation between APTT and the heparin content was observed. While AC proved to be a better linker than HMDI, covalently heparinized EVAL through both HMDI linker and TEPA spacer gave a significantly higher biological activity.
3.2. Immobilization of Heparin to Polyurethanes by Plasma Glow Discharge
Kang and coworkers prepared functional group-grafted, heparinized polyurethanes (PU) by employing plasma glow discharge [13]. Polyurethaneurea was subjected first to oxygen plasma glow discharge [14] and then treated (Fig. 6) with acryloylbenzotriazole (AB) dissolved in dry diethyl ether solution and maintained at 20 °C for 90 min. The resulting PU-AB surface was then washed with distilled water and 0.005% (w/w) Triton X-100 aq. solution in an ultrasonic cleaner for 20 min, and dried for 48 h under reduced pressure at room temperature. This step was followed by the introduction of either a carboxyl or amino group for the linking of heparin to the surface. The PU-AB surface was treated with 4N NaOH methanolic solution aq. solution for 3 h at room temperature. The surface was then acidified with 10% aq. citric acid in methanol solution to afford PU-COOH.
Fig. (6).

Formation of PU-AB by oxygen plasma glow discharge and graft polymerization.
PU-NH2 was prepared by immersing PU-AB in aq. ethylene diamine (0.01 mol) for 2 h at room temperature. It was then washed by the same method described for PU-AB. The concentration of carboxyl or amino group immobilized was determined by treating with 4N NaOH methanolic solution for 3 h and released benzotriazole was extracted into dimethylformamide and its absorbance determined at 281 nm. The amount of benzotriazole determined is equal to the concentration of carboxyl or amino group formed.
Heparin was then immobilized to PU through these carboxyl or amino groups. PU-COOH film was treated with 0.42 mM 1-ethyl-3-dimethylaminopropyl carbodiimide (EDC) for 24 h at 4°C followed by washing with deionized water. The film was then treated with 100 ml of 0.001% sodium citrate buffer solution of heparin sodium salt for 24 h at 4 °C to yield PU-C-Hep. This heparin immobilized film was then washed with citric acid, 0.1% Triton X-100 in an aqueous methanol solution, and subsequently rinsed with distilled water. Heparin solution was made by dissolving 100 mg of heparin in 80 ml of 10% sodium citrate aqueous buffer containing EDC. PU-NH2 was then treated with this heparin solution for 24 h at 4 °C to yield PU-N-Hep (Fig. 7).
Fig. (7).

Functional group introduction and heparin immobilization on PU-AB.
The amount of heparin thus immobilized on the surface and its stability were determined (Table 3) by the toluidine blue assay [11]. The sample was immersed in PBS for 1 week. Aliquots were taken at periodic intervals and subjected to the toluidine blue assay to determine the heparin released from the film.
Table 3.
Amount of Heparin Immobilized on Modified Polyurethane Supports
| Surfaces | Amount of Immobilized Heparin (μg/cm2) | |
|---|---|---|
| Distilled Water Washed | Triton X-100 Washed | |
| PU-N-Hep | 3.6 ± 0.04 | 2.0 ± 0.13 |
| PU-C-Hep | 2.5 ± 0.08 | 1.4 ± 0.08 |
The amount of heparin immobilized through the amino group was higher than that through the carboxyl group. However, there was a significant reduction in the amount of immobilized heparin when the film was washed with Triton X-100 instead of distilled water only. This difference suggests that washing the film only with distilled water was not sufficient to remove the physically adsorbed heparin. The stability of covalently immobilized heparin was found to be higher than that of physically adsorbed or ionically immobilized heparin. When immersed in PBS, the film released a small amount of heparin that was physically adsorbed in a few hours.
In 1999, the same research group used acrylic acid and methyl acrylate to control the number of carboxyl groups on the plasma treated PU surfaces. They prepared heparinized PU surfaces using a different linker [15] (Fig. 8).
Fig. (8).

Scheme showing the reaction protocol for the preparation of heparinized PU surfaces.
The oxygen plasma treated PU (6 cm × 6 cm) was incubated in dry acetonitrile (20 ml) containing acrylic acid (AA) and methyl acrylate (MA) at 65 °C for 5 h to form PU-C. The concentration of carboxyl groups was varied by using a range of AA/MA (0.2/0.8) ratios. The film was washed and the concentration of carboxyl groups was determined by the previously described method [16]. For the introduction of free amino groups, a surface grafting of poly(ethylene oxide) (PEO, MWav = 600 & 3360) was used. PU-C was incubated with an aq. solution of 1-ethyl-3-dimethyl amidopropyl carbodiimide (WSC) in sodium citrate solution at 4 °C for 2 h to activate the carboxyl groups. The film was then immersed in an aqueous solution of amino-terminated PEO at 4 °C for 24 h. The carboxyl groups of heparin were activated using the same approach and then incubated in a sodium citrate buffer containing carboxyl-activated heparin at 4 °C for 24 h to afford a heparinized PU surface.
3.3. Heparinized Poly(2-Hydroxyethylmethacrylate) Based Microspheres
Denizli prepared heparinized p(HEMA) based microspheres having a diameter of 150-200 μm [17]. The monomer phase containing 2-hydroxyethyl methacrylate (HEMA), a comonomer AA or dimethylaminoethylmethacrylate (DM-AEMA), the linker – ethylene glycol dimethacrylate (EGD-MA) and the polymerization initiator – 2,2’-azobisisobutyronitrile (AIBN) was added to a saturated aqueous solution of magnesium oxide. The reactions were carried out at 70 °C for 3 h followed by heating to 90 °C for 1 h. The contents were then cooled followed by filtration to recover the microspheres. Impurities, unreacted monomers, and MgO were removed by washing the microspheres in a packed bed column with dilute HCl solution and water-ethanol mixture. The microspheres were washed with 0.5M NaCl solution and water, dried and activated using aqueous cyanogen bromide at pH 11.5 NaOH for 60 min. The activated microspheres were then washed with 0.1 M NaHCO3, ethanolamine and FeCl3 solution for 1 h to remove the impurities, and to deactivate any remaining active groups, and washed with distilled water having 0.5 M NaCl. CNBr-activated microspheres were treated with heparin solution (1-8 mg/ml) at 25 °C for 2 h to optimize the immobilization. The heparinized microspheres were washed with 0.1 M NaHCO3 and distilled water and the amount of heparin immobilized on microspheres was measured by toluidine blue assay [11] (Fig. 9). The amount of heparin immobilized increased with increasing amounts of the activation reagent, CNBr up to 30 mg/ ml, beyond which there was no further increase. P(HEMA) homopolymer microspheres showed the highest level of immobilized heparin. In vitro assays such as coagulation time (CT), APTT and prothrombin time (PT) assays were utilized to measure the biocompatibility of these heparinized p(HEMA) based microspheres.
Fig. (9).

The effect of CNBr conc. on the amount of heparin immobilized (heparin concentration = 2 mg/ml, 25 °C, pH 7.4) (solid circle: p(HEMA), solid square: p(HEMA-AA), empty circle: p(HEMA-DMAEMA), empty square: p(HEMA-MMA).
In all the in vitro assays, the microspheres were pretreated with 0.1 M sodium phosphate buffer (pH 7.4) at room temperature for 24 h and washed with 0.5 M NaCl solution and distilled water on a glass filter. Human plasma was preheated to 37 °C for 2 min. For CT, the preheated plasma was treated with 100 mg pretreated microspheres, and CT was determined by fibrometer [18]. For APTT, partial thromoplastin was added to the plasma followed by microspheres. CaCl2 (0.1 ml, 0.025 M) was added 30 s after the addition of microspheres. APTT was measured by fibrometer [19]. The same method was followed for the measurement of PT, except that the preheated (37 °C for 2 min) thromboplastin was added to the preheated human plasma instead of partial thromboplastin. Fig. 10 shows the results of CT, APTT and PT measurements carried out on various p(HEMA) based surfaces.
Fig. (10).
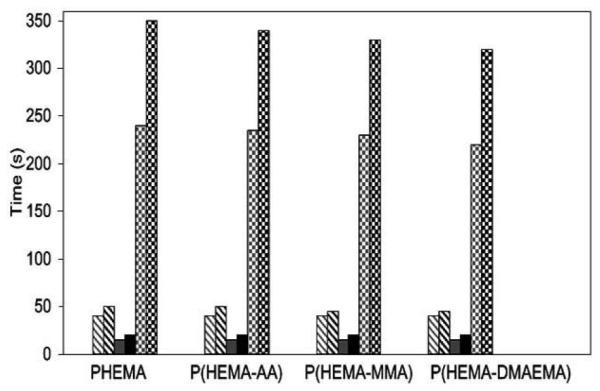
CT, APTT, PT data of various microsphere surfaces (grey: without heparin immobilized, black: with heparin immobilized; diagonal: APTT, solid: PT, checker board: CT).
The clotting times of all the heparinized microspheres were larger than those of unmodified microspheres. P(HEMA) showed the greatest elongation in CT, because of the presence of largest amount of immobilized heparin. These data show that the anticoagulant activity of heparin was retained during the immobilization process.
3.4. Immobilization of Heparin and Highly Sulfated Hyaluronic Acid Onto Polyethylene
Favia and coworkers prepared heparinized poly(ethylene) (PE) and hyaluronic acid (HA) immobilized poly(ethylene) by employing two methods - plasma treatment in O2/H2O radio frequency glow disharges (RFGD) or plasma enhanced chemical vapor deposition (PE-CVD) fed with AA (Fig. 11) [20, 21]. Both methods afforded carboxyl groups on PE surface, which were then utilized in the immobilization (24 h, 4 °C, pH 5-6, morpholine-ethane-sulfonate buffer) of O,O’ bis(2-aminopropyl) poly(ethylene glycol) through the formation of amide bonds (Fig. 11). Activation of carboxyl groups relied on EDC.
Fig. (11).
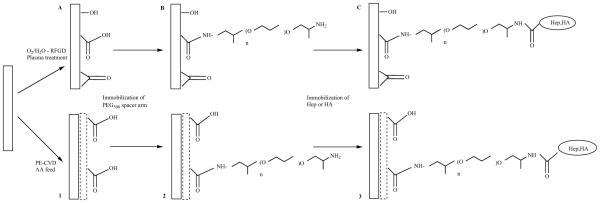
Immobilization of heparin and HA onto poly(ethylene) through O2/H2O RFGD plasma treatment (steps A,B,C) or PE-CVD AA RFGD poly(ethylene) (steps 1,2,3).
These heparinized surfaces were then subjected to thrombin time (TT) experiments. Each substrate surface was placed at the bottom of polystyrene tubes with platelet poor plasma maintained at 37°C for 5 min, human thrombin was added to initiate the coagulation cascade, and clotting time was measured (Fig. 12). The entire PE surface contained immobilized PEG500, 1,2-diaminoethane (DAE), Hep and HA. Immobilization of heparin or HA onto the PE substrates increased the clotting time to about 15 s, when the non-heparinized (or HA) surfaces had a clotting time of only 10 s. This data demonstrates that the anticoagulant property of heparin was preserved on PE surface.
Fig. (12).

Thrombin time measurements of various surface modified PE substrates.
3.5. Surface Macromolecular Microarchitecture Design: Photo-Block-Graft-Copolymerization Using N, N-Diethyldithiocarbamate
Thrombus formation was observed after the complete release of heparin from cationic biomaterials formed by ionic complex with heparin. This is because heparin-free cationic surfaces induce platelet adhesion and aggregation through the electrostatic interactions of anionic cellular components and the cationic surface. Nakayuma and Matsuda minimized this problem by impregnating heparin onto a non-ionic hydrophilic surface, which results in minimal cell adhesion and protein adsorption [22]. They fabricated a heparinized AB-type-block-graft-copolymer with the use of poly(N-[3-(dimethylamino)propyl]acrylamide) (p(DMAPAAm)) and poly(N, N-dimethylacrylamide) (p(DMAAm)) (Fig. 13). The authors showed that the top p(DMAAm) layer acted as a shield against adhesion or adsorption, resulting in minimal thrombus formation even after the complete release of heparin.
Fig. (13).

Schematic representation of the preparation of heparinized p(DAPAAm)-b-(PDMAAm)-block-graft-copolymer (a) Dithiocarbamate-derivatized PST film; (b) p(DMAPAAm) graft polymer; (c) heparin; and (d) p(DMAAm) graft polymer.
3.6. Heparinization of DACRON and PTFE Surfaces
Surface modification of Dacron (poly(ethylene-terephthalate)) and PTFE (poly(tetrafluoroethylene)) vascular grafts was reported by Chandy and co-workers [23]. Collagen and laminin were used as linkers of heparin with the surfaces (Fig. 14). PTFE and Dacron vascular grafts (3 × 5 cm2) were plasma treated (Ar, 150-200 mT, 50 W, 3-5 min), followed by a treatment with 100 mg% collagen IV solution (PBS, pH 7.4, rt, 4 h) to afford a collagen grafted surface. Then these surfaces were treated with 0.6% glutaraldehyde (GA) followed by an incubation in laminin solution at 4 °C to make collagen-laminin-coated Dacron and PTFE.
Fig. (14).

Schematic representation of PTFE surfaces.
The carboxyl groups of collagen-laminin (CL) treated surfaces were activated by treating them with EDC. Heparin was dissolved in sodium phosphate buffer, pH 7.4, and then added to the carboxyl-activated-surfaces and maintained overnight at 4 °C. These heparinized surfaces were then characterized by FTIR-ATR, platelet adsorption and fibrinogen adsorption (Table 4). The number of adhering platelets and the amount of adhered fibrinogen observed on heparinized surfaces were significantly reduced when compared with the respective values of corresponding untreated surfaces.
Table 4.
Biocompatibility Studies of Heparinized PTFE and Dacron Surfaces
| Surfaces | Platelet Adhesion (30 min) (mm2) |
Fibrinogen Adhesion (3 h) (μg/cm2) |
|---|---|---|
| PTFE (untreated) | 62.92 ± 4.9 | 1.08 ± 0.21 |
| CL-PTFE-heparin | 15.00 ± 3.5 | 0.26 ± 0.01 |
| Dacron (untreated) | 48.93 ± 5.7 | 0.86 ± 0.04 |
| CL-Dacron-heparin | 17.50 ± 3.3 | 0.22 ± 0.02 |
3.7. Biocompatible Surfaces by Immobilization of Heparin in Diamond-Like Carbon Films Deposited on Various Substrates
Steffen and co workers reported heparinization of diamond-like carbon (DLC) deposited on various surfaces such as PTFE, PTFE vascular prostheses, polystyrene and silicon wafers [24]. Low molecular weight heparin (MWav 4000-6000) was used for this purpose. Heparin (0.2 mg/ml) was depolymerized using nitrous acid and treated with NaBH3CN in NaCl at pH 3.5 and 50°C for 2 h to achieve a covalent end-point immobilization of heparin [25]. Thrombin tests were accomplished to determine the enhanced blood compatibility of these modified surfaces. Thrombin solution was added to a microdish together with citrated blood plasma, calcium chloride and the DLC-heparin was incubated for 2 min and the clotting time was determined. DLC-heparinization afforded an elongation in clotting time corresponding to an enhancement in their blood compatibilities.
3.8. PET Immobilized with Insulin and/or Heparin Using Plasma Glow Discharge
Kim and coworkers [26] employed oxygen plasma glow discharge to produce peroxides on the surface of poly (ethylene terephthalate) that catalyzed the polymerization of acrylic acid, affording carboxyl-containing PET (PET-AA). Grafting of PEO, followed by reaction with insulin and then heparin yielded the insulin-heparin co-immobilized PET. PET-PEO film was immersed in an aqueous solution of insulin with carboxylic group activation using EDC. Heparin was similarly immobilized by immersing carbodiimide activated PET-PEO film in a sodium citrate buffer solution containing heparin. In both insulin and heparin immobilizations, the contents were maintained at 4°C for 24 h followed by washing with PBS and 0.1% Triton X-100 aq. soln. in an ultrasonic cleaner for 5 min. This step was essential to remove the physically adsorbed insulin and heparin. The amount of immobilized insulin and immobilized heparin were determined using the Coomassie brilliant blue G-250 interaction method [14] and toluidene blue method [27] (Fig. 15). These insulin heparin immobilized PET films were then characterized by attenuated total reflectron FTIR, electron spectroscopy for chemical analysis and a contact angle goniometry. Only 1.8% of the carboxylic acid groups of grafted AA participated in the reaction with the amino groups of PEO. Further, due to the high molecular weight of PEO and steric hindrance of the grafted PEO, only a small percentage of PEOs participated in the reaction with the carboxyl groups on the surfaces. Bioassays such as thrombus formation, plasma recalcification time (PRT), APTT, adhesion and activation of platelets were performed on these samples.
Fig. (15).

Overall process of the immobilization of insulin and /or heparin on PET.
Thrombus formation test, PRT and APTT assays were performed using the previously reported methods [28, 29]. For adhesion and activation of platelets analysis, the citrated blood was centrifuged to obtain the platelet-rich plasma (PRP). PRP was placed on the surface-modified PETs and incubated at 37 °C for 30 and 60 min. PBS was then added to PRP in order to arrest the further platelet adhesion. Finally, the number of platelets adhering to the surface was determined by measuring the lactate dehydrogenase (LDH) activity of cells lysed with Triton X-100 [30-32]. LDH activity was correlated to platelet by counting in a hemocytometer (Fig. 16). The amount of thrombus formed on glass was taken as the standard (100%). PET control and PET-AA resulted in 75% thrombus formation. The introduction of PEO or insulin reduced thrombus formation to 55%. As expected, the introduction of heparin decreased the thrombus formation (28%). The co-immobilization of heparin and insulin reduced thrombus formation to 30%. Plasma recalcification time on surface modified films was compared to glass surface (Fig. 17). Unmodified PET showed prolonged PRT, which increased slightly in the cases of PET-PEO and PET-In. For PET-Hep and PET-I-H surfaces, PRT was significantly prolonged.
Fig. (16).
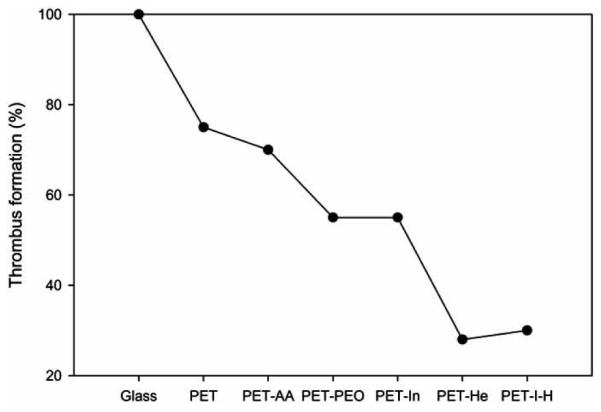
The amount of thrombus formed on PETs with various surface modifications after 30 min incubation (The value corresponding to glass was taken as 100%).
Fig. (17).
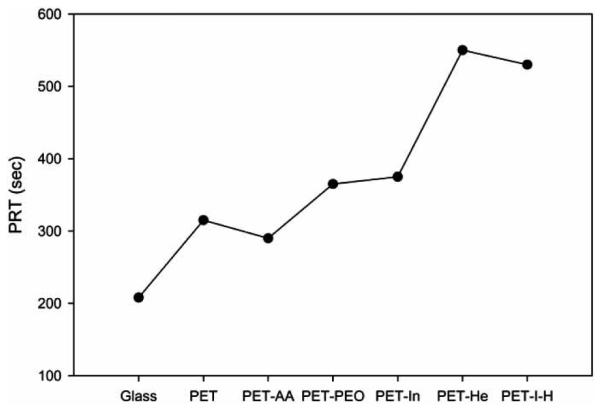
Plasma recalcification time as a function of surface-modified PETs.
PET-AA, PET-PEO and PET-In did not show a significant prolongation in APTT (Fig. 18). However, PET-Hep and PET-I-H showed remarkable increase in the APTT (58 and 55 s respectively) suggesting that the thrombin activity had been suppressed by the binding of antithrombin-III with the immobilized heparin.
Fig. (18).

APTT as a function of surface modifications.
The plot of number of platelets adhering to the surfaces showed that platelet adhesion increased with increased incubation times (Fig. 19). The number was lower for the PET-PEO and PET-In compared to the plain PET film. The number was remarkably decreased when the film contained immobilized heparin and/or insulin. Serotonin release was determined as a function of surface modifications and the presence of imipramine (Fig. 20). For all the surfaces, in the presence of imipramine, the amount of serotonin released was slightly higher when compared the corresponding values in the absence of imipramine. This observation demonstrates imipramine inhibits the re-uptake of released serotonin [32].
Fig. (19).

Amount of platelets adhered on surface modified PETs with the initial platelet concentration as 100. (Circle - 60 min; diamond - 30 min incubation time).
Fig. (20).

Serotonin released from platelets on PETs with various surface modifications (solid square: 30 min without imipramine; solid circle: 30 min with imipramine; empty square: 60 min without imipramine; solid circle: 60 min with imipramine).
Serotonin release increased in proportion to the increase in the number of adhering platelets. Serotonin released using PET accounted for 66% after 60 min incubation in the presence of imipramine. This further decreased with the introduction of PEO (54%), insulin (55%), and most significantly with heparin (36-39%). Salzman and coworkers [33] have reported an acceleration of serotonin release by a material with a lower platelet adhesion. Mori and coworkers have used PEO as a surface-immobilizing molecule to improve the blood compatibility of the polymers [34]. PETs immobilized with heparin and/or insulin, however, offered a better biocompatibility than the PEO grafted PETs in this study. Low molecular weight of PEO and the presence of positive amino group at the end of grafted PEO were believed to be the reasons for the reduced level of biocompatibility for PEO. The oxygen glow discharge technique to initiate the heparin immobilization had been previously employed by Bae and coworkers [15] to make heparinized polyurethanes.
3.9. Immobilization of Heparin to EDC/NHS-Crosslinked Collagen, Characterization and In Vitro Evaluation
Wissink and coworkers [35] reported the immobilization of heparin to the EDC/NHS (N-hydroxy succinimide) crosslinked collagen through carboxyl group activation by EDC/NHS. Collagen has been used as a hemostatic powder or sponge because of its high thrombogenic property [36], Collagen also induces platelet adhesion and activation of intrinsic blood coagulation. Wissink and coworkers tried to improve the blood compatibility of collagen by immobilizing heparin onto it. Collagen films were first crosslinked using EDC and NHS. 0.05 M 2-morpholinoethane sulfonic acid (MES buffer, pH 5.40) was used during the crosslinking to prevent the hydrolysis of EDC. Collagen films were incubated in EDC/NHS in MES buffer solution (1.731 g EDC; 0.415 g NHS in MES buffer solution per gram of collagen) for 4 h followed by the washing of the films with phosphate buffer and water. Heparin sodium salt was tritiated by treating NaB3H4 to heparin dissolved in distilled water (pH 8.0 adjusted by NaOH) for 3 h. Tritiated heparin was purified by dialysis (PBS buffer) followed by lyophilization. The carboxyl groups of the tritiated heparin were then activated by using EDC/NHS. After this activation step, crosslinked collagen was treated with EDC/NHS activated heparin solution for 2 h followed by a washing with 0.1 M sodium phosphate buffer, 4 M NaCl and water. The optimum conditions for the immobilization of heparin were determined to be 10 min - heparin pre-activation time, 2 h - immobilization time, pH 5.60, 2 % heparin solution, a variable molar ratio of EDC-Hep-carboxyl group of 0 to 2.0, and a fixed molar ratio of NHS to EDC of 0.6. Thrombin tests were performed on the surface modified collagen as a function of EDC: Hep-carboxyl group ratio to study the anticoagulant activity of heparin. The optimum EDC: Hep-carboxyl group ratio for the highest anticoagulant activity (~3.6 mU/cm2) was found to be 0.2, while the anticoagulant activity of the nonheparinized, EDC-NHS crosslinked collagen was only 0.25 mU/cm2. Thus, the thrombogenic property of the crosslinked collagen was improved by heparinization. The EDC/NHS crosslinked collagen was also investigated in vivo for its biocompatibility [37]. Heparin-collagen immobilization resulted in variable fibrin formation at day 1 and 2. The elevated levels of vascularization, found in the first 3 weeks, was explained as due to the entrapment of heparin binding growth factors present in the tissue surroundings by the collagen-heparin matrices.
3.10. Preparation and Characterization of A Poly(2-Hydroxyethyl Methacrylate) Biomedical Gel
Duncan and coworkers [38] enhanced the biocompatibility of p(HEMA) by the immobilization of heparin onto the surface in both heterogeneous and homogeneous phases. For the heterogeneous phase reaction, p(HEMA) beads of uniform size distribution (600 μm) were obtained by crushing the p(HEMA) films and passing through a sieve. Glycerol, p(HEMA) beads and heparin were then added to magnesium chloride solution and the contents were mixed for 30 min. Glutaraldehyde (0.5-2 w/v %) was used as a linker for heparin immobilization. The reaction contents were stirred for 24 h. The heparinized p(HEMA) was then obtained by vacuum filtration and washed with distilled water. A homogeneous phase reaction was carried out in dimethyl sulfoxide (DMSO). Magnesium chloride and heparin were added to 10 % p(HEMA) solution in DMSO, and stirred for 30 min, glutaraldehyde (0.5-5 %) was added and the reaction contents were stirred overnight. Heparinized p(HEMA) was obtained by precipitating the DMSO solution slowly in water at room temperature (Fig. 21).
Fig. (21).
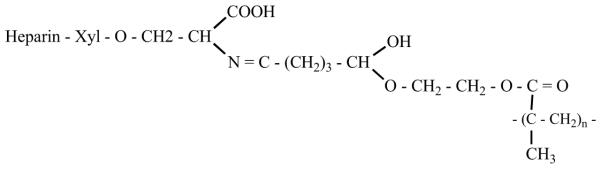
Heparinized PHEMA hydrogel.
The amounts of heparin immobilized by both the methods were determined using toluidine assay [12]. The amount of heparin immobilized by the homogeneous phase reaction was found to be greater than in the heterogeneous phase reaction. The anticoagulant activity of the immobilized heparin was determined by using TT and modified APTT studies. TT and APTT, proportional to the amount of immobilized heparin, showed greater anticoagulant activity for the p(HEMA) gel heparinized by homogeneous phase reaction. Thus, bioactive p(HEMA) gels were prepared by immobilizing heparin by using GA as linker.
3.11. Effects of Heparin Immobilization on the Surface Characteristics of A Biological Tissue Fixed with A Naturally Occurring Crosslinking Agent (Genipin)
An enhanced hydrophilicity, surface tension and the amount of platelet adhesion has been observed by employing the heparinization of the biological tissues [39]. Tsai and coworkers immobilized heparin on biological tissues by various following approaches such as ionic bonding, multipoint covalent bonding and an end-point covalent attachment (Fig. 22) using GA or genipin (GP) as linkers for this immobilization.
Fig. (22).

Schematic representation of the various approaches of heparin immobilization on biological surfaces (a) tissue without heparinization (b) tissue with ionically bound heparin (c) tissue with covalently bound heparin via multi point attachment (d) tissue with covalently bound heparin via end point attachment.
The non-heparinized tissue was used as a control; the pericardium obtained was fixed in 0.625 % GA or GP solution (PBS, pH 7.4) at room temperature for 3 days affording GA-fixed or GP fixed non-heparinized tissue (GA-NH or GP-NP). The tissue with ionically bound heparin (GA-HI or GP-HI) was prepared [40] by first immersing the porcine pericardia in 2.1% protamine sulfate solution for 1 h, followed by fixing with GA or GP solutions followed by a treatment with sodium heparin. The tissue with covalently bound heparin through multi-point attachment (GA-HM or GP-HM) were prepared [41] by treating the pericardia with GA or GP solution as mentioned in ionic bonding, followed by a thorough rinsing and the treatment with sodium heparin for 24 h and subsequently with EDC for 4 h. The pH was adjusted to 5.0 by adding HCl. The heparinized tissues were then fixed with GA or GP solution again for 3 days. The same method [25] used for making heparinized PU was followed to make the tissues with covalently bound heparin through end-point attachment (GA-HE or GP-HE). Blood compatibility studies such as plasma protein adsorption (Table 5) and the platelet adhesion (Table 6) studies were performed using these heparinized surfaces. Heparinization of GA and GP fixed tissue increased their hydrophilicity and reduced their mole ratio of adsorbed fibrinogen to adsorbed albumin and also the amount of adhered platelets. The cellular compatibility of the GP-fixed tissues with or without the immobilization of heparin was greater than the corresponding GA-fixed tissues.
Table 5.
The Study of Competitive Adsorption of Protein Mixtures–Molar Ratio of Adsorbed Fibrinogen to Adsorbed Albumin
| Sample | Fibrinogen/Albumin Mole Ratio |
|---|---|
| GA | 0.28 ± 0.04 |
| GA-HI | 0.20 ± 0.01 |
| GA-HM | 0.22 ± 0.02 |
| GA-HE | 0.14 ± 0.02 |
| GP | 0.26 ± 0.02 |
| GP-HI | 0.21 ± 0.02 |
| GP-HM | 0.23 ± 0.01 |
| GP-HE | 0.18 ± 0.004 |
Table 6.
Amount of Platelets Adhered on the Test Tissue Sample
| Sample | Amount of platelet adhered |
|---|---|
| GA | 312 ± 24 |
| GA-HI | 177 ± 10 |
| GA-HM | 211 ± 6 |
| GA-HE | 185 ± 10 |
| GP | 313 ± 22 |
| GP-HI | 184 ± 14 |
| GP-HM | 219 ± 17 |
| GP-HE | 181 ± 10 |
3.12. Surface Modification and Blood Compatibility of Polyacrylonitrile Membrane with Immobilized Chitosan-Heparin Conjugate
The blood compatibility of polyacrylonitrile (PAN) polymers is insufficient in spite of their very good biocompatibility [42], necessitating the systemic or intravenous administration of heparin during hemodialysis. Yang and Lin prepared heparinized PAN membranes through carboxyl groups (by hydrolysis of PAN using NaOH) and chitosan [43] (Fig. 23). The PAN polymer was first made into flat sheets by mixing 15% (w/w) PAN and 2.5% (w/w) poly (vinylpyrrolidone) in dimethyl formamide at 80 °C for 2 h followed by casting on a glass plate to form a flat membrane. After subsequent washing with water, PAN membrane was incubated in 100 ml of aqueous NaOH solution at 40 °C to introduce the carboxyl groups on the PAN surface (PAN-A). A maximum surface density of carboxyl groups of 88 nmol/cm2 was achieved at 10 min of incubation time.
Fig. (23).

Scheme for the heparinization of PAN through two approaches.
Heparinization of PAN was done using two approaches (1) through EDC linking as mentioned earlier and (2) through chitosan oligosaccharides. In the second approach, the EDC coupled PAN (PAN-EDC) was immersed in phosphate buffer solution pH 7.4 containing 1% (w/w) chitosan at 4 °C for 24 h to make the chitosan coupled PAN (PAN-C) which was then treated with the linker, 0.5% glutaraldehyde solution for 30 min at 50 °C. The resulting membrane was then treated with 20 ml heparin solution (20 IU/ml) in citric buffer pH 4.0 at 50 °C for 2 h to afford the heparinized PAN surface (PAN-C-H). The blood compatibility of the heparinized PAN surfaces was measured by APTT studies (Table 7). The APTT of the plain PAN was almost similar to that of the glass surface. PAN-C-H showed elongated APTT time compared to PAN-A-H, because of higher surface density of heparin present on the PAN-C-H (3.41 μg/cm2) compared to that of PAN-A-H (2.52 μg/cm2). The stability of heparin measured by monitoring the release kinetics of heparin was higher in the case of PAN-C-H than in PAN-A-H.
Table 7.
APTT Measurements of the PAN Surfaces
| Membrane | APTT (s) |
|---|---|
| Glass | 35 ± 2 |
| PAN | 38 ± 3 |
| PAN-A | 44 ± 4 |
| PAN-C | 37 ± 2 |
| PAN-A-H | 84 ± 4 |
| PAN-C-H | 172 ± 7 |
3.13. Covalent Immobilization of Chitosan/Heparin Complex with A Photosensitive Hetero-Bifunctional Crosslinking Reagent on PLA Surface
Chitosan was used as a linker to form heparinized poly (lactic acid) (PLA) surfaces. Zhu and coworkers used 4-azidobenzoic acid, a photosensitive hetero-bifunctional crosslinking reagent to couple chitosan with PLA [44]. Chitosan, EDC and 4-azidobenzoic acid were stirred with N,N,N’,N’-tetramethylethylene diamine (TEMED) in methanol at room temperature for 72 h and filtered to yield 4-azido-benzoic acid bonded chitosan (Az-CS). The powder was then washed with methanol, dried and dissolved on 1% aq. acetic acid/methoxyethanol solution. This solution was cast on a PLA film and dried in a dark dessicator, followed by an irradiation by Hg lamp for 1 min. The surface was then washed with 1% aq. acetic acid, 0.05%NaOH and water and dried (PLA-CS). This film was then immersed in heparin solution (acetate buffer pH 4.5) for 24 h, washed with water and dried to afford heparinized PLA surfaces (PLA-C-H). All these PLA surfaces were then subjected to L929 cell adhesion studies (Table 8). The number of cells adhered increased after chitosan coating and also with subsequent heparinization. This might probably because of the enhanced hydrophilicity of the PLA surface upon chitosan and heparin immobilization.
Table 8.
Number of L929 Cells Adhered on Various PLA Surfaces
| Sample | No. of Cells After 24 h (× 104) |
|---|---|
| PLA | 1.06 ± 0.2 |
| PLA-C | 2.08 ± 0.1 |
| PLA-C-H | 3.68 ± 0.3 |
3.14. Physicochemical and Blood Compatibility Characterization of Heparin Functionalized Polypyrrole Surface
The electrically conductive polypyrrole (PPY) film was heparinized by covalent linkage through cyanuric chloride (CC) activation (Fig. 24) [45]. PPY film was first treated with plasma glow discharge (35 W, Ar pressure 0.6 torr, oscillator frequency 40 kHz) for 10 s. The plasma treated PPY films were then exposed to air to enhance the formation of surface oxides and peroxides. The films were then treated with poly(ethyleneglycol) methacrylate (PEGMA) solutions of concentrations from 0.5 to 10 % (v/v) in water, followed by an exposure to UV radiation for 60 min at 28 °C. The grafted films were water washed at 50 °C for 24 h to remove unreacted monomer and homopolymer to afford PEGMA- PPY film. PEGMA- PPY film was then stirred in NaOH at 0 °C to 4 °C. Ice cold acetone containing 2 g of CC was added dropwise to the above contents and the pH was adjusted to 7.0 by Na2CO3. After 5 h, the film was washed with acetone and water and dried under vacuum (PPY-PEGMA-CC). PPY-PEGMA-CC was then immersed in 3 mg/ml heparin solution in formamide for 3 days. In this approach, heparinization of PPY occurred through the coupling of hydroxyl or amine groups of heparin with the chloride of CC on the PPY surface. The film was then washed thoroughly with acetone and water to remove the unreacted and weakly bound heparin from the surface to afford PPY-PEGMA-CC-heparin. The pristine and the surface modified PPY films were then subjected to PRT studies (Table 9).
Fig. (24).

Heparinization of PPY through PEGMA and CC.
Table 9.
PRT Determination of the Pristine and the Surface Modified PPY Films
| Sample | PRT (s) |
|---|---|
| Glass | 200 |
| PPY-pristine | 270 |
| PPY-CC | 236 |
| PPY-Heparin | 500 |
Heparinization of PPY significantly elongated the PRT (500 s) when compared to the PRT of the pristine PPY (270 s). The introduction of CC, however, decreased the PRT implying that CC activates the coagulation cascade easily. PPY film along with its intrinsic good electrically conductive properties has also good biocompatibility upon heparinization.
3.15. Protein Adsorption and Platelet Adhesion of Polysulfone Membrane Immobilized with Chitosan and Heparin Conjugate
The blood compatibility of polysulfone (PS) membranes was enhanced by the immobilization of heparin through chitosan linkers. Yang and Lin performed [46] this heparinization by a couple of approaches: first directly on the PS surface through acrylic acid linker, second through acrylic acid and chitosan linkers (Fig. 25).
Fig. (25).

Heparinization of polysulfone with/without chitosan linkers.
A casting solution containing 20 % (w/w) PS, 2.5 % (w/w) polyvinylpyrrolidone and 77 % (w/w) N,N-dimethylacetamide was cast as a film on a glass plate. The film was then washed with water for 12 h and dried under vacuum at 50 °C for 6 h. The films were then treated with air containing 3 g/m3 of ozone for various time periods. The ozone treated films were vacuum degassed for 5 min to remove physically adsorbed ozone. The film (2×2 cm2) was then treated with aqueous acrylic acid solution containing FeSO4 (to prevent the auto-polymerization of acrylic acid) at 65 °C for 2 h followed by sonication with 0.1 % (w/w) sodium dodecyl sulfate for 15 min to form PS-A. PS-A was immersed in 20 ml of 0.01 M EDC at 4 °C for 24 h followed by sonication in water to afford PS-A-EDC. PS-A-EDC was then incubated with 0.1 % (w/w) chitosan in 1 % (v/v) acetic acid at 4 °C for 24 h. Chitosan of various molecular weights (1170 (CSO), 160000 (C16), 400000 (C40)) were used in this step. Heparin was immobilized on PS-A (first approach) by immersing PS-A in citric buffer containing 0.01 M EDC at 4 °C for 24 h followed by treating with 10 IU/ml of heparin in citric buffer at 4 °C for 24 h. In the second approach, PS-A-Chitosan was treated with phosphate buffer (pH 7.4) containing 0.5 % GA for 30 min followed by 10 IU/ml of heparin for 2 h. In both the approaches, after the immobilization, the films were washed with concentrated salt solution to remove the physically and ionically bound heparin followed by vacuum drying at 40 °C for 12 h. APTT and platelet adhesion studies were performed in order to evaluate the surface properties of heparinized PS film (Table 10). Platelet adhesion studies were performed with the platelet rich plasma (PRP) of human whole blood. The platelet adhesion (%) was calculated by the equation platelet adhesion (%) = 100 × (n0-nt)/no, where no and nt are the platelet counts before and after contacting the film. In APTT study, the untreated and the ozone PS films behaved similar to the negative control in platelet poor plasma (PPP). Heparinization of the PS film prolonged the APTT, however, with the chitosan linker, the enhancement was better. Higher the chitosan MW, the longer APTT was prolonged, suggesting the better exposure of heparin with longer linkers. The percentage of platelets adhered was also less with the heparinized surfaces, less with longer linkers, in consistent with the APTT studies.
Table 10.
Results of APTT and Platelet Adhesion Studies
| Film | APTT (s) | Platelet Adhered (%) |
|---|---|---|
| PS | 37 ± 3 | 24.5 ± 2.9 |
| PS-A-H | 51 ± 4 | 21.3 ± 3.4 |
| PS-CSO | 41 ± 5 | 23.6 ± 2.1 |
| PS-CSO-H | 79 ± 7 | 19.6 ± 3.3 |
| PS-C16 | 39 ± 3 | 29.2 ± 3.0 |
| PS-C16-H | 218 ± 11 | 16.8 ± 3.5 |
| PS-C40 | 40 ± 5 | 36.7 ± 4.4 |
| PS-C40-H | 435 ± 19 | 14.6 ± 2.6 |
3.16. Simple Method for Immobilization of Bio-Macromolecules Onto Membranes of Different Types
Immobilization of heparin onto various surfaces was performed also through a combination of electrostatic interactions [47]. In this approach, a polyelectrolyte layer was adsorbed on an oppositely charged membrane by electrostatic interaction, resulting to an inversion of the original charges on the membrane. The immobilization of heparin to this charge-modified membrane became straightforward by electrostatic interactions. AN69 and PSU-SPSU (polysulfone of Udel-sulfonated polysulfone) membranes were heparinized by this approach using polyethyleneimine (PEI) as the electrolyte. The first step involved immersing the membrane in a 0.5 % (w/w) PEI solution (pH 8.5-10) for 10 min followed by a thorough washing with water. The final step involved the treating of this PEI coated membrane with an aqueous solution of heparin (5 mg/ml) for 30 min at room temperature. Adsorption of heparin on to the membranes was successfully confirmed by dye binding assays with anionic Red Ponceau S and cationic Brilliant Green dyes at various steps of the immobilization process. The anticoagulant activity of heparin followed by measuring the cephalin-kaolin time and PT, however, was found not significant enough to conclude the presence of heparin in its bioactive form. This absence of anticoagulant activity might probably be due to either an ineffective way of immobilization or inactivation of heparin upon immobilization.
3.17. Covalent Immobilization of Heparin on PLGA Surface
Poly(lactic acid-co-glycolic acid) (PLGA) is known for its excellent biodegradability, good mechanical properties and a degradation time comparable to the natural healing time of the damaged human tissues. Its poor blood-compatibility can be enhanced by heparinization making it a very good choice as a biomaterial for the making of artificial organs. Wang and coworkers achieved the immobilization of heparin on PLGA surfaces through EDC/NHS coupling (Fig. 26) [48]. PLGA film was prepared by pouring a PLGA solution (75:25) in chloroform on a watch glass and drying the chloroform over a current a dry air for 24 h. Thus obtained PLGA film was cross linked with chitosan (0.2 %) in EDC/NHS/MES for 4 h. Heparinization of chitosan cross-linked PLGA surface was achieved by immersing in 1 % heparin solution for 4 h followed by extensive washing with water.
Fig. (26).

Covalent immobilization of heparin on PLGA surface by EDC/NHS coupling.
The bioactivity of immobilized heparin was then evaluated by platelet adhesion and hepatocyte culturing studies. Platelet adhesion studies aided by scanning electron microscopy confirmed the enhancement of the blood compatibility of the PLGA surfaces upon the immobilization of heparin. Heparinization resulted in almost no platelet adhesion on the PLGA surface when compared to the controls (unmodified and chitosan linked PLGA surfaces). Hepatocytes (obtained from adult rat) seeded on chitosan-linked and heparinized-PLGA surfaces showed excellent proliferation compared with the unmodified PLGA surfaces. The number of cells stayed significantly high untill the completion of the experiment in the cases of chitosan-linked and heparinized surfaces, confirming the presence of immobilized heparin in its native bioactive state.
3.18. Hemocompatibility of Polyacrylonitrile Dialysis Membrane Immobilized with Chitosan and Heparin Conjugate
Polyacrylonitrile (PAN) membrane was coated with chitosan (CS)/heparin (HEP) polyelectrolyte complex through covalent attachment in order to make the PAN membrane useful for renal dialysis with reduced immunogenic effects [49]. The scheme for the covalent attachment of the CS/HEP complex with the PAN membrane is shown in Fig. 27. PAN membrane was prepared by membrane casting method, briefly by casting a polymer solution (15 % PAN in DMF) on a glass plate and immersing in deionized water to form a flat membrane. These membranes were then treated with 1 M NaOH for 10 min to convert the cyanide groups to carboxylic groups. The direct heparinization on the PAN membrane (PAN-H) was achieved through EDC in citric buffer (pH 4.8) (Fig. 27 (A)). In another approach, carboxylic-bearing PAN membranes were treated with EDC (in citric buffer solution), followed by CS (0.25 mg/ml in 1 % acetic acid solution) to afford chitosan immobilized PAN (PAN-C).
Fig. (27).

Covalent attachment of chitosan/heparin complex to polyacrylonitrile membrane
PAN-C was then immersed in glutaraldehyde (GA) for 30 min followed by washing with PBS and water. Finally, heparin (1000 IU/ml, PBS solution) was treated with GA-treated PAN-C at 4 C for 30 min. These membranes were then subjected to Soxhlet extraction to remove residual GA, and to afford the CS-HEP conjugated PAN membrane (PAN-C-H).
These two types of surface modified PAN membranes were then evaluated for protein adsorption and platelet adhesion, metabolites permeation and anticoagulant activity. Water contact angle which is a measure of hydrophilicity of a surface increased with heparinization. PAN-C-H showed the highest hydrophilicity followed by PAN-H and then by PAN-C. Human serum albumin (HSA, Mw = 65,000) and human plasma fibrinogen (HPF, Mw = 341,000) were subjected to adsorption studies on the modified PAN surfaces (Fig. 28). The heparinized PAN surfaces (PAN-H and PAN-C-H) showed less plasma protein adsorption. PAN-C-H showed up to 38 % and 26 % reduction of plasma protein adsorption when compared to those of PAN. PAN-C-H showed less protein adsorption than that of PAN-H. The increased adsorption of other surfaces is explained by electrostatic interactions between proteins and surfaces. A similar trend was observed in the platelet adhesion studies as well.
Fig. (28).

Plasma Protein Adsorption on the surface modified PAN membranes (square: HPF; circle: HSA).
PAN-C membrane showed a slightly higher thrombus formation that that of the original PAN membrane, while heparinized PAN membranes reduced the thrombus formation by almost 2/3 of that of the original value. Blood coagulation studies including APTT and PRT showed the excellent blood compatibility of the heparinized membranes. PAN-H and PAN-C-H membranes showed APTT values of around 140 s and 200 s (no clotting) when compared to the 38 s of the original membrane. These studies proved the presence of bioactive heparin immobilized on the dialysis membranes, and thereby facilitating the use of PAN membranes in human dialysis treatment with enhanced blood compatibility.
3.19. Immobilization of Heparin on PVDF Membranes with Microporous Structures
Heparinized poly (vinylidene fluoride) (PVDF) was prepared [50] by Lin and coworkers through plasma induced polymerization of acrylic acid. This approach is similar to that of followed by Kim and coworkers [26]. The only difference in this approach was heparin was directly linked to poly (acrylic acid) through EDC rather than through PEO. In this work, PVDF membranes were prepared with varying surface porosities and used for immobilization of heparin. These heparinized membranes were able to inhibit platelet adhesion proving the enhanced blood compatibility of these membranes.
3.20. Immobilization of Heparin in Cellulose Matrices Using Ionic Liquids
Novel cellulose-based heparinized biocomposites, reported by Linhardt and coworkers, exploited the enhanced dissolution of polysaccharides in room temperature ionic liquids (RTILs) [51]. The ability of RTILs (e.g., 1-ethyl, 3-butylimidazolium benzoate) to dissolve heparin, heparan sulfate, chondroitin sulfate and hyaluronic acid was demonstrated in preliminary studies [52]. A second RTIL 1-butyl, 3-methylimidazolium chloride, reportedly dissolved unmodified cellulose on application of microwave irradiation [53]. By combining these RTILs, various blood compatible cellulose-based biomaterials were fabricated in the form of membranes [51] and fibers [54].
This simple approach involves mixing of two RTIL solutions each containing either cellulose or heparin and making a composite structure out of the mixed solution. A co-solvent ethanol was then used to dissolve both the RTILs leaving a composite containing heparin dispersed uniformly in cellulose matrix. A heparin-cellulose composite membrane prepared by this approach was found to have excellent blood compatible properties when compared to other polymeric surfaces that were heparinized through covalent bonding (Fig. 29).
Fig. (29).

Activated partial thromboplastin time values of celluloseheparin composite in comparison with some other heparinized polymeric surfaces.
The composite membrane was also evaluated for its potential use in kidney dialyzers through an equilibrium dialysis experiments on two substrates, urea and bovine serum albumin (BSA). Urea reached equilibrium within 60 min while BSA did not reach equilibrium for 45 h. This result suggests that the composite membrane has appropriate selectivity for use in kidney dialyzers. The presence of heparin in this composite membrane should avoid the requirement of systemic administration of heparin during renal dialysis, and cellulose will act as the dialyzer.
Cellulose-based blood-compatible fibers were also fabricated from the same system using electrospinning technique [54]. Electrospinning technique involves exposing a drop of solution containing a polymer dissolved in a volatile solvent to a high voltage (~ 15 - 20 KV). Cellulose-heparin fibers formed by this approach were found to prolong the clotting time of human whole blood as measured by Thromboelastography (TEG). TEG measures the overall clotting kinetics of human whole blood. These composite fibers had superior blood compatible properties when compared to mere cellulose fibers. Heparin, a biomolecule, stayed active even after exposure to this high voltage.
3.21. Heparinized Carbon Nanotubes
Various nanomaterials including metallic nanoparticles, dendrimers, carbon nanotubes (CNTs) and fullerenes, and polymeric nanoparticles are being proposed for biomedical in vivo applications including drug delivery matrices, artificial implants, and diagnostic and imaging probes. Linhardt and coworkers introduced an approach to immobilize heparin on to nanomaterials to address blood compatibility issues [55]. As a model study, heparinized carbon nanotubes were prepared, and their blood compatible properties were evaluated. Heparinization involved three simple steps, firstly a coating of CNTs with an amino-polymer, such as PEI. This coating relies on hydrophobic interaction to introduce free amino groups on the surface of the CNTs. Second, the hydroxyl groups present in heparin were activated using cyanogen bromide (Fig. 30) [7]. In the final step, the activated heparin was conjugated to the PEI-coated CNTs. The amount of heparin loaded on to the CNTs through this approach was approximately 30(w/w) as measured by carbazole assay. The blood compatibility of these heparinized CNTs was then evaluated using APTT (Fig. 31).
Fig. (30).

Schematic representation of the activation of heparin towards free amino groups.
Fig. (31).

APTT measurements of modified CNTs (crossed bar: No CNTs; empty bar: Pristine CNTs; black bar: PEI-CNTs; grey bar: Hep-PEI-CNTs).
Pristine CNTs yielded APTT values similar to the control (no CNTs). PEI-CNTs, however, prolonged the APTT slightly probably because of their increased hydrophilicity upon PEI coating. Heparinized CNTs significantly improved the APTT values, demonstrating that heparin retained its anticoagulant activity even after its immobilization on to CNTs. These heparinized CNTs are proposed as potential building blocks for constructing nanodevices meant for in vivo biomedical applications including targeted drug delivery, artificial implants, and imaging and diagnostics.
4. CONCLUSIONS
Heparin, a major biomolecular drug, has great significance in regulating many biological pathways including cell-cell recognition, signal transduction, growth processes, coagulation cascade and the cellular interaction of growth factors. The half-life of heparin in the body is too short for many of these potential applications. Heparin’s pharmacokinetics and pharmacodynamics can be improved by attaching it to solid supports, reducing the rate of its metabolic breakdown. This review has examined various immobilization approaches that have proven useful in incorporating heparin on to a variety of solid supports useful in biomedical applications. These immobilization techniques and the wide range of solid supports (from polymers to nanomaterials) have been examined. The types of supports and immobilization chemistries have been evolving with the rapid changes taking place in the cutting edge technologies driving new biological and biomedical applications. The future of heparinized biomaterials in medicine and medical devices is ensured by these developments.
ABBREVIATIONS
- GAG
Glycosaminoglycans
- LMW
Low molecular weight
- DVT
Deep venous thrombosis
- AT-III
Antithrombin III
- EVAL
Ethylenevinyl alcohol
- APC
Adipoyl chloride
- HMDI
Hexamethylene diisocyanate
- PBS
Phosphate buffered saline
- TEPA
Tetraethylene-pentamine
- p(HEMA)
Poly(2-hydroxyethyl) methacrylate
- AC
Acrolyl chloride
- ATR/FTIR
Attenuated total reflection/ Fourier transform infrared
- DED
N,N-diethylethylenediamine
- DEDQ
Quarternized DED
- APTT
Activated partial thromboplastin time
- PU
Polyurethane
- AB
Acryloylbenzotriazole
- EDC
1-Ethyl-3-dimethylaminopropyl carbodiimide
- Hep
Heparin
- AA
Acrylic acid
- MA
Methyl acrylate
- PEO
Poly(ethylene oxide)
- MMA
Methyl methacrylate
- DMAEMA
Dimethylaminoethylmethacrylate
- EGDMA
Ethylene glycol dimethacrylate
- AIBN
2,2’-Azobisisobutyronitrile
- CT
Coagulation time
- PT
Prothrombin time
- HA
Hyaluronic acid
- RFGD
Radio frequency glow disharges
- PE
Poly(ethylene)
- PE-CVD
Plasma enhanced chemical vapor deposition
- TT
Thrombin time
- PEG
Poly(ethylene glycol)
- p(DMAPAAm)
dPoly(N-[3-(dimethylamino) propyl] acrylamide)
- p(DMAAm)
Poly(N, N-dimethylacrylamide)
- PTFE
(Poly(tetrafluoroethylene)
- GA
Glutaraldehyde
- CL
Collagen-laminin
- DLC
Diamond-like carbon
- PET
Poly (ethylene terephthalate)
- PRT
Plasma recalcification time
- PRP
Platelet-rich plasma
- LDH
Lactate dehydrogenase
- MES
2-Morpholinoethane sulfonic acid
- NHS
N-hydroxy succinimide
- DMSO
Dimethyl sulfoxide
- GP
Genipin
- PAN
Polyacrylonitrile
- PLA
Poly(lactic acid)
- TEMED
N,N,N’,N’-tetramethylethylene diamine
- Az-CS
4-Azidobenzoic acid bonded chitosan
- PPY
Polypyrrole
- CC
Cyanuric chloride
- PEGMA
Poly(ethyleneglycol) methacrylate
- PS
Polysulfone
- PPP
Platelet poor plasma
- PSU-SPSU
Polysulfone of Udel-sulfonated polysulfone
- PEI
Polyethyleneimine
- PLGA
Poly(lactic acid-co-glycolic acid)
- HSA
Human serum albumin
- HPF
Human plasma fibrinogen
- PVDF
Poly(vinylidene fluoride)
- RTIL
Room temperature ionic liquid
- BSA
Bovine serum albumin
- TEG
Thromboelastography
- CNT
Carbon nanotube
5. REFERENCES
- [1].Rydberg E, Westfall MJ, Nicholas RA. Low Molecular weight heparin in preventing and treating DVT. Am. Fam. Physician. 1999:1607. [PubMed] [Google Scholar]
- [2].Sanchez J, Elgue G, Riesenfeld J, Olsson P. Studies of adsorption, activation, and inhibition of factor XII on immobilized heparin. Thromb. Res. 1998;89:41–50. doi: 10.1016/s0049-3848(97)00310-1. [DOI] [PubMed] [Google Scholar]
- [3].Jackson RL, Busch SJ, Cardin ADW. Physiol. Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- [4].Danishefsky I, Tzeng F, Ahrens M, Klien S. Thromb. Res. 1976;8:131–140. doi: 10.1016/0049-3848(76)90256-5. [DOI] [PubMed] [Google Scholar]
- [5].Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- [6].Linhardt RJ. Perspective: 2003 Claude S. Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity. J. Med. Chem. 2003;46:2551–2554. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- [7].Yafuso M, Linhardt RJ. 1996. US Patent 5583213.
- [8].Mulloy B, Forster MJ, Jones C, Davies DB. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993;293:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Islam T, Butler M, Sikkander SA, Toida T, Linhardt RJ. Further evidence that periodate cleavage of heparin occurs primarily through the antithrombin binding site. Carbohydr. Res. 2002;337:2239–2243. doi: 10.1016/s0008-6215(02)00229-x. [DOI] [PubMed] [Google Scholar]
- [10].Sgouras D, Duncan R. Methods for the evaluation of biocompatibility of soluble synthetic polymers which have potential for biomedical use: 1-use of the tetrazolium-based colorimetric assay as a preliminary screen for evaluation of in-vitro cytotoxicity. J. Mater. Sci. Mater. Med. 1990;1:61–68. [Google Scholar]
- [11].Marconi W, Benvenuti F, Piozzi A. Covalent bonding of heparin to a vinyl copolymer for biomedical applications. Biomaterials. 1997;18:885–890. doi: 10.1016/s0142-9612(97)00015-x. [DOI] [PubMed] [Google Scholar]
- [12].Marconi W, Barontini P, Martinelli A, Piozzi A. New polyurethane compositions containing high amounts of covalently bonded heparin. Makromol. Chem. 1993;194:1347. [Google Scholar]
- [13].Kang IK, Kwon OH, Lee YM, Sung YK. Preparation and surface characterization of functional group-grafted and heparin-immobilized polyurethanes by plasma glow discharge. Biomaterials. 1996;17:841–847. doi: 10.1016/0142-9612(96)81422-0. [DOI] [PubMed] [Google Scholar]
- [14].Kang I-K, Kwon BK, Lee JH, Lee HB. Immobilization of proteins on poly(methyl methacrylate) films. Biomaterials. 1993;14:787–792. doi: 10.1016/0142-9612(93)90045-4. [DOI] [PubMed] [Google Scholar]
- [15].Bae J-S, Seo E-J, Kang I-K. Synthesis and characterization of heparinized polyurethanes using plasma glow discharge. Biomaterials. 1999;20:529–537. doi: 10.1016/s0142-9612(98)00204-x. [DOI] [PubMed] [Google Scholar]
- [16].Kang I-K, Baek BK, Lee YM, Sung YK. Synthesis and Surface characterization of heparin-immobilized polyurethanes. J. Polym. Sci. Polym. Chem. 1998;36:2331–2338. [Google Scholar]
- [17].Denizli A. Heparin-immobilized poly(2-hydroxyethyl methacrylate)-based microspheres. J. Appl. Polym. Sci. 1999;74:655–662. [Google Scholar]
- [18].Doumas BR, Watson W, Biggs H. Albumin standards and the measurement of serum albumin with bromocresol green. Clin. Chem. Acta. 1971;31:87. doi: 10.1016/0009-8981(71)90365-2. [DOI] [PubMed] [Google Scholar]
- [19].Lagergren M, Olsson P, Swedenborg J. Inhibited platelet adhesion: a non-thrombogenic characteristic of a heparin-coated surface. J. Surgery. 1974;75:643. [PubMed] [Google Scholar]
- [20].Favia P, Palumbo F, D'Agostino R, Lamponi S, Magnani A, Barbucci R. Immobilization of heparin and highly-sulfated hyaluronic acid onto plasma-treated polyethylene. Plasmas Polym. 1998;3:77–96. [Google Scholar]
- [21].Favia P, Stendardo MV, d' Agostino R. Selective grafting of amine groups on polyethylene by means of NH3-H2 RF glow discharges. Plasmas Polym. 1996;1:91. [Google Scholar]
- [22].Nakayama Y, Matsuda T. Surface Macromolecular Microarchitecture Design: Biocompatible Surfaces via Photo-Block-Graft-Copolymerization Using N,N-Diethyldithiocarbamate. Langmuir. 1999;15:5560–5566. [Google Scholar]
- [23].Chandy T, Das GS, Wilson RF, Rao GHR. Use of plasma glow for surface-engineering biomolecules to enhance blood compatibility of Dacron and PTFE vascular prosthesis. Biomaterials. 2000;21:699–712. doi: 10.1016/s0142-9612(99)00231-8. [DOI] [PubMed] [Google Scholar]
- [24].Steffen HJ, Schmidt J, Gonzalez-Elipe A. Biocompatible surfaces by immobilization of heparin on diamond-like carbon films deposited on various substrates. Surface and Interface Analysis. 2000;29:386–391. [Google Scholar]
- [25].Larm O, Larsson R, Olsson P. A new non-thrombogenic surface prepared by selective covalent binding of heparin via a modified reducing terminal residue. Biomater. Med. Devices Artif. Organs. 1983;11:161–173. doi: 10.3109/10731198309118804. [DOI] [PubMed] [Google Scholar]
- [26].Kim YJ, Kang IK, Huh MW, Yoon SC. Surface characterization and in vitro blood compatibility of poly(ethylene terephthalate) immobilized with insulin and/or heparin using plasma glow discharge. Biomaterials. 2000;21:121–130. doi: 10.1016/s0142-9612(99)00137-4. [DOI] [PubMed] [Google Scholar]
- [27].Kang I-K, Kwon OH, Lee YM, Sung YK. Preparation and surface characterization of functional group-grafted and heparin-immobilized polyurethanes by plasma glow discharge. Biomaterials. 1996;17:841–847. doi: 10.1016/0142-9612(96)81422-0. [DOI] [PubMed] [Google Scholar]
- [28].Kang IK, Kwon OH, Kim MK, Lee YM, Sung YK. In vitro blood compatibility of functional group-grafted and heparin-immobilized polyurethanes prepared by plasma glow discharge. Biomaterials. 1997;18:1099–1107. doi: 10.1016/s0142-9612(97)00035-5. [DOI] [PubMed] [Google Scholar]
- [29].Rhodes NP, Williams DF. Plasma recalcification as a measure of contact phase activation and heparinization efficacy after contact with biomaterials. Biomaterials. 1994;15:35–37. doi: 10.1016/0142-9612(94)90194-5. [DOI] [PubMed] [Google Scholar]
- [30].Rubin K, Kjellan L, Obrink B. Intercellular adhesion between juvenile liver cells. Exp. Cell. Res. 1977;109:413–422. doi: 10.1016/0014-4827(77)90021-0. [DOI] [PubMed] [Google Scholar]
- [31].Kang IK, Ito Y, Sisido M, Imanishi Y. Serotonin and β-thromboglobulin release reaction from platelet as triggered by interaction with polypeptide derivatives. J. Biomed. Mater. Res. 1988;22:595–611. doi: 10.1002/jbm.820220702. [DOI] [PubMed] [Google Scholar]
- [32].Park SD, Kang IK, Kim KH, Lee YM, Sung YK. Synthesis and physical properties of biocompatible and biodegradable polyetherurethaneurea: II. In vitro blood compatibility of poly etherurethaneurea containing polydimethylsiloxane segments. Polymer (Korea) 1994;18:868–876. [Google Scholar]
- [33].Salzman EW, Rosenberg RD, Smith MH, Lindon J, NFavreau L. Effect of heparin and heparin fractions on platelet aggregation. J. Clin. Invest. 1980;65:64–73. doi: 10.1172/JCI109661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mori Y, Nagaoka S, Takiuchi H, Kikuchi T, Noguchi N, Tanzawa H, Noishiki Y. A new antithrombogenic material with long polyethylene oxide chains. Trans. Am. Soc. Artif. Intern. Organs. 1982;28:459–463. [PubMed] [Google Scholar]
- [35].Wissink MJ, Beernink R, Pieper JS, Poot AA, Engbers GH, Beugeling T, van Aken WG, Feijen J. Immobilization of heparin to EDC/NHS-crosslinked collagen. Characterization and in vitro evaluation. Biomaterials. 2001;22:151–163. doi: 10.1016/s0142-9612(00)00164-2. [DOI] [PubMed] [Google Scholar]
- [36].Stemberger A, Ascherl R, Blumel G. Kollagen, ein Biomaterial in der Medizin. Hamostaseologie. 1990;10:164–176. [Google Scholar]
- [37].Van Wachem PB, Plantinga JA, Wissink MJB, Beernink R, Poot AA, Engbers GHM, Beugeling T, Van Aken WG, Feijen J, Van Luyn MJA. In vivo biocompatibility of carbodiimide-crosslinked collagen matrices: effects of crosslink density, heparin immobilization, and bFGF loading. J. Biomed. Mater. Res. 2001;55:368–378. doi: 10.1002/1097-4636(20010605)55:3<368::aid-jbm1025>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- [38].Duncan AC, Boughner D, Campbell G, Wan WK. Pre-paration and characterization of a poly(2-hydroxyethyl methacrylate) biomedical hydrogel. Eur. Polym. J. 2001;37:1821–1826. [Google Scholar]
- [39].Tsai CC, Chang Y, Sung HW, Hsu JC, Chen CN. Effects of heparin immobilization on the surface characteristics of a biological tissue fixed with a naturally occurring crosslinking agent (genipin): an in vitro study. Biomaterials. 2001;22:523–533. doi: 10.1016/s0142-9612(00)00206-4. [DOI] [PubMed] [Google Scholar]
- [40].Noishiki Y, Miyata T. A Simple method to heparinize biological materials. J. Biomed. Mater. Res. 1986;20:337–346. doi: 10.1002/jbm.820200306. [DOI] [PubMed] [Google Scholar]
- [41].Nimni ME, Cheung D, Strates B, Kodama B, Sheikh K. Bioprosthesis derived from cross-linked and chemically modified collagen tissues. Collagen. 1988;III:1–38. [Google Scholar]
- [42].Cases A, Reverter JC, Escolar G, Sanz C, Lopez-Pedret J, Revert L, Ordinas A. Platelet activation on hemodialysis: influence of dialysis membranes. Kidney Intl. Suppl. 1993;41:S217–220. [PubMed] [Google Scholar]
- [43].Yang MC, Lin WC. Surface modification and blood compatibility of polyacrylonitrile membrane with immobilized chitosan-heparin conjugate. J. Polym. Res. 2002;9:201–206. [Google Scholar]
- [44].Zhu A, Zhang M, Wu J, Shen J. Covalent Immobilization of chitosan/heparin complex with a photosensitive hetero-bifunctional crosslinking reagent on PLA surface. Biomaterials. 2002;23:4657–4665. doi: 10.1016/s0142-9612(02)00215-6. [DOI] [PubMed] [Google Scholar]
- [45].Li Y, Neoh Koon G, Cen L, Kang E-T. Physicochemical and blood compatibility characterization of polypyrrole surface functionalized with heparin. Biotechnol. Bioeng. 2003;84:305–313. doi: 10.1002/bit.10757. [DOI] [PubMed] [Google Scholar]
- [46].Yang MC, Lin WC. Protein adsorption and platelet adhesion of polysulfone membrane immobilized with chitosan and heparin conjugate. Polym. Adv. Technol. 2003;14:103–113. [Google Scholar]
- [47].Nguyen QT, Ping Z, Nguyen T, Rigal P. Simple method for immobilization of bio-macromolecules onto membranes of different types. J. Memb. Sci. 2003;213:85–95. [Google Scholar]
- [48].Wang XH, Li DP, Wang WJ, Feng QL, Cui FZ, Xu YX, Song XH. Covalent immobilization of chitosan and heparin on PLGA surface. Int. J. Biol. Macromol. 2003;33:95–100. doi: 10.1016/s0141-8130(03)00072-2. [DOI] [PubMed] [Google Scholar]
- [49].Lin W-C, Liu T-Y, Yang M-C. Hemocompatibility of polyacrylonitrile dialysis membrane immobilized with chitosan and heparin conjugate. Biomaterials. 2004;25:1947–1957. doi: 10.1016/j.biomaterials.2003.08.027. [DOI] [PubMed] [Google Scholar]
- [50].Lin D-J, Lin D-T, Young T-H, Huang F-M, Chen C-C, Cheng L-P. Immobilization of heparin on PVDF membranes with microporous structures. J. Memb. Sci. 2004;245:137–146. [Google Scholar]
- [51].Murugesan S, Mousa S, Vijayarghavan A, Ajayan PM, Linhardt RJ. Ionic liquid-derived blood-compatible composite membranes for kidney dialysis. J. Biomed. Mater. Res. B: Applied Biomaterials. 2006;79B:298–304. doi: 10.1002/jbm.b.30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Murugesan S, Wiencek JM, Ren RX, Linhardt RJ. Benzoate-based room temperature ionic liquids-thermal properties and glycosaminoglycan dissolution. Carbohydr. Polym. 2006;63:268–271. [Google Scholar]
- [53].Swatloski RP, Spear SK, Holbrey JD, Rogers RD. Dissolution of cellulose with Ionic Liquids. J. Am. Chem. Soc. 2002;124:4974–4975. doi: 10.1021/ja025790m. [DOI] [PubMed] [Google Scholar]
- [54].Viswanathan G, Murugesan S, Pushparaj V, Nalamasu O, Ajayan PM, Linhardt RJ. Preparation of Biopolymer Fibers by Electrospinning from Room Temperature Ionic Liquids. Biomacromolecules. 2006;7:415–418. doi: 10.1021/bm050837s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Murugesan S, Park T-J, Yang H, Mousa S, Linhardt RJ. Blood compatible carbon nanotubes - Nano-based Neoproteoglycans. Langmuir. 2006;22:3461–3463. doi: 10.1021/la0534468. [DOI] [PMC free article] [PubMed] [Google Scholar]